

Abstract
Introduction
Transforming growth factor (TGF)-β suppresses breast cancer formation by preventing cell cycle progression in mammary epithelial cells (MECs). During the course of mammary tumorigenesis, genetic and epigenetic changes negate the cytostatic actions of TGF-β, thus enabling TGF-β to promote the acquisition and development of metastatic phenotypes. The molecular mechanisms underlying this conversion of TGF-β function remain poorly understood but may involve signaling inputs from integrins.
Methods
β3 Integrin expression or function in MECs was manipulated by retroviral transduction of active or inactive β3 integrins, or by transient transfection of small interfering RNA (siRNA) against β3 integrin. Altered proliferation, invasion, and epithelial-mesenchymal transition (EMT) stimulated by TGF-β in control and β3 integrin manipulated MECs was determined. Src involvement in β3 integrin mediated alterations in TGF-β signaling was assessed by performing Src protein kinase assays, and by interdicting Src function pharmacologically and genetically.
Results
TGF-β stimulation induced αvβ3 integrin expression in a manner that coincided with EMT in MECs. Introduction of siRNA against β3 integrin blocked its induction by TGF-β and prevented TGF-β stimulation of EMT in MECs. β3 integrin interacted physically with the TGF-β receptor (TβR) type II, thereby enhancing TGF-β stimulation of mitogen-activated protein kinases (MAPKs), and of Smad2/3-mediated gene transcription in MECs. Formation of β3 integrin:TβR-II complexes blocked TGF-β mediated growth arrest and increased TGF-β mediated invasion and EMT. Dual β3 integrin:TβR-II activation induced tyrosine phosphorylation of TβR-II, a phosphotransferase reaction mediated by Src in vitro. Inhibiting Src activity in MECs prevented the ability of β3 integrin to induce TβR-II tyrosine phosphorylation, MAPK activation, and EMT stimulated by TGF-β. Lastly, wild-type and D119A β3 integrin expression enhanced and abolished, respectively, TGF-β stimulation of invasion in human breast cancer cells.
Conclusion
We show that β3 integrin alters TGF-β signaling in MECs via Src-mediated TβR-II tyrosine phosphorylation, which significantly enhanced the ability of TGF-β to induce EMT and invasion. Our findings suggest that β3 integrin interdiction strategies may represent an innovative approach to re-establishing TGF-β mediated tumor suppression in progressing human breast cancers.
Introduction
Transforming growth factor (TGF)-β is a powerful tumor suppressor that prevents the uncontrolled proliferation of epithelial, endothelial, and hematopoietic cells. In doing so, TGF-β initiates transmembrane signaling by activating its type I and type II serine/threonine kinase receptors (TGF-β receptor TβR-I and TβR-II, respectively). Following its transphosphorylation and stimulation by TβR-II, TβR-I then binds, phosphorylates, and activates the intracellular effectors Smad2 and Smad3, which subsequently complex with Smad4 and translocate to the nucleus to regulate target gene transcription [1]. Although the Smad pathway is by far the most characterized TGF-β activated pathway, TGF-β also governs cell physiology through activation of mitogen-activated protein kinases (MAPKs; including extracellular signal-regulated kinase [ERK]1/2, p38, and c-Jun amino-terminal kinase) and of phosphoinositol-3 kinase (PI3K) [2-5]. Aberrant activation of MAPKs and PI3K often is associated with cancer development in humans. Precisely how TGF-β activates these alternative pathways and how these signals are integrated into the biology and pathology of TGF-β remain to be elucidated fully [6].
TGF-β plays a dual role during mammary tumorigenesis [7-10]. For instance, TGF-β normally prohibits mammary epithelial cell (MEC) cell cycle progression, and consequently suppresses MEC tumorigenesis. However, during the course of mammary tumorigenesis, TGF-β signaling becomes dysregulated and uncoupled from regulation of cell cycle progression. More importantly, altered TGF-β signaling actively contributes to the acquisition and development of metastatic phenotypes, in part through its ability to stimulate epithelial-mesenchymal transitions (EMTs) in cancerous MECs [11]. Indeed, recent evidence suggests that TGF-β suppresses tumorigenesis largely via Smad2/3-mediated growth arrest [1,6,8,12,13], whereas its ability to promote tumorigenesis and EMT occurs via the integration of Smad2/3 signals with those arising in response to activation of RhoA, MAPKs (e.g. ERK1/2 and p38 MAPK), and PI3K pathways [2,3,14-16]. Thus, breast cancer cells have developed effective strategies for circumventing the tumor suppressing activities of TGF-β, while simultaneously selecting for, or even enhancing, its tumor promoting activities [8,17].
Apart from playing a prominent role in preventing cell cycle progression, TGF-β also is a major regulator of cell microenvironments and extracellular matrix (ECM) remodeling. TGF-β alters cell microenvironments in part through its ability to induce the expression of unique subsets of integrins as well as that of their ECM ligands [18]. In doing so, TGF-β enables malignant cells to undergo EMT and, consequently, to escape their tissue of origin [11].
Integrins are heterodimeric transmembrane receptors that bind ECM ligands and couple cells to their microenvironments [19]. Most integrins initiate transmembrane signaling by activating focal adhesion kinase (FAK) and Src family kinases at adhesive sites. FAK and Src further recruit and activate various downstream effectors, such as PI3K and members of the Ras and Rho families of small GTPases [20-22]. Integrins also interact with and couple to receptor tyrosine kinases (RTKs) to promote cell survival, proliferation, and migration in response to soluble growth factors and cytokines [23,24]. One such integrin, namely αvβ3, binds to arginine-glycine-aspartic acid (RGD) amino acid containing components of the ECM, such as vitronectin, fibronectin, and osteopontin, and mediates RTK activation of MAPKs and cell invasion [23-25]. Similar to the effects of tumorigenesis on TGF-β signaling, integrin expression is altered during tumorigenesis, including developing tumors of the breast [26,27]. In particular, altered αvβ3 integrin expression correlates with mammary tumorigenesis, particularly the processes of breast cancer cell invasion and metastasis [25,27-31], raising the possibility that differential integrin expression may contribute to the tumor promoting activities of TGF-β. Indeed, TGF-β stimulated EMT is abrogated by treatments that inhibit MEC integrin adhesion, suggesting a need for integrins in mediating TGF-β signaling [32,33].
To further investigate the role of altered integrin expression in regulating the MEC response to TGF-β, we determined the effects of β3 integrin expression on the ability of TGF-β to regulate NMuMG (normal murine mammary gland) cell proliferation, invasion, and EMT. We found that treatment of NMuMG cells with TGF-β induced their expression of αvβ3 integrin, an event that coincided with TGF-β stimulation of EMT. Accordingly, β3 integrin deficiency abolished the ability of TGF-β to induce EMT in MECs. Moreover, we found that β3 integrin interacted physically with TβR-II at the cell surface, leading to conversion of TGF-β from a suppressor of NMuMG cell growth to a promoter of their invasiveness and EMT. Mechanistically, activated β3 integrin recruited Src to β3 integrin:TβR-II complexes, where it tyrosine phosphorylated TβR-II, leading to enhanced activation of MAPKs and induction of EMT stimulated by TGF-β. Importantly, abolishing Src activity or expression in NMuMG cells prevented β3 integrin-mediated tyrosine phosphorylation of TβR-II and, consequently, EMT stimulated by TGF-β. Finally, we found that the acquisition of a metastatic phenotype in MCF10A derivatives, which serve as a model of human breast cancer progression regulated by TGF-β [9], coincided with upregulated β3 integrin and FAK expression. Similar to its effects in NMuMG cells, β3 integrin expression significantly enhanced TGF-β mediated stimulation of cell invasion in benign MCF10A cells as well as in their highly metastatic counterparts MCF10CA1a cells. Importantly, the expression of D119A β3 integrin in these metastatic cells completely abolished the ability of TGF-β to induce MCF10CA1a cell invasion.
Taken together, our findings identify a novel convergence point in MECs that enables β3 integrins to override the tumor suppressing activities of TGF-β, suggesting that integrin interdiction strategies may one day represent an innovative approach to re-establishing TGF-β mediated tumor suppression in progressing human breast cancers.
Materials and methods
Retroviral plasmids and expression
The cDNAs encoding wild-type (WT) human β3 integrin, as well as its inactive mutant D119A (D→A point mutation at position 119), were generously provided by Dr Mark H Ginsberg (University of California at San Diego, San Diego, CA, USA [34]). Retroviral β3 integrin vectors were synthesized by PCR amplification using oligonucleotides containing BglII (amino terminus) and XhoI (carboxyl terminus) restriction sites, and subsequently ligated into identical sites immediately upstream of the IRES in the bicistronic retroviral vector pMSCV-IRES-GFP (plasmid murine stem cell virus-internal ribosomal entry site-green fluorescent protein) or pMSCV-IRES-YFP (yellow fluorescent protein). [35] All β3 integrin inserts were sequenced in their entirety on an Applied Biosystems 377A DNA sequencing machine (Applied Biosystems, Foster City, CA USA).
Full-length human c-Src cDNA was PCR amplified from IMAGE clone 4871614 (Invitrogen, Carlsbad, CA, USA) using oligonucleotides containing HindIII (amino terminus) and XbaI (carboxyl terminus) restriction sites, respectively. The resulting PCR product was ligated into corresponding sites in pcDNA3.1/Myc-His B vector (Invitrogen) to carboxyl-terminally tag c-Src with a Myc and (His)6-tag. Kinase dead (Lys295Met) or constitutively active (Tyr530Phe) c-Src mutants were generated by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). Afterward, Myc-His tagged WT and mutant c-Src cDNAs were amplified by PCR and ligated into EcoRI (amino terminus) and BglII (carboxyl terminus) restriction sites in pMSCV-IRES-GFP. All c-Src inserts sequenced in their entirety on an Applied Biosystems 377A DNA sequencing machine.
NMuMG cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS) and 10 μg/ml insulin. MCF10A and MCF10CA1a cells were cultured as previously described [9]. Stable expression of individual β3 integrin subunits or c-Src derivatives in NMuMG and MCF10A cells was accomplished by their overnight infection with control (i.e. pMSCV-IRES-GFP or pMSCV-IRES-YFP), WT or D119A β3 integrin, or mutant c-Src retroviral supernatants produced by EcoPac2 retroviral packaging cells (Clontech, San Diego, CA, USA), as described previously [36]. Cells expressing GFP, YFP, or both fluorescent proteins were isolated and collected 48 hours later on a MoFlo cell sorter (Cytomation, Fort Collins, CO, USA), and subsequently were expanded to yield stable polyclonal populations of control (i.e. GFP or YFP), β3 integrin (i.e. WT or D119A), or mutant c-Src (i.e. kinase-dead or constitutively active) expressing cells. Expression of recombinant β3 integrins in individual NMuMG cell lines was monitored by immunoblotting detergent solubilized whole cell extracts with antibodies against the extracellular domain of β3 integrin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), whereas expression of mutant c-Src protein kinases was detected by immunoblotting with either anti-Src (1:1000 dilution; Santa Cruz Biotechnology) or anti-Myc (1:1000 dilution; Covance, Princeton, New Jersey USA) antibodies.
Fluorescence-activated cell sorting analysis
Control (GFP), WT, or D119A β3 integrin expressing NMuMG cells were cultured in the absence or presence of TGF-β1 (5 ng/ml) for 36 hours to stimulate EMT. Afterward, 1 × 106 cells were trypsinized, washed, and incubated in fluorescence-activated cell sorter (FACS) buffer (1% bovine serum albumen in phosphate-buffered saline [PBS]) supplemented with a 1:20 dilution of either PE-conjugated anti-mouse αv integrin (BD Pharmingen, San Diego, CA, USA) or PE-conjugated anti-human β3 integrin antibodies (BD Pharmingen). After a 30 min incubation on ice, the cells were washed twice in PBS and immediately fixed with 1% paraformaldehyde before fluorescence-activated cell sorting (FACS) analysis of αv or β3 expression in GFP-positive NMuMG cells.
Immunofluorescence studies
The ability of TGF-β to alter actin cytoskeletal architecture was monitored essentially as described previously [37,38]. Briefly, control or β3 integrin expressing NMuMG cells (30,000 cells/well) were plated onto gelatin coated (0.1% in PBS) glass coverslips in 24-well plates. The cells were stimulated with TGF-β1 (5 ng/ml) for 0–36 hours at 37°C. In some experiments, control or β3 integrin-expressing NMuMG cells were stimulated with TGF-β1 in the absence or presence of the Src kinase inhibitor PP2 (10 μmol/l; Calbiochem, Temecula, CA, USA) or its inactive counterpart PP3 (10 μmol/l; Calbiochem). Upon completion of agonist stimulation, the cells were washed in PBS, fixed in 4% paraformaldehyde, and permeabilized by Triton X-100 (0.2% in PBS). The cells were then blocked in PBS supplemented with 1.5% FBS, followed by incubation with TRITC-phalloidin or FITC-phalloidin (0.25 μmol/l). For αvβ3 integrin staining, the cells were blocked in goat β-globulin (200 μg/ml; Jackson Immunoresearch, West Grove, PA, USA) before sequential incubations with anti-αvβ3 LM609 antibody (1:250 dilution; BD Biosciences, San Jose, CA USA), followed by biotinylated goat anti-mouse antibody (5 μg/ml; Jackson Immunoresearch) and finally by Alexa-streptavidin (1.2 μg/ml; Invitrogen). All images were captured on a Nikon Diaphot microscope.
RNA interference studies
NMuMG cells lacking either β3 integrin or c-Src were generated using SMARTpool small interfering RNAs (siRNAs), in accordance with the manufacturer's recommendations (Dharmacon, Lafayette, CO, USA). Briefly, NMuMG cells (30,000 cells/well) were plated either onto plastic or gelatin coated (0.1% in PBS) glass coverslips in 24-well plates and cultured overnight in antibiotic-free media. Fresh media was added the following morning and the cells were transiently transfected with DharmaFECT One reagent (Dharmacon) supplemented with β3 integrin or c-Src siRNAs (100 nmol/l). Thirty-six hours after transfection, the cells were treated with TGF-β1 (5 ng/ml) for varying times at 37°C. Upon completion of agonist stimulation, the cells were harvested and prepared for immunoblotting and immunofluorescence analyses as above. NMuMG cell transfection efficiency was monitored by co-transfection with siGLO RISC-Free siRNA (Dharmacon), which was visualized under fluorescent microscope.
Cellular phosphorylation assays
Control or β3 integrin-expressing NMuMG cells were cultured onto 24-well plates (30,000 cells/well) and allowed to adhere overnight. The next morning, the cells were washed twice in PBS and placed in Dulbecco's modified Eagle's medium supplemented with 0.5% FBS for 4 hours before stimulation with TGF-β1 (5 ng/ml) for 0–120 min. In some experiments, the cells were stimulated with Prolactin (100 ng/ml; generously provided by National Hormone and Peptide Program, US National Institutes of Health), 4α-phorbol 12-myristate 13-acetate (PMA; 10 ng/ml; Sigma, St. Louis, MO, USA), or epidermal growth factor (EGF; 100 ng/ml; Upstate, Charlottesville, VA). Afterward, the cells were washed in ice-cold PBS and lysed in 200 μl of buffer H/1% Triton X-100 [36]. Detergent solubilized whole cell extracts were prepared, clarified by microcentrifugation, and subsequently concentrated by acetone precipitation. Recovered proteins were fractionated through 10% SDS-PAGE gels, immobilized electrophoretically to nitrocellulose membranes, and subsequently probed with a 1:250 dilution of either anti-phospho-Smad2, -ERK1/2, or -p38 MAPK polyclonal antibodies (Cell Signaling Technology, Beverly, MA, USA). The resulting immunocomplexes were visualized by enhanced chemiluminescence. Differences in protein loading could not be readily monitored by b-actin immunoreactivity because stable β3 integrin expression and TGF-β stimulation significantly elevated b-actin expression in MECs (data not shown [5,32,39]). Thus, differences in protein loading were instead monitored by reprobing stripped membranes with anti-ERK1/2 antibodies (1:2500 dilution; Upstate, Charlottesville, VA USA), whose expression in MECs was unaltered by TGF-β treatment [5,32,39].
Cell biological assays
The effect of WT and D119A β3 integrin expression on various TGF-β stimulated activities in MECs was determined as follows: cell proliferation using 10,000 cells/well in a [3H]thymidine incorporation assay, as described elsewhere [35]; cell invasion induced by 10% serum using 350,000 cells/well in a modified Boyden-chamber coated with Matrigel matrices (diluted 1:25 in serum-free Dulbecco's modified Eagle's medium), as described elsewhere [35,40]; and gene expression using 30,000 cells/well in a synthetic pSBE-luciferase reporter gene assay, as described previously [35].
Iodinated transforming growth factor-β1 radioligand binding and cross-linking assay
Control and β3 integrin-expressing NMuMG cells were plated onto 10 cm plates and grown until they reached 90% confluency. The radioligand binding and cross-linking of [125I]TGF-β1 (200 pmol/l) to NMuMG cells was performed as described previously [41]. Afterward, cytokine:receptor complexes contained in detergent-solubilized whole cell extracts were isolated by immunoprecipitation with anti-TβR-II antibodies, as described elsewhere [41]. Immunocomplexes were subsequently fractionated through 7.5% SDS-PAGE and then immobilized electrophoretically to nitrocellulose and probed with anti-β3 integrin antibodies. Iodinated TGF-β1 bound to cell surface TβR-I and TβR-II was visualized by exposure of the dried nitrocellulose membranes to a phosphor screen, which was developed 1–3 days later on a Molecular Dynamics Typhoon Scanner (GE Healthcare Bio-Sciences Corp., Piscataway, NJ USA).
Integrin:TβR-II co-immunoprecipitation assays
Control (2.5 × 106) or β3 integrin (7 × 105) expressing NMuMG cells were cultured onto 10 cm plates and subsequently stimulated with TGF-β1 (5 ng/ml) for varying times in the absence or presence of the Src inhibitor PP2 (10 μmol/l). In some cases, NMuMG cells were held in suspension and replated onto culture dishes previously coated with vitronectin (1 ng/ml; Chemicon, Temecula, CA, USA). Following agonist stimulation, the cells were washed twice in ice-cold PBS and disrupted in Nonidet P-40 lysis buffer [42] (Nonidet P-40; Sigma). The resulting detergent-solubilized whole cell extracts were clarified by microcentrifugation and subjected to the following immunoprecipitation conditions: anti-β1 integrin antibodies (Santa Cruz Biotechnology) using 1 mg whole cell extract; anti-β3 integrin antibodies using 1 mg whole cell extract; anti-phosphotyrosine 4G10 antibodies (Upstate) using 1 mg whole cell extract; or anti-TβR-I or -TβR-II antibodies using 2 mg whole cell extract as described previously [43]. All immunoprecipitations were incubated for 16 hours at 4°C with slow rotation. The resulting immunocomplexes were collected by microcentrifugation, washed, fractionated through 10% SDS-PAGE gels, and transferred electrophoretically to nitrocellulose membranes, which subsequently were probed with anti-β3 integrin (1:1000), anti-TβR-II (1:1500; Santa Cruz Biotechnology), or anti-phosphotyrosine 4G10 (1:1500; Upstate) antibodies.
In vitro tyrosine kinase phosphorylation assays
Tyrosine phosphorylation of TβR-II was determined using an in vitro protein kinase assay that measured the ability of either active FAK or Src to phosphorylate recombinant glutathione S-transferase (GST)-TβR-II, which contained only the cytoplasmic domain (i.e. the carboxyl terminal 380 amino acids) of TβR-II fused to GST. Protein kinase reactions were performed in a final volume of 30 μl, consisting of 1 μg of GST-TβR-II with either 0.2 μg of FAK or 1 Unit of Src (Cell Signaling Technology). Reactions were initiated by addition of 10 μl of 4× assay buffer [44] and were incubated for 30 min at 30°C. Afterward, the reactions were stopped by addition of 4× sample buffer [44] and boiled for 5 min. Reactions were diluted by addition of 1 ml buffer H/Triton X-100 and immunoprecipitated overnight with anti-TβR-II antibodies. The resulting immunocomplexes were collected, washed, and resuspended in 1X sample buffer. GST fusion proteins were fractionated through 10% SDS-PAGE before their immobilization to nitrocellulose membranes. Tyrosine phosphorylation of GST-TβR-II was visualized by immunoblotting nitrocellulose membranes with anti-phosphotyrosine antibodies. Differences in immunoprecipitation efficiency and loading were monitored by reprobing stripped membranes with anti-TβR-II antibodies.
Recombinant GST-Smad3 phosphorylation assay
The phosphorylation of recombinant Smad3 by activated TβR complexes was performed essentially as described previously [45]. Briefly, quiescent NMuMG cells were pretreated with 10 μmol/l of either PP2 or SU6656 (EMD Biosciences, La Jolla, CA, USA) for 1 hour at 37°C, and subsequently were stimulated with TGF-β1 (5 ng/ml) for 30 min at 37°C. Cytokine stimulations were terminated by washing the cells twice in ice-cold PBS, which then were lysed and solubilized on ice in buffer H/1% Triton X-100. The resulting cell extracts (1 mg/tube) were clarified by microcentrifugation, and subsequently were immunoprecipitated with anti-TβR-II antibodies for 2 hours at 4°C. TβR immunocomplexes were recovered by brief centrifugation and subsequently were washed twice in PBS. TβR phosphotransferase activity against recombinant GST-Smad3 was measured for 30 min at 30°C in a final reaction volume of 40 μl consisting of 30 μl of TGF-β receptor complexes, 5 mg GST-Smad3, and 10 μl of 4X assay buffer [43]. Phosphotransferase reactions were stopped by the addition of 4X sample buffer and were boiled for 5 min before their fractionation through 10% SDS-PAGE. Fractionated proteins were immobilized electrophoretically to nitrocellulose membranes, which subsequently were probed with anti-phospho-Smad2/3 antibodies to visualize phosphorylated GST-Smad3. Differences in immunoprecipitation efficiency and loading were monitored by reprobing stripped membranes with anti-TβR-II antibodies.
Results
TGF-β1 mediated EMT increases cell surface αvβ3 integrin expression and β3 integrin:TβR-II complex formation in NMuMG cells
Alterations in cell microenvironments and stromal compartments play critical roles in regulating the progression of tumors from indolent to aggressive states [46,47]. Differential integrin expression [27] and dysregulated TGF-β signaling [48] both contribute to the remodeling of cancer cell microenvironments, particularly those of the breast. Because integrins regulate MEC response to TGF-β [32], we hypothesized that differential integrin expression induced by TGF-β would contribute to its regulation of MEC proliferation and EMT. To test this hypothesis, we stimulated normal mouse mammary epithelial (NMuMG) cells with TGF-β1 and monitored changes in actin stress fiber formation by immunofluorescence. As we have shown previously [38], resting NMuMG cells exhibited typical cuboidal epithelial cell morphology characterized by strong cortical acting staining (Figure 1a). When stimulated with TGF-β1, these cells underwent EMT and exhibited significant formation of actin stress fibers that emanated from focal adhesions (Figure 1a).
Figure 1.
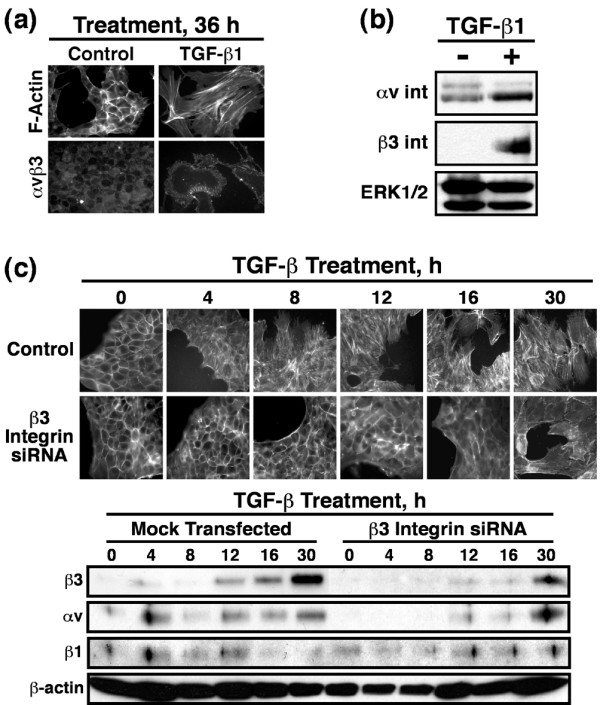
TGF-β1 mediated EMT increases cell surface αvβ3 integrin expression in NMuMG cells. NMuMG cells were incubated in the absence or presence of TGF-β1 (5 ng/ml) for 0–36 hours and assayed for EMT. (a) Alterations in actin cytoskeletal architecture were visualized by direct rhodamine-phalloidin immunofluorescence and αvβ3 integrin by indirect immunofluorescence using anti-αvβ3 integrin (LM609) antibodies. Shown are representative images from a single experiment that was repeated twice with identical results. (b) Detergent-solubilized whole cell extracts (50 μg/lane) were prepared to analyze expression of the αv and β3 integrin subunits by immunoblotting. Differences in protein loading were monitored by reprobing striped blots with anti-ERK1/2 antibodies because, unlike the increase in β-actin expression observed (data not shown) ERK1/2 expression was unaltered in NMuMG cells in response to TGF-β1 treatment. (c) β3 Integrin deficient NMuMG cells were generated by siRNA transfection. The effects of β3 integrin deficiency on TGF-β mediated EMT was visualized by direct FITC-phalloidin immunofluorescence. Shown are representative images from a single experiment that was repeated twice with identical results. Detergent-solubilized whole cell extracts (50 μg/lane) were prepared to analyze expression of the αv, β3, and β1 integrin subunits by immunoblotting. Differences in protein loading were monitored by reprobing striped blots with anti-β-actin antibodies. EMT, epithelial-mesenchymal transitions; ERK, extracellular signal-regulated kinase; siRNA, small interfering RNA; TGF, transforming growth factor.
Because αvβ3 integrin expression is elevated in breast cancers and enhances cancer cell invasion [27,30,31], we monitored the ability of TGF-β to alter αvβ3 integrin expression in NMuMG cells. Immunofluorescence analysis showed that TGF-β1 treatment of NMuMG cells significantly induced their cell surface expression of the αvβ3 integrin (Figure 1a). Western blot analysis confirmed the upregulation of αvβ3 integrin expression upon TGF-β1 mediated EMT (Figure 1b). The association of upregulated αvβ3 integrin expression with TGF-β stimulation of EMT suggested that αvβ3 integrins may play an essential role in facilitating EMT stimulated by TGF-β. We tested this hypothesis by transiently transfecting NMuMG cells with siRNAs directed against β3 integrin and subsequently monitored their ability to undergo EMT in response to TGF-β. Figure 1c shows that β3 integrin deficiency abolished the ability of TGF-β to stimulate EMT in NMuMG cells. More importantly, NMuMG cells transfected with β3 integrin siRNAs only exhibited rudimentary EMT characteristics coordinate with the expression of αvβ3 integrins, which exhibit significantly delayed expression in response to TGF-β (Figure 1c). Moreover, β3 integrin deficiency failed to alter NMuMG cell expression of β1 integrins (Figure 1c), indicating that signals arising from β1 integrin were insufficient in mediating EMT by TGF-β.
Collectively, these findings show that TGF-β induces EMT in NMuMG cells, and that upregulated expression of αvβ3 integrin is essential for this process to occur. The findings also raise the possibility that β3 integrins may play a direct role in regulating various MEC responses to TGF-β3.
Integrins regulate growth factor signaling in part via formation of integrin:RTK complexes [23,24]. Based on our findings presented in Figure 1, we hypothesized that β3 integrins interact physically with cell surface TβRs. In testing this hypothesis, NMuMG cells were stimulated with TGF-β for various periods of time, whereupon whole cell extracts were prepared and immunoprecipitated with anti-β3 integrin antibodies. Probing the resulting immunocomplexes with anti-TβR-II antibodies showed that β3 integrin did indeed interact with TβR-II (Figure 2a), and that EMT was necessary to induce this event. In addition, detergent-solubilized whole cell extracts prepared from control and TGF-β stimulated NMuMG cells were immunoprecipitated with antibodies against either β1 or β3 integrins, and the resulting immunocomplexes were probed for TβR-II. Figure 2b shows that β1 integrins interacted constitutively with TβR-II, whereas EMT was again required to promote the association of β3 integrin with TβR-II. Collectively, these findings indicate that the formation of β3 integrin:TβR-II complexes is an EMT-specific phenomena that may play an important role in regulating TGF-β stimulation of EMT in NMuMG cells.
Figure 2.
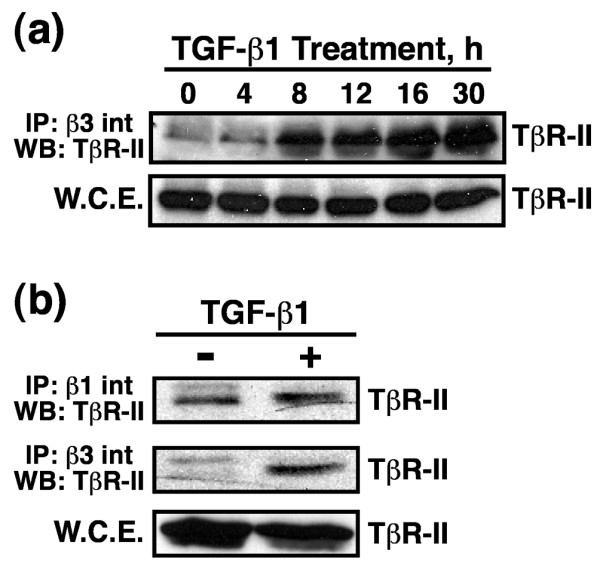
TGF-β1 mediated EMT induces β3 integrin:TβR-II complex formation on the cell surface of NMuMG cells. NMuMG cells were incubated in the absence or presence of TGF-β1 (5 ng/ml) for 0–30 hours to induce EMT. Detergent-solubilized whole cell extracts (1 mg/tube) were immunoprecipitated with (a,b) anti-β3 or (b) anti-β1 integrin antibodies, and subsequently immunoblotted with anti-TβR-II antibodies. Shown are representative immunoblots from a single experiment that was repeated twice with identical results. EMT, epithelial-mesenchymal transitions; TβR; TGF receptor; TGF, transforming growth factor; WCE, whole cell extracts.
β3 Integrin overexpression in NMuMG cells
To further explore the role of β3 integrin in regulating MEC function and TGF-β biology, we used bi-cistronic retroviral transduction to stably express β3 integrin or its inactive counterpart D119A-β3 integrin [34] in NMuMG cells. The D119A-β3 integrin contains a point mutation in the conserved RGD integrin binding motif and therefore fails to bind RGD-containing ligands, such as fibronectin, vitronectin, and osteopontin [49,50]. Recombinant β3 integrin expression in NMuMG cells was confirmed by immunoblotting whole cell extracts from basal or TGF-β1 treated cells (Figure 3a). As observed previously, TGF-β stimulation of EMT in NMuMG cells induced their expression of αv integrin (Figures 1 and 3a). Interestingly, WT β3 integrin expression induced a dramatic compensatory upregulation of αv integrin expression in NMuMG cells (Figure 3a). This β3 integrin response, which was independent of TGF-β administration, was mimicked moderately by expression of its inactive D119A counterpart (Figure 3a). Although D119A β3 integrin is considered to be functionally inactive, its heterologous expression in cells can elicit partial biologic activity in part through its ability to bind non-RGD-containing ligands, such as plasminogen [51]. Thus, these findings suggest that signals arising from activated β3 integrins induce αv integrin expression in MECs.
Figure 3.
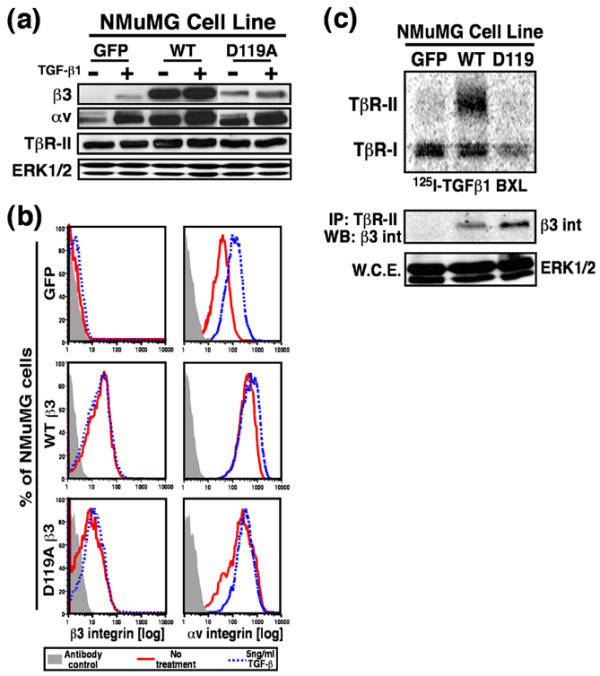
β3 Integrin overexpression in NMuMG cells. NMuMG cells were engineered by bi-cistronic retroviral transduction to stably express GFP alone (control), WT β3 integrin (WT) and GFP, or its inactive mutant D119A and GFP. (a) Detergent-solubilized whole cell extracts (50 μg/lane) were prepared to analyze expression of the αv and β3 integrin subunits, and of TβR-II. Differences in protein loading were monitored by immunoblotting ERK1/2, whose expression in NMuMG cells was not altered by TGF-β treatment. (b) The percentage of NMuMG cells expressing human recombinant β3 integrin or endogenous αv integrin was determined by FACS analysis of cells incubated with either anti-human β3 integrin or anti-mouse αv integrin antibodies. Histograms depict β3 and αv expression only in GFP-positive NMuMG cells and are from a representative experiment that was repeated twice with similar results. (c) Iodinated TGF-β1 was bound and chemically cross-linked to control (GFP), WT, and D119A β3 integrin expressing cells. Cytokine:receptor complexes were isolated by immunoprecipitation with anti-TβR-II antibodies, fractionated through 7.5% SDS-PAGE gels, and immobilized electrophoretically to nitrocellulose membranes. The upper panel shows a representative phosphor image of iodinated TGF-β1 bound to TβR-I and TβR-II. The middle panel shows that β3 integrin was present in isolated iodinated TGF-β1:receptor complexes as detected by anti-β3 integrin Western blot analysis of anti-TβR-II immunocomplexes. Differences in protein loading were monitored by probing detergent-solubilized whole cell extracts (50 μg/lane) with anti-ERK1/2 antibodies (bottom panel). BXL, binding and cross-linking; ERK, extracellular signal-regulated kinase; GFP, green fluorescent protein; WT, wild type; TβR, TGF receptor; TGF, transforming growth factor; WCE, whole cell extracts.
To confirm that recombinant β3 integrins were expressed on the cell surface of NMuMG cells, control or TGF-β1 treated cells were incubated with PE-conjugated anti-human β3 integrin antibodies, and subsequently analyzed by FACS analysis. As expected, retrovirally infected NMuMG cells expressed WT and D119A β3 integrins on their cell surfaces (Fig. 3b, left). In addition, β3 integrin expressing cells were also incubated with PE-conjugated anti-mouse αv integrin antibodies, which confirmed the compensatory upregulation of αv integrins and showed their ability to form functional complexes with cell surface β3 integrin (Figure 3b, right).
Finally, iodinated TGF-β1 radioligand binding and cross-linking assays were performed to confirm that β3 integrins did indeed interact with TβR-II at the cell surface. As shown in Figure 3c, WT and D119A β3 integrins both interacted with TβR-II at the cell surface, indicating that formation of this complex is independent of β3 integrin activation. Quite surprisingly, WT β3 integrin expression, but not that of D119A, increased cell surface expression of TβR-II (Figure 3c). Indeed, the ratio of TβR-II:TβR-I detected at the cell surface in WT β3 integrin expressing NMuMG cells (1.31 ± 0.14; n = 4) was significantly higher (P = 0.009, Student's t-test) than those observed in their GFP β3 integrin (0.76 ± 0.05; n = 4) and D119A β3 integrin (0.72 ± 0.31; n = 4) expressing counterparts. Interestingly, semiquantitative real-time PCR analyses failed to detect differences in TβR-II transcript expression between these individual NMuMG cell lines (data not shown), suggesting that signals arising from β3 integrins alter TβR-II translocation to and/or internalization from the cell surface of MECs.
Collectively, these findings show that retrovirally expressed human β3 integrins were translocated to the cell surface of NMuMG cells, where they formed active heterodimers with αv integrin, and more importantly they interacted physically with TβR-II at the plasma membrane. In addition, these findings and those presented in Figures 1 and 2 illustrate a unique and essential role for β3 integrin in promoting EMT, particularly that mediated by TGF-β, and suggest that the formation of β3 integrin:TβR-II complexes may alter the response of MECs to TGF-β by regulating its intracellular signaling systems.
β3 Integrin expression enhances MAPK activation by TGF-β in NMuMG cells
The coupling of integrins to RTKs often is associated with activation of MAPKs [23,52]. This is noteworthy because the activation of MAPKs is essential for the ability of TGF-β to promote EMT [2,15,16]. We therefore determined whether β3 integrin expression altered the ability of TGF-β to activate MAPKs in NMuMG cells. Although expression of β3 integrin in NMuMG cells had negligible effects on TGF-β stimulated Smad2 phosphorylation (Figure 4a) and nuclear translocation (data not shown), its expression significantly enhanced the activation of ERK1/2 and p38 MAPK by TGF-β (Figure 4a). Integrin expression can elicit dramatic changes in cell adhesion and morphology [23], potentially resulting in global alterations in intracellular signaling systems. To determine the specificity of β3 integrin expression to TGF-β signaling, we monitored the activation status of p38 MAPK in control and β3 integrin expressing NMuMG cells before and after their stimulation with TGF-β1, prolactin, PMA, or EGF. Figure 4b shows that β3 integrin expression selectively regulated the coupling of TGF-β, but not that of prolactin, PMA, or EGF, to p38 MAPK activation in NMuMG cells. Thus, these findings implicate β3 integrins as unique mediators of TGF-β signaling in MECs.
Figure 4.
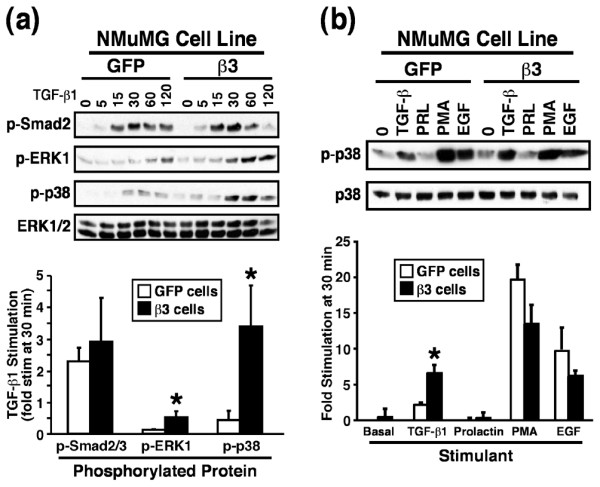
β3 Integrin enhances MAPK activation by TGF-β in NMuMG cells. (a) Control or β3 integrin expressing NMuMG cells were serum starved for 4 hours prior to TGF-β1 stimulation (5 ng/ml) for 0–120 min. Afterward, the activation status of Smad2, ERK1/2, and p38 MAPK was determined by immunoblot analysis using phospho-specific antibodies. Reprobing stripped membranes with anti-ERK1/2 antibodies monitored differences in protein loading. (b) Control or β3 integrin-expressing NMuMG cells were serum starved for 4 hours before stimulation with TGF-β1 (5 ng/ml), prolactin (100 ng/ml), PMA (10 ng/ml), or EGF (100 ng/ml) for 30 min. Afterward, the activation status of p38 MAPK was determined by immunoblot analysis using phospho-specific antibodies. Reprobing stripped membranes with anti-pan p38 MAPK antibodies was used to monitor differences in protein loading. Accompanying graphs depict the mean (± standard error) fold increase of p38 MAPK stimulation observed in three independent experiments (*P < 0.05). EGF, epidermal growth factor; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; PMA, 4α-phorbol 12-myristate 13-acetate; TGF, transforming growth factor.
β3 Integrin expression blocked TGF-β stimulated growth arrest but enhanced TGF-β mediated invasion and EMT in NMuMG cells
Based upon the ability of β3 integrins to enhance MAPK activation by TGF-β, we next sought to determine whether β3 integrins could alter the ability of TGF-β to regulate MEC cell proliferation, invasion, and EMT. We first measured the effects of β3 integrin expression on the rate of DNA synthesis in resting NMuMG cells. As compared with GFP expressing NMuMG cells, those expressing WT β3 integrin exhibited significantly increased rates of DNA synthesis (by 78 ± 0.12%; n = 3; P = 0.002), whereas those expressing D119A β3 integrin exhibited significantly retarded rates of DNA synthesis (by 68 ± 4.0%; n = 3; P = 0.00007). Despite the relative growth rate differences observed between these β3 integrin expressing NMuMG cell derivatives, the response of GFP and D119A expressing NMuMG cells to TGF-β mediated cytostasis was surprisingly similar (Figure 5a). In contrast, WT β3 integrin expression significantly reduced the ability of TGF-β to induce growth arrest in NMuMG cells (Figure 5a), indicating that β3 integrin expression significantly reduces cytostasis mediated by TGF-β, thereby circumventing its tumor suppressor function in MECs.
Figure 5.
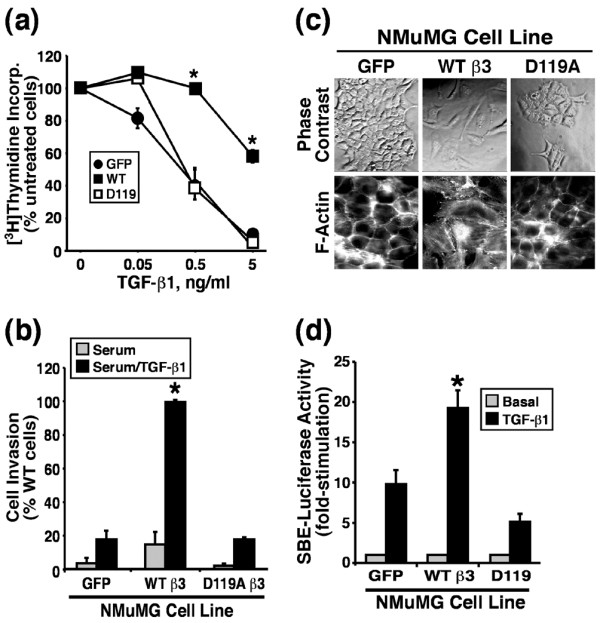
β3 Integrin expression blocks TGF-β stimulated growth arrest but enhances TGF-β stimulated invasion and EMT in NMuMG cells. (a) Control (GFP), WT, or D119A β3 integrin expressing NMuMG cells were stimulated with increasing concentrations of TGF-β1 for 48 hours. Cellular DNA was radiolabeled with [3H]thymidine and quantified by scintillation counting. Data are the means (± standard error) of three independent experiments presented as the percentage of [3H]thymidine incorporation normalized to untreated cells. (b) Control (GFP) and β3 integrin expressing NMuMG cells were allowed to invade through Matrigel matrices in the absence or presence of TGF-β1 (5 ng/ml) for 36 hours. Values are the mean (± standard error) of three independent experiments presented as the percentage invasion relative to TGF-β stimulated β3 integrin expressing NMuMG cells. (c) The cell morphology of control (GFP), WT, and D119A β3 integrin expressing NMuMG cells was monitored by phase contrast microscopy, and alterations in actin cytoskeletal architecture was visualized by direct rhodamine-phalloidin immunofluorescence. Shown are representative images from a single experiment that was repeated twice with identical results. (d) Control (GFP), WT, or D119A β3 integrin expressing NMuMG cells were transiently transfected with pSBE-luciferase and pCMV-β-gal cDNAs, and subsequently were stimulated with TGF-β1 (5 ng/ml) for 24 hours. Afterward, luciferase and β-gal activities contained in detergent-solubilized cell extracts were measured. Values are the mean (± standard error) luciferase activities observed in three independent experiments normalized to maximal reporter gene expression measured in unstimulated cells. EMT, epithelial-mesenchymal transitions; GFP, green fluorescent protein; TGF, transforming growth factor; WT, wild type; SBE, Smad binding element.
We also determined whether β3 integrins are capable of enhancing the tumor promoting functions of TGF-β. In doing so, we compared the ability of β3 integrin expressing NMuMG cell lines to invade through synthetic basement membranes. As shown in Figure 5b, GFP expressing NMuMG cells exhibited minimal invasion through Matrigel matrices in response to serum stimulation, a response that was enhanced insignificantly by TGF-β treatment. Similar to its inability to alter growth regulation by TGF-β, expression of the D119A β3 integrin in NMuMG cells failed to affect their TGF-β regulated invasiveness (Figure 5b). In contrast, NMuMG cells expressing β3 integrin exhibited a trend toward increased invasiveness to serum as compared with control cells (Figure 5b); however, when stimulated with TGF-β1 these cells exhibited significantly enhanced invasion through synthetic basement membranes (Figure 5b). Because increased invasiveness is often associated with EMT [11], we examined the morphology and actin cytoskeleton of NMuMG cells expressing β3 integrins. Consistent with our previous results, only those NMuMG cells that expressed WT β3 integrin acquired an elongated, fibroblast-like morphology (Figure 5c), complete with the formation of actin stress fibers (Figure 5c). Taken together, these findings suggest that although both WT and D119A β3 integrins can form complexes with TβR-II at the cell surface, only functional β3 integrins are capable of inducing EMT and invasion in NMuMG cells.
Lastly, we monitored changes in TGF-β stimulated gene expression by measuring differences in luciferase expression driven by the synthetic SBE (Smad binding element) promoter. Figure 5d shows that only WT β3 integrin expression significantly enhanced TGF-β stimulated gene expression as compared with control cells or those expressing its inactive β3 integrin counterpart.
Collectively, these findings indicate that, unlike its nonfunctional counterparts, expression of WT β3 integrin in MECs diminished TGF-β mediated growth arrest, enhanced TGF-β mediated invasion and EMT, and augmented TGF-β mediated gene expression. Moreover, our results suggest that β3 integrin expression translates TGF-β from an inhibitor of MEC cell growth to a stimulator of their invasion and EMT.
Dual receptor activation induces Src-mediated TβR-II tyrosine phosphorylation
Our findings thus far show that β3 integrin and TβR-II interact upon TGF-β stimulation, β3 integrin expression is essential for EMT stimulated by TGF-β, and β3 integrin expression influences the MEC response to TGF-β. To further explore the consequences of β3 integrin:TβR-II complex formation, GFP expressing NMuMG cells were incubated in the absence or presence of TGF-β for 36 hours, and subsequently were held in suspension before re-plating onto plastic or vitronectin-coated wells in the absence or presence of TGF-β1 for 5 min. Afterward, the phosphorylation status of TβR-II and its ability to form complexes with β3 integrin were monitored by co-immunoprecipitation analyses. Figure 6a shows that following TGF-β induced EMT, TβR-II was constitutively tyrosine phosphorylated and associated with β3 integrin. Because chronic TGF-β stimulation of NMuMG cells induces β3 integrin expression (Figure 1) and complex formation with TβR-II (Figure 2), it was difficult to assess the role of TGF-β receptor and integrin stimulation in the formation of β3 integrin:TβR-II complexes in NMuMG cells. To circumvent this difficulty, resting WT β3 integrin expressing NMuMG cells were dissociated and subsequently replated and stimulated for 5 min, as above. Figure 6b shows that TβR-II only interacted with β3 integrins upon TGF-β1 stimulation and, more importantly, that this response was enhanced significantly upon β3 integrin adhesion. In addition, only upon dual receptor activation (i.e. β3 integrin and TβRs) was TβR-II phosphorylated on tyrosine residues (Figure 6b). Thus, β3 integrin regulates TGF-β signaling by targeting TβR-II for tyrosine phosphorylation in a manner reminiscent of integrin actions on RTK signaling [23].
Figure 6.
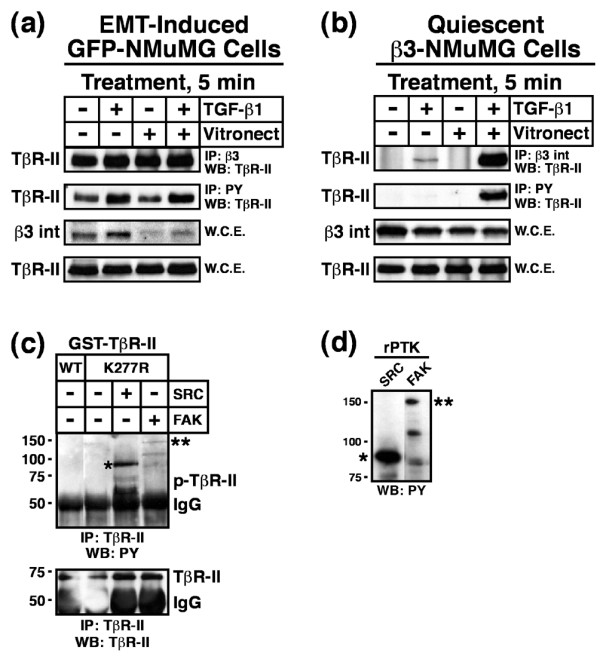
Dual receptor activation induces Src mediated TβR-II tyrosine phosphorylation. (a) GFP-expressing NMuMG cells were stimulated with TGF-β1 (5 ng/ml) for 36 hours to induce β3 integrin expression and EMT. Afterward, the cells were dissociated and re-plated either onto plastic or vitronectin-coated wells in the absence or presence of TGF-β1 (5 ng/ml) for 5 min. Afterward, detergent-solubilized whole cell extracts were prepared (1 mg/tube) and subsequently immunoprecipitated with anti-phosphotyrosine or anti-β3 integrin antibodies as indicated. The presence of TβR-II in precipitated immunocomplexes was determined by Western blotting with anti-TβR-II antibodies. Differences in protein loading were monitored by immunoblotting whole cell extracts (50 mg/lane) for β3 integrin and TβR-II. Shown are representative immunoblots from a single experiment that was repeated twice with similar results. (b) Resting WT β3 integrin-expressing NMuMG cells were dissociated and re-plated as above. Afterward, TβR-II tyrosine phosphorylation and complex formation with β3 integrin was determined by immunoblotting as above. Shown are representative immunoblots from a single experiment that was repeated three times with identical results. (c) Active recombinant Src (1 unit/reaction) or FAK (0.2 μg/reaction) kinases were added to protein kinase reaction mixtures containing 1 μg/tube of either kinase active (WT) or inactive (K277R) GST-TβR-II. Phosphorylation reactions were stopped after 30 min and the phosphorylation status of TβR-II was determined by immunoprecipitation of diluted protein kinase reaction mixtures with anti-TβR-II antibodies. Afterward, immobilized immunocomplexes were probed sequentially with anti-phosphotyrosine antibodies, followed by anti-TβR-II antibodies as indicated (*autophosphorylated recombinant Src; **autophosphorylated recombinant FAK). Data are from a representative experiment that was repeated three times with similar results. (d) Recombinant Src and FAK were incubated in protein kinase assay buffer for 30 min at 30°C. The data show the autophosphorylation on tyrosine residues of recombinant Src (*) and FAK (**) as determined by immunoblotting with anti-phosphotyrosine antibodies. Data are from a representative experiment that was repeated at least three times with similar results. EMT, epithelial-mesenchymal transitions; FAK, focal adhesion kinase; GST, glutathione S-transferase; TβR, TGF-β receptor; TGF, transforming growth factor; WCE, whole cell extracts; WT, wild type.
Although integrins lack intrinsic protein tyrosine kinase (PTK) activity, they facilitate tyrosine phosphorylation of target proteins via their recruitment of FAK and Src PTKs to focal adhesions [22]. We therefore hypothesized that FAK or Src are potential PTKs responsible for phosphorylating TβR-II on tyrosine residues in NMuMG cells. We tested this hypothesis by utilizing an in vitro PTK assay that measured the ability of purified, active FAK or Src to phosphorylate GST fusion proteins containing catalytically active or inactive (i.e. K277R) versions of the cytoplasmic domain of TβR-II. As shown in Figure 6c, Src phosphorylated the cytoplasmic domain of TβR-II on tyrosine residues. Although active and capable of undergoing autophosphorylation on tyrosine residues (Figure 6d), FAK was unable to phosphorylate TβR-II in vitro (Figure 6c), suggesting that Src phosphorylates TβR-II in response to β3 integrin expression.
To further examine the functional link between Src and TβR-II, we attempted to inhibit Src activity or function in NMuMG cells and subsequently monitor the consequences of these manipulations on TβR-II tyrosine phosphorylation. Recent work conducted by Maeda and coworkers [53] showed that the broad spectrum protein kinase inhibitor PP1 antagonizes TGF-β signaling by inhibiting TβR-I and TβR-II protein kinase activity. As such, we monitored the effects of two Src inhibitors, PP2 and SU6656, on the ability of immunoprecipitated TGF-β receptor complexes to phosphorylate recombinant GST-Smad3 in vitro. As expected, TGF-β stimulation greatly enhanced the phosphorylation of recombinant Smad3 (Figure 7a), a reaction that was reduced markedly by SU6656 administration (Figure 7a). In contrast, PP2 treatment failed to inhibit TβR activity and instead appeared to elicit agonist independent receptor activation (Figure 7a). Based on its inactivity against TβRs, PP2 appeared to be a suitable pharmacological agent to inhibit Src activity in MECs. Accordingly, β3 integrin-expressing NMuMG cells treated with PP2 abrogated the tyrosine phosphorylation of TβR-II in TGF-β stimulated NMuMG cells (Figure 7b), indicating that Src activity does indeed mediate tyrosine phosphorylation of TβR-II in β3 integrin expressing NMuMG cells. Along these lines, transient expression of constitutively active Src in β3 integrin expressing NMuMG cells resulted in significantly elevated tyrosine phosphorylation of TβR-II, an event that was not recapitulated by expression of dominant negative Src (Figure 7c). Taken together, these findings indicate that β3 integrin-mediated recruitment of Src facilitates the tyrosine phosphorylation of TβR-II in MECs, and that this phosphorylation event may act to enhance their proliferation, invasion, and EMT by augmenting the ability of TGF-β to activate MAPKs.
Figure 7.
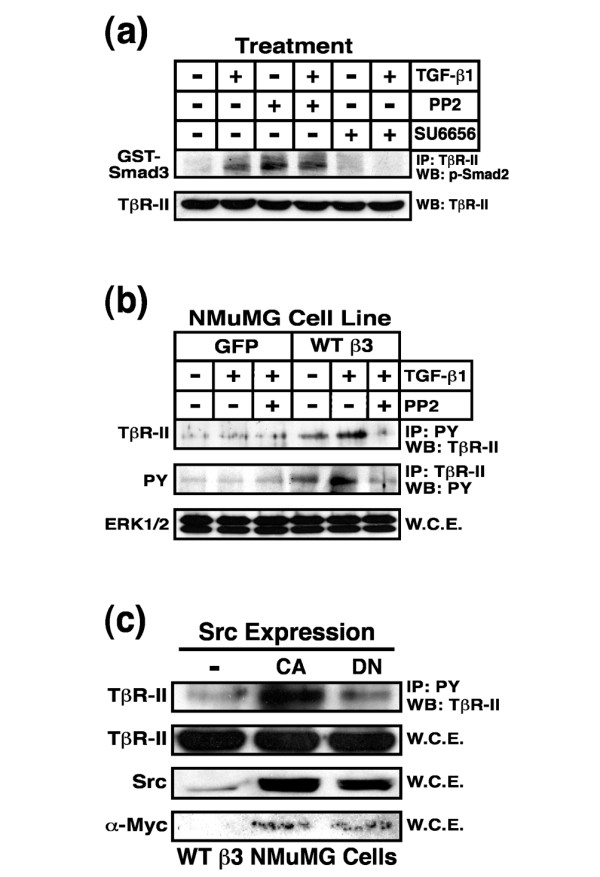
Src inhibition blocks TβR-II tyrosine phosphorylation in NMuMG cells. (a) NMuMG cells were pretreated for 60 min with either PP2 (10 μmol/l) or SU6656 (10 μmol/l). Afterward, the cells were stimulated with TGF-β1 (5 ng/ml) for 20 min, and subsequently were lysed and immunoprecipitated (1 mg/tube) with anti-TβR-II antibodies. The resulting immunocomplexes were used to phosphorylate recombinant GST-Smad3, which was visualized by immunoblotting with anti-phospho-Smad2 antibodies. Data are from a representative experiment that was repeated three times with similar results. (b) Control or β3 integrin expressing NMuMG cells were stimulated with TGF-β1 (5 ng/ml) in the absence or presence the Src inhibitor PP2 (10 μmol/l), as indicated. Afterward, detergent-solubilized whole cell extracts were prepared, immunoprecipitated, and subjected to Western blot analysis either with anti-phosphotyrosine (2 mg WCE/tube) or anti-TβR-II (1 mg WCE/tube) antibodies as indicated. Differences in protein loading were monitored by probing detergent-solubilized whole cell extracts (50 μg/lane) with anti-ERK1/2 antibodies (bottom panel). (c) WT β3 integrin expressing NMuMG cells were transiently transfected with cDNAs encoding either constitutively active (CA) or dominant negative (DN) Src. Afterward, detergent solubilized whole cell extracts were prepared, immunoprecipitated with anti-phosphotyrosine (1 mg WCE/tube), and subjected to WB analysis with anti-TβR-II antibodies. Total Src expression was monitored by probing detergent solubilized whole cell extracts (50 μg/lane) with anti-Src antibodies, whereas exogenous Src expression was confirmed by reprobing stripped membranes with anti-Myc antibodies. Differences in protein loading were monitored by probing stripped membranes with anti-TβR-II antibodies. ERK, extracellular signal-regulated kinase; GST, glutathione S-transferase; IP, immunoprecipitated; PY, phosphotyrosine; TβR, TGF-β receptor; TGF, transforming growth factor; WB, Western blot; WCE, whole cell extracts; WT, wild type.
Src inhibition blocks β3 integrin and TGF-β mediated MAPK activation, EMT, and invasion in NMuMG cells
We next explored the functional importance of Src in regulating various MEC responses to TGF-β. To do so, NMuMG cells were pretreated with either PP2 or its inactive counterpart PP3 and subsequently stimulated with TGF-β to activate ERK1/2 and p38 MAPK. As expected, β3 integrin expression significantly increased ERK1/2 and p38 MAPK activation following TGF-β1 stimulation (Figure 8a). This response to TGF-β was unaffected by PP3 administration, but it was reduced significantly by PP2 treatment of NMuMG cells (Figure 8a). More importantly, PP2 administration completely reverted the EMT phenotype induced by expression of β3 integrin in NMuMG cells, and nearly abolished the ability of TGF-β to stimulate EMT in NMuMG cells (Figure 8b). We also engineered NMuMG cells to stably express dominant negative Src and subsequently measured its effect on the ability of TGF-β to induce EMT and invasion in MECs. As expected, GFP expressing NMuMG cells readily underwent EMT when stimulated by TGF-β (Figure 8c). In stark contrast, NMuMG cells expressing dominant negative Src were stunted in their ability to undergo EMT in response to TGF-β (Figure 8c), and instead formed small, disorganized stress fibers that contrasted sharply with those produced in their GFP expressing counterparts.
Figure 8.
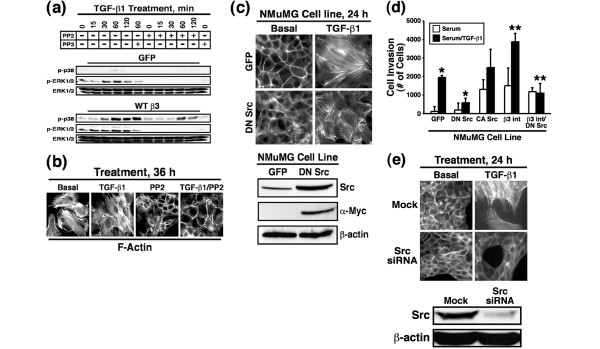
Src inhibition blocks β3 integrin and TGF-β mediated MAPK activation, EMT, and invasion in NMuMG cells. (a) Control or β3 integrin expressing NMuMG cells were serum starved for 4 hours before TGF-β1 (5 ng/ml) stimulation in the absence or presence of either PP2 (10 μmol/l) or its inactive counterpart PP3 (10 μmol/l). Afterward, the activation status of ERK1/2 and p38 MAPK was determined by immunoblot analysis using phospho-specific antibodies. Re-probing stripped membranes with anti-ERK1/2 antibodies monitored differences in protein loading. Data are from a representative experiment that was repeated twice with similar results. (b) β3 Integrin expressing NMuMG cells were stimulated with TGF-β1 (5 ng/ml) for 36 hours in the absence or presence of PP2 (10 μmol/l). Afterward, alterations in actin cytoskeletal architecture were visualized by direct rhodamine-phalloidin immunofluorescence. Shown are representative images from a single experiment that was repeated twice with identical results. (c) Control (GFP) or DN Src-expressing NMuMG cells were stimulated with TGF-β1 (5 ng/ml) for 24 hours. Afterward, alterations in actin cytoskeletal architecture were visualized by direct rhodamine-phalloidin immunofluorescence. Total Src expression was monitored by probing detergent-solubilized whole cell extracts (50 μg/lane) with anti-Src antibodies, whereas exogenous Src expression was visualized using anti-Myc antibodies. Immunoblotting stripped membranes with anti-β-actin antibodies served to monitor differences in protein loading. Shown are representative images from a single experiment that was repeated twice with identical results. (d) Control (GFP), DN Src, CA Src, β3 integrin, and β3 integrin/DN Src expressing NMuMG cells were allowed to invade through Matrigel matrices in the absence or presence of TGF-β1 (5 ng/ml) for 36 hours. Values are the mean (± standard error) of three independent experiments presented as the number of cells invaded per well. (e) Src-deficient NMuMG cells were generated by siRNA transfection. The effects of Src deficiency on TGF-β mediated EMT were visualized by direct FITC-phalloidin immunofluorescence. Shown are representative images from a single experiment that was repeated twice with identical results. Detergent-solubilized whole cell extracts (50 μg/lane) were prepared to monitor Src expression by immunoblotting with anti-Src antibodies. Differences in protein loading were monitored by re-probing striped blots with anti-β-actin antibodies. CA, constitutively active; DN, dominant negative; ERK, extracellular signal-regulated kinase; GFP, green fluorescent protein; MAPK, mitogen-activated protein kinase; TGF, transforming growth factor; WB, Western blot; WT, wild type.
Proper organization of cell polarity is essential for cancer cell invasion and metastasis [54]. Based on the aberrant EMT response observed in dominant negative Src expressing NMuMG cells, we suspected that their invasion through synthetic basement membranes would also be impaired. Accordingly, dominant negative Src expression significantly inhibited the ability of TGF-β to induce NMuMG cell invasion (Figure 8d). Interestingly, singular expression of either constitutively active Src or β3 integrin in NMuMG cells significantly enhanced tonic and TGF-β stimulated cell invasion (Figure 8d). More importantly, introducing dominant negative Src into β3 integrin expressing NMuMG cells completely abolished the ability of TGF-β to stimulate cell invasion in MECs (Figure 8d). Thus, activity of both β3 integrin and Src is necessary for MEC invasion stimulated by TGF-β.
Finally, the findings above suggest that Src may play an essential role in facilitating EMT stimulated by TGF-β. We tested this hypothesis by transiently transfecting NMuMG cells with siRNAs directed against Src and subsequently monitored their ability to undergo EMT in response to TGF-β. Figure 8e shows that Src deficiency did indeed abolish the ability of TGF-β to stimulate EMT in NMuMG cells, a response reminiscent of that observed in NMuMG cells deficient in β3 integrin (Figure 1c). Collectively, these findings indicate that Src expression and activity are essential for the ability of TGF-β to stimulate MAPKs, and to induce EMT and cell invasion in MECs.
β3 Integrin expression increases in malignant human MECs and regulates invasion stimulated by TGF-β
Our findings have thus far solely investigated the relationship between β3 integrins and TGF-β in normal MECs. It should be noted that cells undergoing neoplastic transformation, including those of the breast [26,27], exhibit altered integrin expression profiles during tumorigenesis, suggesting that integrins could play a key role in the acquisition of neoplastic phenotypes. The human MCF10A system consists of a series MEC cell lines that share a common ancestry and represent distinct stages of breast cancer development, ranging from normal to highly invasive and metastatic [55]; they also represent a model system for studying the conversion of TGF-β from a tumor suppressor to a tumor promoter [9].
Interestingly, breast cancer progression was associated with elevated expression of β3 integrin and its effector, FAK, which correlated with the conversion of TGF-β from a suppressor to a promoter of breast cancer progression (Figure 9a). Moreover, exogenous β3 integrin expression in normal, poorly invasive MCF10A cells significantly enhanced their invasion through synthetic basement membranes in response to TGF-β (Figure 9b). In contrast, the malignant, metastatic MCF10CA1a cells readily invaded Matrigel matrices when stimulated with TGF-β, a response that was augmented significantly by WT β3 integrin expression (Figure 9c). Importantly, D119A β3 integrin expression completely abolished the ability of TGF-β to stimulate MCF10ACA1a cell invasion (Figure 9c). Collectively, our findings suggest that mammary tumorigenesis alters β3 integrin expression in neoplastic MECs, and that β3 integrins may play a crucial role in translating TGF-β from a suppressor to a promoter of breast cancer growth and invasion.
Figure 9.
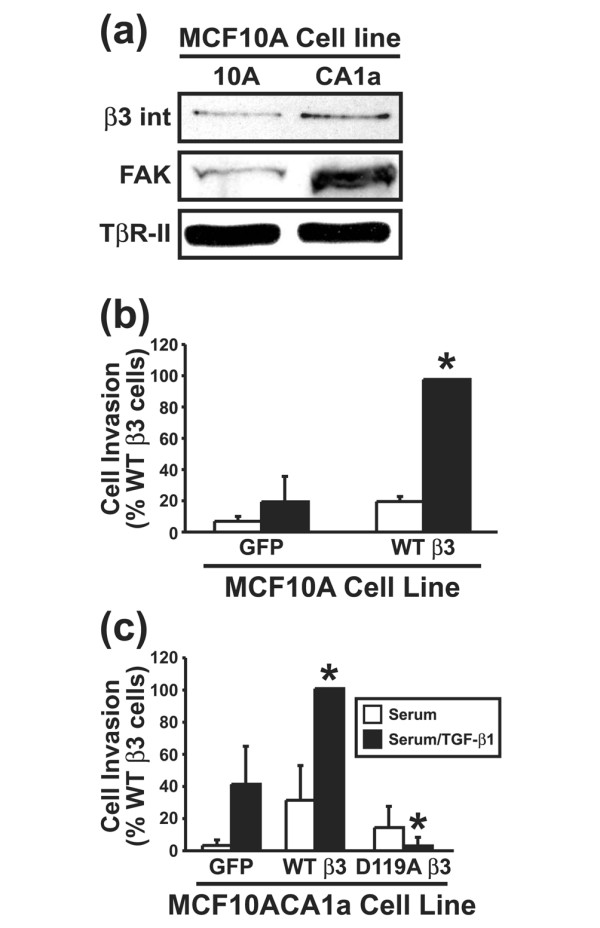
β3 Integrin expression increases in malignant human MECs and regulates invasion stimulated by TGF-β. (a) Detergent-solubilized whole cell extracts (100 μg/lane) of MCF10A and MCF10CA1a cells were prepared to analyze expression of the β3 integrin subunit and FAK by immunoblotting. Differences in protein loading were monitored by probing striped blots with anti-TβR-II antibodies. Control (GFP) and β3 integrin expressing (b) MCF10A cells or (c) MCF10CA1a cells were allowed to invade through Matrigel matrices in the absence or presence of TGF-β1 (5 ng/ml) for 36 hours. Values are the mean (± standard error) of three independent experiments presented as the percentage invasion relative to TGF-β stimulated WT β3 integrin expressing cells. FAK, focal adhesion kinase; MEC, mammary epithelial cell; TβR, TGF-β receptor; TGF, transforming growth factor; WT, wild type.
Discussion
The process of EMT recently has been subject to intense research investigation because of its importance in mediating cancer cell motility, invasion, and metastasis [11,48]. TGF-β is a major regulator of both developmental and pathological EMT, particularly that in diseased MECs, which frequently exhibit decreased cytostasis and increased invasiveness in response to TGF-β [56]. Unfortunately, the molecular mechanisms underlying the conversion of TGF-β from an inhibitor of MEC proliferation to an inducer of their EMT are poorly understood. Because cell microenvironmental changes regulate cancer development and progression, including the acquisition of EMT, and because integrins and TGF-β are major regulators of cell microenvironments, we hypothesized that differential integrin expression induced by TGF-β would contribute to its regulation of MEC proliferation and EMT.
To this end, we now show that β3 integrin expression alters the response of MECs to TGF-β, particularly its ability to regulate cell proliferation, invasion, and EMT. Indeed, β3 integrin expression appears essential for TGF-β stimulation of EMT (Figure 1c). Moreover, we demonstrate that β3 integrin directly couples to the TGF-β signaling system by interacting physically with TβR-II (Figure 2), resulting in enhanced MEC proliferation, invasion, and EMT when stimulated with TGF-β (Figure 5). More importantly, β3 integrin promotes Src mediated tyrosine phosphorylation of TβR-II (Figure 6), which we show to be essential for the ability of TGF-β to activate MAPKs (Figure 8) and, consequently, to induce EMT in MECs (Figure 8). More importantly, we show for the first time that induction of β3 integrin expression during mammary tumorigenesis correlates with metastasis development, and that antagonizing β3 integrin signaling abolishes TGF-β stimulation of breast cancer cell invasion (Figure 9). Collectively, we identified a potentially important mechanism whereby β3 integrin expression selectively facilitates TGF-β stimulation of MEC invasion and EMT.
The joint activation of integrin and RTK signaling systems is essential for the positional control of cells. More importantly, cell attachment to the ECM also determines the nature and context of how cells interpret and respond to cytokine and growth factor binding [52]. For instance, the ability of EGF and platelet-derived growth factor receptors to stimulate fibroblast migration requires RTK receptor association with integrins and FAK [24]. Similarly, Scaffidi and coworkers [42] showed that αvβ3 integrin bound TβR-II in lung fibroblasts, thereby enhancing fibroblast proliferation stimulated by TGF-β. Those authors further speculated that the formation of αvβ3 integrin:TβR-II complexes may exacerbate TGF-β mediated wound healing and fibrotic reactions [42], processes reminiscent of the ability of β3 integrin:TβR-II complexes to drive TGF-β stimulation of EMT in MECs. Most recently, the adapter protein Dab2, which mediates Smad2/3 activation by TβRs [57], was shown to participate in TGF-β stimulated EMT by interacting with integrins and preventing MEC apoptosis [58]. These findings, together with those presented herein, indicate an important and underappreciated role for integrins in regulating cellular response to TGF-β. Future studies in MECs must determine the relationship between TGF-β stimulated expression of β3 integrin and Dab2, as well as β3 integrin stimulated TβR-II expression in MECs; whether Dab2 links β3 integrin to TβR-II during EMT; and the ability of additional integrins to influence, either positively or negatively, MEC response to TGF-β.
A particularly novel finding of our study was the demonstration that β3 integrin induced TβR-II tyrosine phosphorylation via Src kinase. Indeed, we show for the first time that Src activity mediates tyrosine phosphorylation of TβR-II in MECs, leading to TGF-β mediated MAPK activation, and to MEC invasion and EMT. Previous studies established that TβR-II is phosphorylated predominantly on Ser and Thr residues [8,13,59], which, depending on the site of phosphorylation, either augments or attenuates TβR-II protein kinase activity [60]. However, tyrosine phosphorylation of TβR-II is not without precedent. Indeed, Lawler and coworkers [61] showed that TβR-II is a dual-specificity protein kinase that autophosphorylates not only on Ser/Thr residues but also on tyrosine 259, 336, and 424. Moreover, although Phe substitution at these positions significantly reduced TβR-II protein kinase activity, these mutations had little affect on the ability of TGF-β to induce gene expression in lung epithelial cells [61], and as such the role of tyrosines 259, 336, and 424 in mediating TGF-β action remain to be clarified. We too have converted tyrosines 259, 336, and 424 to phenylalanine in all possible combinations (i.e. single, double, and triple mutations), all of which failed to alter Src mediated tyrosine phosphorylation of GST-TβR-II (K277R) in vitro (Galliher AJ, Schiemann WP, unpublished data). Sequence analysis of the cytoplasmic domain of TβR-II revealed two possible Src phosphorylation consensus motifs at tyrosines 284 and 470. These two sites were individually mutated to phenylalanines, and preliminary data suggest that Src phosphorylates TβR-II at tyrosine 284 (Galliher AJ, Schiemann WP, unpublished data). Further analysis of this site in mediating TGF-β stimulation of EMT in MECs is currently under investigation.
Activation of MAPKs, particularly that of ERK1/2 [5] and p38 MAPK [2,32], are necessary for TGF-β to stimulate EMT. We also found that TGF-β stimulation of ERK1/2 and p38 MAPK are necessary for induction of EMT in MECs. More importantly, we show for the first time that Src activity is essential for activation of MAPKs by TGF-β and for its stimulation of EMT. Indeed, our results indicate that β3 integrin expression and Src activity are sufficient in overcoming TGF-β mediated cytostasis in MECs. Src activity also has been associated with protecting hepatocytes from apoptosis induced by TGF-β [62-64] and with the ability of TGF-β to stimulate ovarian cancer cell invasion [65]. More recently, TGF-β treatment of MECs was shown to induce their expression of RPTPκ, a protein tyrosine phosphatase that activates Src and mediates the antiproliferative and the promigratory effects of TGF-β [66]. These findings, together with those presented herein, implicate Src as an important player operant in dictating the MEC response to TGF-β; they also suggest that Src inhibition, similar to integrin interdiction, may one day be used to enhance the tumor suppressing activities of TGF-β in breast cancer cells.
Finally, based on the fact that phosphotyrosine residues often create binding motifs that couple receptors to various signaling pathways [67], including the MAPK cascade, it is tempting to speculate that Src-mediated tyrosine phosphorylation of TβR-II functions similarly as a receptor docking site to recruit SH2 and/or PTB containing signaling molecules to β3 integrin:TβR-II complexes. If correct, then such a mechanism could account for the augmented ability of TGF-β to activate MAPKs in β3 integrin expressing MECs, and for the shift in MAPK and Smad2/3 signaling that favored EMT over cytostasis in response to TGF-β. Indeed, the ability of β3 integrin to increase Smad2/3 transcriptional activity without altering Smad2/3 phosphorylation suggests that other integrin or MAPK stimulated nuclear factors converge on TGF-β targeted promoters and synergize with Smad2/3 in coordinating gene expression regulated by TGF-β. This notion is wholly consistent with previous work demonstrating the ability of Smad2/3 to synergize with activated TβR complexes in mediating EMT [37,68].
Conclusion
In summary, we present evidence that the differential expression of β3 integrin induced by TGF-β functions to convert this cytokine from an inhibitor of MEC proliferation to a stimulator of their EMT and invasion. More importantly, we show that β3 integrin regulates TGF-β signaling by interacting physically with TβR-II and stimulating its phosphorylation on tyrosine residues by Src. The net effect of these events results in enhanced TGF-β stimulation of MAPKs and, consequently, induction of EMT and invasion in MECs. Indeed, our findings suggest that integrin interdiction may prevent tumor development and progression by maintaining, reinforcing, or re-establishing the tumor suppressing activities of TGF-β.
Abbreviations
ECM = extracellular matrix; EGF = epidermal growth factor; EMT = epithelial-mesenchymal transitions; ERK = extracellular signal-regulated kinase; FACS = fluorescence-activated cell sorter; FAK = focal adhesion kinase; FBS = fetal bovine serum; GFP = green fluorescent protein; GST = glutathione S-transferase; IRES = internal ribosomal entry site; MAPK = mitogen-activated protein kinase; MEC = mammary epithelial cell; PBS = phosphate-buffered saline; PCR = polymerase chain reaction; PI3K = phosphoinositol-3 kinase; PMA = 4α-phorbol 12-myristate 13-acetate; pMSCV = plasmid murine stem cell virus; PTK = protein tyrosine kinase; RGD = arginine-glycine-aspartic acid; RTK = receptor tyrosine kinase; siRNA = small interfering RNA; TβR = TGF-β receptor; TGF = transforming growth factor; WT = wild type; YFP = yellow fluorescent protein.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AJG participated in designing the study, acquiring data, interpreting data, analyzing data, and preparing the manuscript. WPS participated in designing the study, interpreting data, and preparing the manuscript.
Acknowledgments
Acknowledgements
TGF-β1 was generously provided by R&D Systems Inc. Prolactin was generously provided by the National Hormone and Peptide Program (NHPP) at the National Institutes of Health (NIH). Members of the Schiemann Laboratory are thanked for critical reading of the manuscript. We also thank William Townend, Shirley Sobus, and Joshua Loomis for expertise and help provided on studies performed in the Cytometry Core Facility at the National Jewish Medical and Research Center. A special thanks goes to Ian Hardy for help in the analysis of FACS data. Support was provided in part by the National Institutes of Health (CA095519 and CA114039) and the Concern Foundation to WPS.
Contributor Information
Amy J Galliher, Email: Amy.Galliher@UCHSC.edu.
William P Schiemann, Email: Bill.Schiemann@uchsc.edu.
References
- Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002;115:3193–3206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor b-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA. TGF-β-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat Cell Biol. 2001;3:708–714. doi: 10.1038/35087019. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Bottinger EP. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc Natl Acad Sci USA. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Bottinger EP, Jakubczak JL, Haines DC, Bagnall K, Wakefield LM. Transgenic mice overexpressing a dominant-negative mutant type II transforming growth factor β receptor show enhanced tumorigenesis in the mammary gland and lung in response to the carcinogen 7,12-dimethylbenz-[a]-anthracene. Cancer Res. 1997;57:5564–5570. [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, Wakefield LM. TGF-β switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116–1124. doi: 10.1172/JCI200318899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YA, Dukhanina O, Tang B, Mamura M, Letterio JJ, MacGregor J, Patel SC, Khozin S, Liu ZY, Green J, et al. Lifetime exposure to a soluble TGF-β antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109:1607–1615. doi: 10.1172/JCI200215333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. BioEssays. 2001;23:912–923. doi: 10.1002/bies.1132. [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS. TGFβ signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/S0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K, Janda E, Pierreux CE, Rytèomaa M, Schulze A, McMahon M, Hill CS, Beug H, Downward J. Raf induces TGFβ production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev. 2000;14:2610–2622. doi: 10.1101/gad.181700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-β1-induced EMT in vitro. Neoplasia. 2004;6:603–610. doi: 10.1593/neo.04241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield LM, Roberts AB. TGF-β signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–29. doi: 10.1016/S0959-437X(01)00259-3. [DOI] [PubMed] [Google Scholar]
- Ignotz RA, Heino J, Massague J. Regulation of cell adhesion receptors by transforming growth factor-β. Regulation of vitronectin receptor and LFA-1. J Biol Chem. 1989;264:389–392. [PubMed] [Google Scholar]
- Cary LA, Han DC, Guan JL. Integrin-mediated signal transduction pathways. Histol Histopathol. 1999;14:1001–1009. doi: 10.14670/HH-14.1001. [DOI] [PubMed] [Google Scholar]
- Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Src kinase activation by direct interaction with the integrin β cytoplasmic domain. Proc Natl Acad Sci USA. 2003;100:13298–13302. doi: 10.1073/pnas.2336149100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary LA, Guan JL. Focal adhesion kinase in integrin-mediated signaling. Front Biosci. 1999;4:D102–113. doi: 10.2741/cary. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–478. doi: 10.1016/S0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- Gui GP, Wells CA, Yeomans P, Jordan SE, Vinson GP, Carpenter R. Integrin expression in breast cancer cytology: a novel predictor of axillary metastasis. Eur J Surg Oncol. 1996;22:254–258. doi: 10.1016/S0748-7983(96)80013-8. [DOI] [PubMed] [Google Scholar]
- Mizejewski GJ. Role of integrins in cancer: survey of expression patterns. Proc Soc Exp Biol Med. 1999;222:124–138. doi: 10.1046/j.1525-1373.1999.d01-122.x. [DOI] [PubMed] [Google Scholar]
- Berry MG, Goode AW, Puddefoot JR, Vinson GP, Carpenter R. Integrin β1-mediated invasion of human breast cancer cells: an ex vivo assay for invasiveness. Breast Cancer. 2003;10:214–219. doi: 10.1007/BF02966720. [DOI] [PubMed] [Google Scholar]
- Dabrowska K, Opolski A, Wietrzyk J, Switala-Jelen K, Boratynski J, Nasulewicz A, Lipinska L, Chybicka A, Kujawa M, Zabel M, et al. Antitumor activity of bacteriophages in murine experimental cancer models caused possibly by inhibition of β3 integrin signaling pathway. Acta Virol. 2004;48:241–248. [PubMed] [Google Scholar]
- Felding-Habermann B, O'Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, Hughes PE, Pampori N, Shattil SJ, Saven A, et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci USA. 2001;98:1853–1858. doi: 10.1073/pnas.98.4.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furger KA, Allan AL, Wilson SM, Hota C, Vantyghem SA, Postenka CO, Al-Katib W, Chambers AF, Tuck AB. β3 integrin expression increases breast carcinoma cell responsiveness to the malignancy-enhancing effects of osteopontin. Mol Cancer Res. 2003;1:810–819. [PubMed] [Google Scholar]
- Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin β1 signaling is necessary for transforming growth factor-b activation of p38MAPK and epithelial plasticity. J Biol Chem. 2001;276:46707–46713. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- Diaz-Gonzalez F, Forsyth J, Steiner B, Ginsberg MH. Trans-dominant inhibition of integrin function. Mol Biol Cell. 1996;7:1939–1951. doi: 10.1091/mbc.7.12.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann WP, Blobe GC, Kalume DE, Pandey A, Lodish HF. Context-specific effects of fibulin-5 (DANCE/EVEC) on cell proliferation, motility, and invasion: fibulin-5 is induced by TGF-β and affects protein kinase cascades. J Biol Chem. 2002;277:27367–27377. doi: 10.1074/jbc.M200148200. [DOI] [PubMed] [Google Scholar]
- Schiemann BJ, Neil JR, Schiemann WP. SPARC inhibits epithelial cell proliferation in part through stimulation of the transforming growth factor-β-signaling system. Mol Biol Cell. 2003;14:3977–3988. doi: 10.1091/mbc.E03-01-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P. TGF-β type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci. 1999;112:4557–4568. doi: 10.1242/jcs.112.24.4557. [DOI] [PubMed] [Google Scholar]
- Sokol J, Neil J, Schiemann B, Schiemann W. The use of cystatin C to inhibit epithelial-mesenchymal transition and morphological transformation stimulated by transforming growth factor-β. Breast Cancer Res. 2005;7:R844–R853. doi: 10.1186/bcr1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Hebert MC, Zhang YE. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albini A, Iwamoto Y, Kleinman HK, Martin GR, Aaronson SA, Kozlowski JM, McEwan RN. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res. 1987;47:3239–3245. [PubMed] [Google Scholar]
- DeCoteau JF, Knaus PI, Yankelev H, Reis MD, Lowsky R, Lodish HF, Kadin ME. Loss of functional cell surface transforming growth factor β (TGF-β) type 1 receptor correlates with insensitivity to TGF-beta in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 1997;94:5877–5881. doi: 10.1073/pnas.94.11.5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi AK, Petrovic N, Moodley YP, Fogel-Petrovic M, Kroeger KM, Seeber RM, Eidne KA, Thompson PJ, Knight DA. αvβ3 integrin interacts with the transforming growth factor β (TGFβ) type II receptor to potentiate the proliferative effects of TGFβ1 in living human lung fibroblasts. J Biol Chem. 2004;279:37726–37733. doi: 10.1074/jbc.M403010200. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Lin HY, Henis YI, Plamondon J, O'Connor-McCourt MD, Lodish HF. The transforming growth factor β receptors types I, II, and III form hetero-oligomeric complexes in the presence of ligand. J Biol Chem. 1993;268:22215–22218. [PubMed] [Google Scholar]
- Schiemann WP, Graves LM, Baumann H, Morella KK, Gearing DP, Nielsen MD, Krebs EG, Nathanson NM. Phosphorylation of the human leukemia inhibitory factor (LIF) receptor by mitogen-activated protein kinase and the regulation of LIF receptor function by heterologous receptor activation. Proc Natl Acad Sci USA. 1995;92:5361–5365. doi: 10.1073/pnas.92.12.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann WP, Pfeifer WM, Levi E, Kadin ME, Lodish HF. A deletion in the gene for transforming growth factor b type I receptor abolishes growth regulation by transforming growth factor b in a cutaneous T-cell lymphoma. Blood. 1999;94:2854–2861. [PubMed] [Google Scholar]
- Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70:537–546. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fata JE, Werb Z, Bissell MJ. Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast Cancer Res. 2004;6:1–11. doi: 10.1186/bcr634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Moses HL. Tumor-stroma interactions. Curr Opin Genet Dev. 2005;15:97–101. doi: 10.1016/j.gde.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JO, Bankston LA, Arnaout MA, Liddington RC. Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure. 1995;3:1333–1340. doi: 10.1016/S0969-2126(01)00271-4. [DOI] [PubMed] [Google Scholar]
- Liddington RC, Ginsberg MH. Integrin activation takes shape. J Cell Biol. 2002;158:833–839. doi: 10.1083/jcb.200206011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus J, Smith J, Ginsberg M. Integrin-mediated cell adhesion: the extracellular face. J Biol Chem. 1994;269:25235–25238. [PubMed] [Google Scholar]
- Giancotti FG, Tarone G. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu Rev Cell Dev Biol. 2003;19:173–206. doi: 10.1146/annurev.cellbio.19.031103.133334. [DOI] [PubMed] [Google Scholar]
- Maeda M, Shintani Y, Wheelock MJ, Johnson KR. Src activation is not necessary for transforming growth factor (TGF)-β-mediated epithelial to mesenchymal transitions (EMT) in mammary epithelial cells. PP1 directly inhibits TGF-β receptors I and II. J Biol Chem. 2006;281:59–68. doi: 10.1074/jbc.M503304200. [DOI] [PubMed] [Google Scholar]
- Bose R, Wrana JL. Regulation of Par6 by extracellular signals. Curr Opin Cell Biol. 2006;18:206–212. doi: 10.1016/j.ceb.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Santner SJ, Dawson PJ, Tait L, Soule HD, Eliason J, Mohamed AN, Wolman SR, Heppner GH, Miller FR. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65:101–110. doi: 10.1023/A:1006461422273. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-b in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Hocevar BA, Smine A, Xu XX, Howe PH. The adaptor molecule Disabled-2 links the transforming growth factor β receptors to the Smad pathway. EMBO J. 2001;20:2789–2801. doi: 10.1093/emboj/20.11.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunier C, Howe PH. Disabled-2 (Dab2) is required for transforming growth factor β-induced epithelial to mesenchymal transition (EMT) J Biol Chem. 2005;280:17540–17548. doi: 10.1074/jbc.M500974200. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-β receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Luo K, Lodish HF. Positive and negative regulation of type II TGF-β receptor signal transduction by autophosphorylation on multiple serine residues. EMBO J. 1997;16:1970–1981. doi: 10.1093/emboj/16.8.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler S, Feng XH, Chen RH, Maruoka EM, Turck CW, Griswold-Prenner I, Derynck R. The type II transforming growth factor-β receptor autophosphorylates not only on serine and threonine but also on tyrosine residues. J Biol Chem. 1997;272:14850–14859. doi: 10.1074/jbc.272.23.14850. [DOI] [PubMed] [Google Scholar]
- Murillo MM, del Castillo G, Sanchez A, Fernandez M, Fabregat I. Involvement of EGF receptor and c-Src in the survival signals induced by TGF-β1 in hepatocytes. Oncogene. 2005;24:4580–4587. doi: 10.1038/sj.onc.1208664. [DOI] [PubMed] [Google Scholar]
- Park SS, Eom YW, Kim EH, Lee JH, Min do S, Kim S, Kim SJ, Choi KS. Involvement of c-Src kinase in the regulation of TGF-β1-induced apoptosis. Oncogene. 2004;23:6272–6281. doi: 10.1038/sj.onc.1207856. [DOI] [PubMed] [Google Scholar]
- Valdes F, Murillo MM, Valverde AM, Herrera B, Sanchez A, Benito M, Fernandez M, Fabregat I. Transforming growth factor-β activates both pro-apoptotic and survival signals in fetal rat hepatocytes. Exp Cell Res. 2004;292:209–218. doi: 10.1016/j.yexcr.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Wakahara K, Kobayashi H, Yagyu T, Matsuzaki H, Kondo T, Kurita N, Sekino H, Inagaki K, Suzuki M, Kanayama N, et al. Transforming growth factor-β-dependent activation of Smad2/3 and up-regulation of PAI-1 expression is negatively regulated by Src in SKOV-3 human ovarian cancer cells. J Cell Biochem. 2004;93:437–453. doi: 10.1002/jcb.20160. [DOI] [PubMed] [Google Scholar]
- Wang SE, Wu FY, Shin I, Qu S, Arteaga CL. Transforming growth factor β (TGF-β)-Smad target gene protein tyrosine phosphatase receptor type κ is required for TGF-β function. Mol Cell Biol. 2005;25:4703–4715. doi: 10.1128/MCB.25.11.4703-4715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z. Recognition and regulation of primary-sequence motifs by signaling modular domains. Prog Biophys Mol Biol. 1999;71:359–372. doi: 10.1016/S0079-6107(98)00045-5. [DOI] [PubMed] [Google Scholar]
- Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16:1987–2002. doi: 10.1091/mbc.E04-08-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]