

Abstract
Background
Conventional resuscitation (CR) from hemorrhagic shock (HS) often restores and maintains hemodynamics but fails to restore intestinal perfusion. Post-CR intestinal ischemia has been implicated in the initiation of a gut-derived exaggerated systemic inflammatory response and in the progressive organ failure following HS. We propose that intestinal ischemia can be prevented with hypertonic saline resuscitation (HTSR).
Methods
Anesthetized male Sprague-Dawley rats (200 to 215 g) were hemorrhaged to 50% of mean arterial pressure (MAP) for 60 minutes and randomly assigned to 1 of the resuscitation groups (n = 7 each): Group I: sham operation and no HS; Group II: HS + CR with the return of the shed blood + 2 volumes of normal saline (NS); Group III: HS + return of the shed blood + hypertonic saline (HTS); (7.5 % NaCl, 4 ml/kg); Group IV: HS + HTS, then return of the shed blood after 60 minutes; Group V: HS + HTS, then 1 volume of NS after 60 minutes. Microvascular diameters of inflow (A1) and proximal and distal premucosal arterioles (A3) in terminal ileum and flow in A1 were measured using in vivo videomicroscopy and optical Doppler velocimetry. Hematocrit, plasma osmolarity, and electrolytes were measured in Groups II and III.
Results
HS caused a selective vasoconstriction in A1 arterioles that was not seen in the premucosal arterioles. CR restored and maintained MAP and caused generalized, progressive vasoconstriction at all intestinal arteriolar levels that is associated with hypoperfusion. HTSR failed to restore or maintain MAP or intestinal A1 arteriolar blood flow until the shed blood was returned. However, HTSR prevented the post-resuscitation, premucosal vasoconstriction and produced an insidious selective vasodilation in the A3 arterioles, which was most significant with early blood return (Group III). This selective arteriolar vasoactivity was associated with a significant improvement of endothelial cell function. Plasma hyperosmolality and hypernatremia persisted during the entire 2 hours post-resuscitation with HTS.
Conclusions
Small-volume HTSR can be used as a resuscitation regimen at the trauma scene and for selective clinical conditions where hypotensive resuscitation is indicated. HTSR improves intestinal perfusion by selective vasodilation of the precapillary arterioles even at MAP close to shock levels.
Massive blood loss due to trauma is the leading cause of death of trauma patients and military combatants. HS causes a disproportionate vasoconstriction and redistribution of the cardiac output, and with resuscitation, the reperfused gut triggers an exaggerated systemic inflammatory response that promotes local splanchnic and remote organ injuries. Conventional management of trauma patients with HS consists of rapid restoration of the intravascular volume with isotonic Ringer’s solution and blood transfusions. Theoretically, replacement of the vascular volume deficit with crystalloid fluid and blood resuscitation from HS should improve cardiac filling and output, and lessen the need for pharmacologic treatment of the peripheral resistance to sustain and maintain an effective arterial pressure. Two clinical studies document that, despite normalization of blood pressure, heart rate, and urine output, tissue hypoperfusion persists in 80% to 85% of patents, as evidenced by lactic acidemia and decreased mixed venous oxygen saturation.1,2 Other clinical studies have shown that the level and rate of normalization of serum lactate (index of tissue oxygen utilization correlated with mortality in degree of elevation and in the time-dependent rate of normalization3,4). Systemic base deficit (index of tissue perfusion) also shows a similar predictive pattern of mortality.5 However, interventions that focus on correction of this oxygen debt by driving oxygen transport variables, such as cardiac index or oxygen delivery index following conventional resuscitation, to supernormal levels fails to reduce mortality in severely injured patients.6,7 Similarly, in animal models of shock/resuscitation in which splanchnic vasculature and blood flow were directly determined, progressive vasoconstriction and hypoperfusion were observed even with adequate resuscitation that restored and maintained central hemodynamics.8–10 In general, the goal of resuscitation should be the restoration of splanchnic and peripheral tissue perfusion not central hemodynamics.
Over the past few decades, there has been an increasing interest in small-volume resuscitation with intravenous hypertonic saline (HTS) (7.5%, 2400 mOsm/L). Hypertonic saline resuscitation (HTSR) was reported to offer immunomodulation potentials over traditional lactated Ringer’s.11–13 HTSR also requires a significantly lower infusion volume to restore cardiac output and blood pressure.11–14 Despite restoration and maintenance of central hemodynamics by HTSR, splanchnic and peripheral tissue perfusion were restored to pre-hemorrhage levels only transiently after resuscitation. This was followed by a progressive decrease to levels comparable with those during shock 2 hours post-resuscitation.10,15–18 Therefore, these experiments and others demonstrate that HTS alone or in combination with dextran promptly restore central hemodynamics and tissue perfusion for a short period following resuscitation. Indeed, individual and meta-analysis of 8 double-blinded, randomized controlled trials with HTS/dextran and 6 trials with HTS alone failed to provide evidence of improved resuscitation outcome in terms of improved survival. HTSR therapy is based on an osmotic-driven mobilization of interstitial and intracellular fluids to the vascular compartment. In addition, it is generally assumed that HTSR improves tissue perfusion by selective arteriolar vasodilation and by decreasing the swelling of the endothelial and red blood cells.19 Because there is no experimental data to support this assumption, the present study was designed to determine the effects of HTSR on the gut’s perfusion by repeated and simultaneous direct in vivo measurements of the intestinal microvascular diameters and blood flow with intravital microscopy.
MATERIALS AND METHODS
Male Sprague-Dawley rats (Harlan, Indianapolis, Ind) (200–215 g) were used for this study. Animals were maintained in a facility approved by the American Association for the Accreditation of Laboratory Animal Care. Animals were acclimated for 2 weeks prior to the experimental use; they received a standard rat chow (15 g/day) and water ad libitum. The experimental protocol was approved by the Institutional Animal Care and Use Committee and Biohazard Safety Committee at the Veteran’s Administration Hospital, Louisville, Ky.
Prior to the surgical preparation, all animals received 2 ml of normal saline subcutaneously to compensate for body fluid loss during the surgical preparation and equilibration. Anesthesia was induced with intraperitoneal pentobarbital (50 mg/kg) and supplemental subcutaneous injections equal to 25% of the initial dose as required to maintain a surgical plane of anesthesia throughout the experimental protocol. Body temperature was maintained at 37°C ± 0.5°C with a rectal probe and a servo-controlled heating pad. Surgery was performed after loss of blink and withdrawal reflexes. Tracheostomy was performed to reduce airway resistance, and the animals were allowed to breathe spontaneously. PE-50 catheters were inserted into the right femoral artery and vein for the blood withdrawal and administration of the resuscitation fluids, respectively. The carotid artery was cannulated for the continuous monitoring and recording of blood pressure on a pressure measurement system (Digi-Med, Louisville, Ky).
Intestinal microvascular preparation
The peritoneal cavity was exposed through the midline abdominal incision ( ~ 1.5 cm), and a 2- to 3-cm segment of the terminal ileum was gently withdrawn from the peritoneal cavity with its neurovascular supply intact. Two ligatures were placed on the ileum at a distance of 1 to 1.5 cm, and the ileum was opened along the antimesenteric border with electrocautery. Enteric contents were gently washed from the mucosal surface, and animals were placed on a specially designed polyurethane board. The opened ileum was suspended, serosal side up, over a viewing port while submerged in a tissue bath containing nonvasoactive modified Krebs solution (6.92 g/L sodium chloride, 0.44 g/L potassium chloride, 0.37 g/L calcium chloride, and 2.1 g/L sodium bicarbonate, pH of 7.4, osmolality of 286 mOsm/L) with 4-0 silk sutures. The bathing solution was maintained at 37°C and bubbled with carbon dioxide and nitrogen to maintain the tissue bath pH at 7.35 to 7.4 throughout the experiment. Isoproterenol was added to the bathing solution at a very low concentration (0.01 μg/ml) to retard the peristalsis. This dose of isoproterenol is below the threshold affecting the vascular smooth-muscle tone.20 The animal was positioned on the stage of a trinocular microscope for the direct intravital videomicroscopy. Microvascular images were transmitted through the microscope to a photodiode array in an optical Doppler velocimeter (Microcirculation Research Institute, Texas A&M University, College Station, Tx) to measure centerline red blood cell velocity for the calculation of the intestinal microvascular blood flow. The microvascular image was then transmitted to a digital camera (models K-P-D51/D50; Hitachi Denshi, Tokyo, Japan) and displayed in a calibrated computer monitor. Microvascular digital images were stored as streamline video in the computer hard drive for later measurement of microvascular diameter using calipers. Criteria for the intestinal preparation acceptable for the intravital videomicroscopy included a baseline mean arterial pressure (MAP) >90 mmHg, red blood cell velocity in first order arterioles >20 mm/sec, and an active vasomotion in the arteriolar system.
The standard nomenclature for intestinal microvessels was applied.20 In brief, first-order arterioles (A1) arise from a mesenteric arcade artery, traverse the mesenteric border of the bowel wall, and penetrate through the muscle layers to the submucosal layer. In the submucosa, second-order arterioles (A2) arise from A1 and run along the longitudinal axis of the bowel. First- and second-order venules parallel the A1 and A2 arterioles. Third-order arterioles (A3) branch at right angles from A2 arterioles and continue on to terminate in the mucosa as central villus arterioles. Along their course, the A3 arterioles also give rise to smaller arterioles that supply the seromuscular layers of the bowel wall. Centerline red blood cell velocity in A1 arterioles was measured with optical Doppler velocimetry. The maximal velocity signal, displayed digitally, was used to calculate A1 blood flow according to the formula: (V/1.6) × (R2 × 0.001), where V is the centerline flow velocity, 1.6 is a correction factor that converts centerline velocity to average cross-sectional velocity, R is the intraluminal microvascular radius measured in μm, and 0.001 is a conversion factor to express flow measured in nl/sec. This equation assumes a parabolic flow velocity and a circular conduit. Studies have identified 1.58 to 1.60 as the ideal correction factor for a wide range of microvessels.
Plasma electrolytes (Na+, K+, Cl−, HCO3−), blood gases (PO2, PCO2), acid-base parameters (pH, Cbase, anion gap) in arterial blood were measured using ABL 700 series (Radiometer Medical, Brønshøj, Denmark). Plasma osmolarity was measured using Osmette (Precision Systems, Inc, Natick, Mass). Hematocrit was measured with hemocytometer using standard procedure.
Experimental protocol
The timeline for the experimental protocol is shown in Fig 1. After surgical preparation, 60 minutes were allowed for gut equilibration and recovery of the animal from surgical stress. Throughout the experiment, the exteriorized ileum was continuously suffused with Krebs solution in the tissue bath. Blood pressure, heart rate, rectal and bath temperatures, and bath pH were continuously monitored and recorded every 5 minutes (Digi-Med). Baseline microvascular measurements were taken at the end of the equilibration period when the variability in the measurements was <5% and included MAP, heart rate, rectal and bath temperatures, and bath pH. Also measured were diameters of the intestinal inflow A1 arteriole, A3 premucosal arteriole proximally pA3 and distally dA3, and centerline red cell velocity in the A1 arteriole. HS was initiated and maintained according to protocol. HS was achieved by blood withdrawal from the femoral artery into a syringe containing 1 ml of normal saline and 8 units of heparin (minimal amount to prevent blood-clotting) at a rate of 1 ml/min. Blood withdrawal continued until 50% of baseline MAP was attained. The nominal 50% MAP was maintained for 60 minutes with further blood withdrawal or reinfusion as required. On average, the total volume of the withdrawn blood was 5.3 ± 0.6 ml. After 60 minutes of HS, animals were randomly assigned to conventional resuscitation (CR) with the return of the shed blood over 5 minutes, followed by normal saline (2X the shed blood volume) infused intravenously over the subsequent 25 minutes. HTSR was performed according to protocol. Microvascular and hemodynamic data acquisition was continued at 20-minute intervals during shock and a subsequent 120 minutes of resuscitation. At 120 minutes following resuscitation, a single dose of acetylcholine (Ach) (10−5 M), an endothelium-dependent, receptor-dependent agonist, was added to the tissue bath to determine endothelial cell function.
Fig 1.
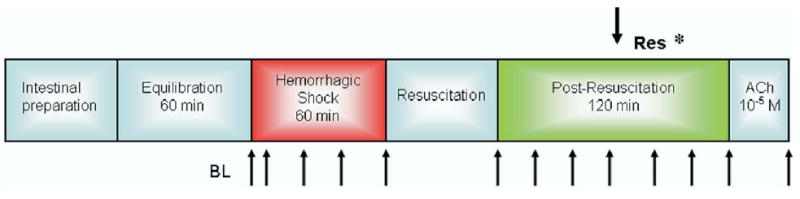
Timeline of the experimental protocol.Res, resuscitation; *, timing of initiation of second phase of resuscitation in Groups IV and V; Ach, acetylcholine; BL, baseline; ↑, timing of the measurements.
Experimental groups
Animals were randomized according to the after groups (n = 7 animals/group): (1) Group I: sham operation (vessel cannulation and intestinal preparation only) and no HS; (2) Group II: HS + CR with the return of the shed blood + 2 volumes of normal saline (NS); (3) Group III: HS + return of the shed blood + HTS (7.5 % NaCl, 4 ml/kg); (4) Group IV: HS + HTS (7.5 % NaCl, 4 ml/kg), then return of the shed blood after 60 minutes; (5) Group V: HS + HTS (7.5 % NaCl, 4 ml/kg), then 1 volume of NS after 60 minutes.
Statistical analysis
All data is presented as mean ± SEM unless stated otherwise. Microvascular diameter data was normalized as a percentage of the baseline or percentage of the maximal dilation to Ach as indicated. Differences among groups were assessed by 2-way analysis of variance (ANOVA) and Bonferroni post-test. A result was considered significant when the probability of type 1 error was < 5% (P < 0.05).
RESULTS
Mean arterial pressure (MAP)
As expected, HS produced a 50% reduction in MAP in all hemorrhaged groups. There was no significant difference in the level of hypotension between the 4 hemorrhaged groups (Fig 2). Resuscitation restored and maintained MAP to pre-HS baseline only in Groups II and III where the shed blood was returned; in Groups IV and V, MAP remained significantly lower than the pre-HS baseline during the post-resuscitation period. However, in Group IV, MAP reached 70% of baseline after HTS infusion and fully restored to pre-HS baseline after the return of the shed blood. In contrast, in Group V, infusion of normal saline 60 minutes after HTS infusion only transiently improved MAP.
Fig 2.
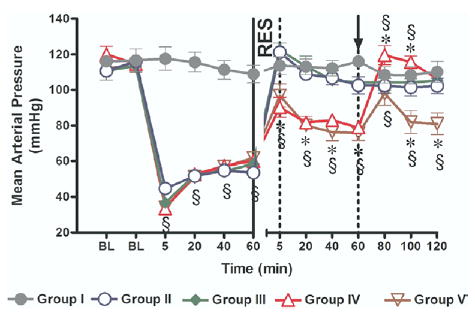
Mean arterial pressure (MAP). Group I = sham operation and no hemorrhagic shock (HS); Group II = HS + conventional resuscitation (CR) with the return of the shed blood + 2 volumes of normal saline (NS); Group III = HS + return of the shed blood + hypertonic saline (HTS) (7.5 % NaCl, 4 ml/kg); Group IV = HS + HTS (7.5 % NaCl, 4 ml/kg), then return of the shed blood after 60 min; Group V = HS + HTS (7.5 % NaCl, 4 ml/kg), then 1 volume of NS after 60 min. BL, baseline; RES, resuscitation; ↑, timing of initiation of second phase of resuscitation in Groups IV and V; §, P < 0.05 vs sham (Group I); *, P < 0.05 vs CR (Group II) by 2-way ANOVA and Bonferroni post-test.
Intestinal microvascular diameter
HS caused a differential response in intestinal microvascular reactivity. There was a significant constriction of the inflow A1 arterioles in all hemorrhaged groups, which was not seen in the A3 premucosal arterioles (Fig 3). CR in Group II caused a progressive post-resuscitation vasoconstriction in all arteriolar levels. With the exception of a brief restoration of A1 arteriolar diameter toward baseline in Group III, all resuscitation regimes in the present study failed to restore A1 arteriolar diameter to baseline pre-HS values during the entire 2 hours post-resuscitation. In contrast, HTSR prevented vasoconstriction in the premucosal A3 arterioles and caused an insidious time-dependent vasodilation in both proximal and distal A3 in Groups III to V (P < 0.001 vs CR [Group II] at 120 minutes post-resuscitation). Such A3 vasodilation was most significant in the dA3 arteriole in Group III ( ± 29.4% vs baseline), where HTSR was combined with the early return of the shed blood.
Fig 3.
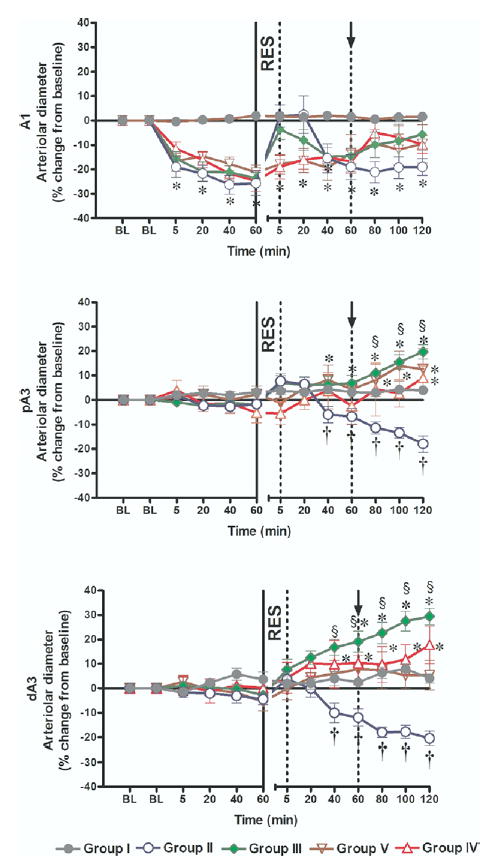
Intestinal microvascular diameters. Group I = sham operation and no hemorrhagic shock (HS); Group II = HS + conventional resuscitation (CR) with the return of the shed blood + 2 volumes of normal saline (NS); Group III = HS + return of the shed blood + hypertonic saline (HTS) (7.5 % NaCl, 4 ml/kg); Group IV = HS + HTS (7.5 % NaCl, 4 ml/kg), then return of the shed blood after 60 min; Group V = HS + HTS (7.5 % NaCl, 4 ml/kg), then 1 volume of NS after 60 min. BL, baseline; RES, resuscitation; ↑, timing of initiation of second phase of resuscitation in Groups IV and V; *, P < 0.05 vs sham; §, P < 0.05 vs CR (Group II); †, P < 0.05 vs baseline by 2-way ANOVA and Bonferroni post-test.
Blood flow
HS caused a marked reduction in intestinal arteriolar A1 blood flow. Such a reduction was similar in hemorrhaged groups (Fig 4). In Groups II and III, A1 arteriolar blood flow was briefly restored to supranormal levels immediately after resuscitation. This was followed by a progressive deterioration of A1 blood flow in Group II that received CR. In contrast, with HTSR in Group III, there was a modest increase in A1 blood flow that averaged 16% ± 5% (P < 05, n = 7) of baseline pre-HS value at 2 hours post-resuscitation. In Groups IV and V, A1 blood flow slightly improved after the first phase of resuscitation (HTS) such that the return of the shed blood in Group IV allowed further improvement and restoration of blood flow to the baseline. In Group V, an additional infusion of normal saline failed to restore baseline pre-HS A1 flow.
Fig 4.
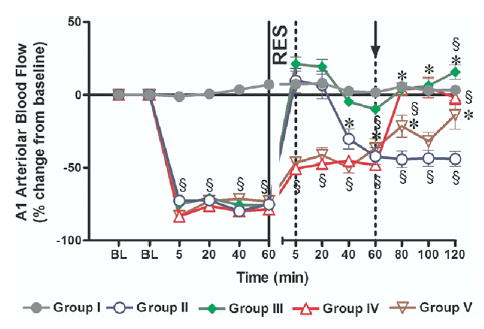
Intestinal A1 blood flow. Group I = sham operation and no hemorrhagic shock (HS); Group II = HS + conventional resuscitation (CR) with the return of the shed blood + 2 volumes of normal saline (NS); Group III = HS + return of the shed blood + hypertonic saline (HTS) (7.5 % NaCl, 4 ml/kg); Group IV + HS + HTS (7.5 % NaCl, 4 ml/kg), then return of the shed blood after 60 min; Group V = HS + HTS (7.5 % NaCl, 4 ml/kg), then 1 volume of NS after 60 min. BL, baseline, RES, resuscitation; ↑, timing of initiation of second phase of resuscitation in Groups IV and V; §, P < 0.05 vs sham; * P < 0.05 vs CR (Group II) by 2-way ANOVA and Bonferroni post-test.
Endothelial cell function
Endothelium-dependent arteriolar dilation to Ach (10−5 M) was impaired in the proximal and distal premucosal A3 arterioles after CR (P < 0.001 vs sham [Group 1]) (Fig 5), indicating endothelium cell dysfunction after HS with CR. HTSR in Groups III to V reversed this dysfunction, more significantly in Group III where shed blood + HTS were the initial resuscitation fluids. Responses in A1 arterioles had a tendency to decrease but were not significantly different between Groups II to IV vs sham. However, in Group V, A1 responses were markedly impaired (P < 0.05 vs sham), which indicated that blood return is essential for the restoration of endothelial function.
Fig 5.
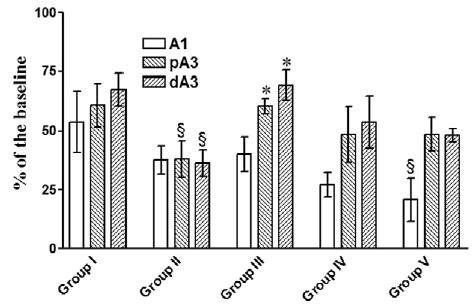
Response to ACh (10−5M) at 120 min post-resuscitation. A1, inflow arteriole; pA3, proximal premucosal arteriole; dA3, distal premucosal arteriole. Group I = sham-operation and no hemorrhagic shock (HS); Group II = HS + conventional resuscitation (CR) with the return of the shed blood + 2 volumes of normal saline (NS); Group III = HS + return of the shed blood + hypertonic saline (HTS) (7.5 % NaCl, 4 ml/kg); Group IV = HS + HTS (7.5 % NaCl, 4 ml/kg), then return of the shed blood after 60 min; Group V = HS + HTS (7.5% NaCl, 4 ml/kg), then 1 volume of NS after 60 min. §, P < 0.05 vs sham; *, P < 0.05 vs CR (Group II) by 2-way ANOVA and Bonferroni post-test.
Hematocrit, plasma electrolytes, and osmolarity after HS/HTSR
Serum electrolytes, plasma osmolality, blood hematocrit, blood gases, and acid-base parameters were measured in Groups II and III at baseline, and at 5 and 120 minutes post-resuscitation. As shown in Fig 6, panel A, CR (Group II) did not cause any significant alterations in sodium concentration and plasma osmolality. In contrast, HTSR (Group III) caused a significant increase in serum sodium to 150.4 ± 1.2 mmol/l and 145 ± 0.3 mmol/l vs baseline, respectively, at 5 and 120 minutes post-resuscitation ( P < 0.01 vs Group II, and P < 0.01 vs baseline). On average, the calculated sodium load delivered with CR was 1.54 to 1.85 mEq/l, which is higher than the 1.06 to 1.09 mEq/l of sodium delivered with HTSR in Group III. However, concentration of chloride was increased in Groups II and III (Fig 6, B). As expected, CR (Group II) was not associated with significant changes in plasma osmolality (Fig 6, C). In contrast, HTSR markedly increased plasma osmolality to 326 ± 5 mOsm/l and 315 ± 3 mOsm/l at 5 and 120 minutes post-resuscitation (P < 0.01 vs Group II, and P < 0.01 vs baseline).
Fig 6.
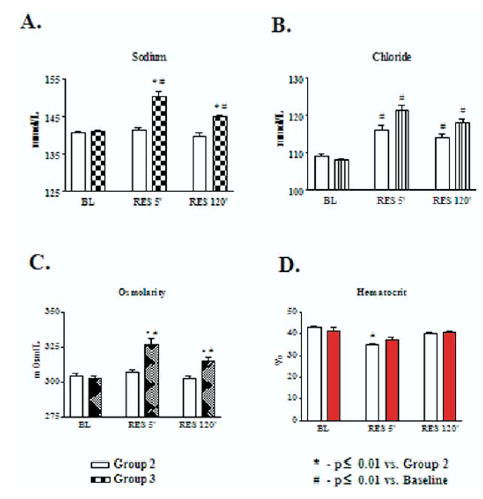
Blood chemistry. Serum sodium concentrations (A), chloride concentration (B), plasma osmolality (C), and blood hematocrit (D) were measured in Groups II and III at the baseline, and at 5 and 120 min post-resuscitation. * P < 0.05 vs conventional resuscitation (CR) (Group II); #, P < 0.05 vs baseline by 2-way ANOVA and Bonferroni post-test.
As expected, CR (Group II) decreased hematocrit at 5 minutes post-resuscitation (Fig 6, C). However, in spite of the significant hemodilution occurring with the infusion of the large volume of saline in Group II, hematocrit was restored to baseline at 120 minutes post-resuscitation. There was a modest decrease in serum K+ and Ca2+ associated with metabolic acidosis at 5′minutes after resuscitation, followed by restoration to baseline values at 120′ post-resuscitation (data not shown). There were no significant differences in blood gases between the groups (data not shown).
DISCUSSION
The present study is the first to investigate the effects of HTSR on the intestinal microcirculation and blood flow after nonlethal HS. Our studies demonstrate the following: (1) HS causes a selective vasoconstriction of the intestinal inflow A1 arterioles, which is not seen in the premucosal A3 arterioles; (2) CR with blood and saline causes a generalized and progressive vasoconstriction at all levels of the intestinal microvasculature, associated with progressive hypoperfusion; (3) HTSR alone does not restore or maintain hemodynamics and intestinal blood flow unless supplemented with the return of the shed blood; (4) HTSR preferentially restores the diameters of the intestinal premucosal arterioles and significantly ameliorates the endothelial dysfunction of these vessels; (5) Early administration of blood with HTSR is essential for the full restoration of central hemodynamics and tissue perfusion.
The body’s early response to traumatic injury and HS is characterized by a series of physiologic compensatory adjustments aimed at saving the life of the organism, such that neurohumoral reflexes mediated by catecholamines, vasopressin, and angiotensin II, promote vasoconstriction in certain vascular beds to ensure that an adequate fraction of the cardiac output supplies oxygen and nutrients to vital organs.21,22 This preferential redistribution of the cardiac output occurs at the expense of other vascular beds like the gut.23 With continued peripheral circulatory insufficiency, tissue hypoperfusion becomes self-destructive as blood flow falls below the metabolic needs of the parenchymal cells and vital organs. With continued curtailments of the cardiac output, vital organs will develop signs of irreversible tissue damage. The change in cell membrane potential and fluidity prompts a paradoxical cellular swelling, which further aggravates capillary blood flow and tissue hypoperfusion. With resuscitation, a reperfusion injury is introduced to cause obligatory fluid sequestration and enhance a gut-derived exaggerated systemic inflammatory response.24 Given this interrelated pathophysiology, it is reasonable to suggest that the goal of resuscitation should be the restoration of the splanchnic blood flow. In our previous studies in rats, we have shown that the shock-induced intestinal microvascular derangements and endothelial cell dysfunction can only partially be attenuated with a resuscitation regimen containing blood. We also have demonstrated that large-volume crystalloid resuscitation is associated with the most deleterious effects on the gut’s microcirculation and endothelial cell function.9 This is supported by the data from Group IV in the current study, in which the return of the shed blood 60 minutes after the initial HTSR treatment promptly restored and maintained MAP and intestinal blood flow. This data suggest that HTSR is clearly an attractive, alterative initial treatment at the trauma scene. In addition, initial HTSR causes a modest increase in MAP above the threshold level known to cause renal injury. This observation suggests that HTSR might be considered in clinical conditions where hypotensive resuscitation is indicated. Thus, resuscitation with a small volume of HTS that does not promptly correct the intravascular volume deficit or restore central hemodynamics can restore intestinal perfusion by an osmolality-dependent preferential and selective premucosal arteriolar vasodilation. This precapillary vasodilation determines the number of perfused capillaries, thereby increasing the capillary surface area available for O2 and nutrient exchange. It is likely that this HTSR-mediated vascular reactivity is a temporary response due to the rapid dissipation of the osmotic gradient, suggesting the importance of restoring the intravascular deficit preferably with blood rather than crystalloid resuscitation.
Few studies have specifically addressed the vascular effects of HTSR.15,16 In these studies, laser Doppler flow probes were placed locally on major arteries to monitor regional splanchnic blood flow. In contrast, one study (by Scalia et al10) provided direct measurement of the intestinal A1 microvessels after HTSR supplemented with dextran. In this study, the authors reported that the A1 arteriolar diameter of the terminal ileum observed directly with intravital microscopy was restored equally with HTSR and 2 or 3 volumes of Ringer’s lactate at 20 minutes after the resuscitation.10 In a second study, Diebel et al25 used indirect methods to simultaneously monitor superior mesenteric and splanchnic mucosal blood flow. The authors found that the superior mesenteric artery blood flow was returned to baseline level, whereas mucosal blood flow remained only 64% of baseline level after crystalloid resuscitation.25 Although there were differences in experimental protocols between the cited literature and the current study, the data collectively demonstrates that crystalloid resuscitation, which effectively restores central hemodynamics and regional blood flow, does not preclude an ongoing splanchnic microvascular vasoconstriction and tissue hypoperfusion. The selective intestinal premucosal arteriolar vasodilation by HTSR in the present study is remarkable. It should be mentioned that this selective vasodilation occurred despite a significant upstream constriction of the inflow feeding A1 arteriole. It is well established that the precapillary arterioles modulate capillary recruitment and define the number of perfused capillaries. Therefore, the selective vasodilation of the small premucosal arterioles might have markedly increased the local mucosal tissue perfusion without a significant increase in the overall regional blood flow. A provision for this to occur is the restoration of capillary flow and function. Indeed, in low flow states such as HS there is increased capillary plugging by activated neutrophils,26 significant narrowing of capillary lumen caused by capillary endothelial cell swelling,27–29 and increased compression on the capillary wall from interstitial fluid accumulation. If capillary blood flow is proportional to the fourth power of the radius, then any condition that alters the capillary cross-sectional area can deleteriously affect capillary blood flow. A number of studies have shown that HTSR ameliorates the hemorrhage-induced capillary narrowing.30,31 Clearly, the microvascular effects of HTSR are exerted distally on the precapillary and the true capillaries of the gut, and results in improved endothelial cell function, vasodilation, and tissue perfusion.
The antiinflammatory and immune effects of small-volume HTSR have been well studied. Data of HS obtained from in vitro human leukocyte studies and animal models demonstrate that hypertonicity decreases neutrophil priming/adherence,11,32–34 stimulates lymphocyte proliferation,35 inhibits proinflammatory, and yet stimulates antiinflammatory cytokine production by monocyte/macrophages.36–38 These immunomodulation effects have recently been reproduced in trauma patients.39 Despite the immunomodulation and the selective microvascular effects of HTSR, meta-analysis of well-controlled studies has failed to demonstrate a significant change in treatment outcome. The reason for this conclusion is not known. Clearly more multicenter human studies are needed to better define the possible benefits and risks of HTSR.
In summary, intravascular resuscitation from HS promptly restores and maintains hemodynamics but is associated with a generalized and progressive intestinal vasoconstriction and hypoperfusion. HTSR alone does not restore or maintain MAP unless the shed blood is returned. HTSR improves intestinal perfusion by exerting selective vasodilation of the small precapillary arterioles even at low MAP.
DISCUSSION
Dr William J. Flynn (Buffalo, NY): This study also shows that resuscitation with hypertonic saline produces dilation of premucosal arterioles. Does this mean that hypertonic cells directly dilate these vessels or is this effect produced through some intermediary mechanism?
Are you proposing that hypertonic saline should be used as an adjunct to standard resuscitation, or should it be used in place of it?
Finally, are central hemodynamics an unreliable indicator of adequate resuscitation?
Dr. Charles E. Lucas (Detroit, Mich): I currently teach my residents that the capillary is a dumb tube following the orders placed upon it by the three-dimensional configuration of the interstitial space. As you know, the nutrients flow from the capillary to the cell and metabolites move from the cell back to the capillary. Both must move through the interstitial space matrix. I would like you to speculate as to whether, in your opinion, the hypertonic saline is producing these changes by causing alterations in the endothelium of the capillary or alterations in the stearic relationships of the interstitial space matrix.
Dr. Bruce A. Harms (Madison, Wis): In the pulmonary circulation, which is what our lab in Madison has looked at, we see tremendous maldistribution at the alveolar level with any kind of hemorrhagic shock. Is the vasoconstriction that you see in your model uniform at the A3 level in the villus, or do you see maldistribution that is difficult to reverse and regulated by unclear mechanisms?
Many years ago, Dr George Kramer showed that you can’t sustain hemodynamic effects with hyper-tonic saline without a larger molecule. That is why he put dextran in the hypertonic saline solution. What causes the maintenance of that vasoconstrictor effect? You gave back the shed blood, but can you get the effect if you use other molecules?
Have you looked at other mechanisms of regulation at the mucosal level? Thromboxine analogs have also been shown to decrease the vasoconstrictor effect, at least in the micropulmonary circulation.
Dr Palmer Q. Bessey (New York, NY): It seems to me that the effects that you observed with your resuscitation could be due to enhanced intravascular volume from the hypertonic saline in the blood; it could also be an indirect cellular effect, in that if there were less endothelial cell swelling, there would be less resistance in the microcirculation. There might be an alteration in the tone or reactivity of the vasoconstrictor muscles within the vessels. Do you have a hunch which of those might be operative here?
The second question has to do with the return of the shed blood. It is striking that the effect is sustained, and in fact dependent, upon the return of the shed blood, which of course is autologous blood. Do you see a similar effect if you gave allogeneic blood that had been stored for some time before you give it for resuscitation?
Dr Jonathan M. Saxe (Dayton, Ohio): Did you give equivalent amounts of salt? It didn’t appear to me that your salts were equivalent and that you may have been giving more in the hypertonic saline group.
Dr E. R. Zakaria: Hyperosmolality is known to be vasoactive. The mechanisms as to how hyperosmolality dilates the premucosal arterioles have not been defined. However, ongoing research in our lab suggests that hyperosmolality-induced vasodilation is endothelium-dependent and involves extrusion of cellular water through the trans-endothelium water-only pathways named aquaporin-1 (AQP-1), which are channel-forming integral membrane proteins. Our data clearly demonstrate that hypertonic saline resuscitation alone does not restore and maintain splanchnic perfusion unless the shed blood is returned, and therefore it should be used as an adjunctive therapy. Measuring central hemodynamics is an unreliable indicator of adequate resuscitation. Restoration and maintenance of splanchnic tissue perfusion is. Therefore, adequate resuscitation should be redefined by the ability of the resuscitation regimen to restore and maintain splanchnic perfusion.
In shock, there is no capillary blood flow due to the change in the pre-to-post capillary resistance ratio, which results in no O2, nutrient and fluid exchange across the capillary wall, and this dysfunction could totally account for the pathophysiology of shock. In addition, shock/resuscitation alters the transcapillary Starling forces, which govern fluid exchange across the capillary wall. Intravascular hypertonic saline infusion can theoretically add an absorption force that can be very effective in restoring vascular volume through interstitial water absorption via the aquaporins. We suspect that this mechanism is limited by the rapid diffusion of NaCl into the interstitium. This rapid dissipation of the osmotic gradient explains the inability of hypertonic saline resuscitation alone to maintain vascular volume and central hemodynamics over time.
Dr. Harms, the vasoconstriction and hypoperfusion of the gut, which follows adequate resuscitation that restores and maintains hemodynamics is generalized and progressive. This explains the maldistribution you see in the pulmonary and splanchnic vascular beds. The only strategy to reverse the pathophysiology of shock is to restore capillary perfusion. We do not have data with combined hypertonic saline and colloid (dextran) resuscitation. The pathogenesis of the post-resuscitation vasoconstriction is multifactorial and involves neurohormonal reflexes, complement activation, and hypoxia, as well as endothelial cell dysfunction. Previous experiments have demonstrated that pentoxyfylline, dextran sulfate, and complement inhibition can all prevent the post-resuscitation vasoconstriction. For various reasons these basic science data did not translate into clinical application.
The premucosal arteriolar vasodilation that we see with adjunctive hypertonic saline resuscitation is not due to restoration or expansion of the intra-vascular volume. In our previous studies, we have shown that super-resuscitation with volumes equal to the animal’s own plasma volume is associated with even more severe post-resuscitation vasoconstriction and endothelial cell dysfunction. We now have data to support that the vasodilation we see is due to restoration of endothelial cell function by a hyperosmolality-driven, endothelium-dependent mechanism. Endothelial cell swelling drastically affects capillary blood flow because flow is related to the fourth power of the radius in this very narrow microvessel. We have not done experiments with allogeneic blood.
The salt load was not equivalent between the experimental groups. The hypertonic saline (7.5%) resuscitation volume (4 ml/kg) in fact delivers 31% less sodium load than in the animals that received convention resuscitation.
Footnotes
Presented at the 63rd Annual Meeting of the Central Surgical Association, Louisville, Kentucky, March 9-11, 2006.
Supported by a VA Merit Review grant and by NIH research Grant # R01 HL076160-03, funded by the National Heart, Lung, and Blood Institute and the United States Army Medical Resources and Material.
References
- 1.Scalea TM, Maltz S, Yelon J, Trooskin SZ, Duncan AO, Sclafani SJ. Resuscitation of multiple trauma and head injury: role of crystalloid fluids and inotropes. Crit Care Med. 1994;22:1610–5. [PubMed] [Google Scholar]
- 2.Abou-Khalil B, Scalea TM, Trooskin SZ, Henry SM, Hitch-cock R. Hemodynamic responses to shock in young trauma patients: need for invasive monitoring. Crit Care Med. 1994;22:633–9. doi: 10.1097/00003246-199404000-00020. [DOI] [PubMed] [Google Scholar]
- 3.Abramson D, Scalea TM, Hitchcock R, Trooskin SZ, Henry SM, Greenspan J. Lactate clearance and survival following injury. J Trauma. 1993;35:584–8. doi: 10.1097/00005373-199310000-00014. [DOI] [PubMed] [Google Scholar]
- 4.Broder G, Weil MH. Excess lactate: an index of reversibility of shock in human patients. Science. 1964;143:1457–9. doi: 10.1126/science.143.3613.1457. [DOI] [PubMed] [Google Scholar]
- 5.Rutherford EJ, Morris JA, Jr, Reed GW, Hall KS. Base deficit stratifies mortality and determines therapy. J Trauma. 1992;33:417–23. doi: 10.1097/00005373-199209000-00014. [DOI] [PubMed] [Google Scholar]
- 6.Dunham CM, Siegel JH, Weireter L, Fabian M, Goodarzi S, Guadalupi P, et al. Oxygen debt and metabolic acidemia as quantitative predictors of mortality and the severity of the ischemic insult in hemorrhagic shock. Crit Care Med. 1991;19:231–43. doi: 10.1097/00003246-199102000-00020. [DOI] [PubMed] [Google Scholar]
- 7.Heyland DK, Cook DJ, King D, Kernerman P, Brun-Buisson C. Maximizing oxygen delivery in critically ill patients: a methodologic appraisal of the evidence. Crit Care Med. 1996;24:517–24. doi: 10.1097/00003246-199603000-00025. [DOI] [PubMed] [Google Scholar]
- 8.Fruchterman TM, Spain DA, Wilson MA, Harris PD, Garrison RN. Selective microvascular endothelial cell dysfunction in the small intestine following resuscitated hemorrhagic shock. Shock. 1998;10:417–22. doi: 10.1097/00024382-199812000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Zakaria ER, Spain DA, Harris PD, Garrison RN. Resuscitation regimens for hemorrhagic shock must contain blood. Shock. 2002;18:567–73. doi: 10.1097/00024382-200212000-00014. [DOI] [PubMed] [Google Scholar]
- 10.Scalia SV, Taheri PA, Force S, Ozmen V, Lui D, Fish J, et al. Mesenteric microcirculatory changes in nonlethal hemorrhagic shock: the role of resuscitation with balanced electrolyte or hypertonic saline/dextran. J Trauma. 1992;33:321–5. [PubMed] [Google Scholar]
- 11.Angle N, Hoyt DB, Coimbra R, Liu F, Herdon-Remelius C, Loomis W, et al. Hypertonic saline resuscitation diminishes lung injury by suppressing neutrophil activation after hemorrhagic shock. Shock. 1998;9:164–70. doi: 10.1097/00024382-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Coimbra R, Hoyt DB, Junger WG, Angle N, Wolf P, Loomis W, Evers MF. Hypertonic saline resuscitation decreases susceptibility to sepsis after hemorrhagic shock. J Trauma. 1997;42:602–6. doi: 10.1097/00005373-199704000-00004. [DOI] [PubMed] [Google Scholar]
- 13.Coimbra R, Junger WG, Hoyt DB, Liu FC, Loomis WH, Evers MF. Hypertonic saline resuscitation restores hemorrhage-induced immunosuppression by decreasing prostaglandin E2 and interleukin-4 production. J Surg Res. 1996;64:203–9. doi: 10.1006/jsre.1996.0329. [DOI] [PubMed] [Google Scholar]
- 14.Angle N, Hoyt DB, Cabello-Passini R, Herdon-Remelius C, Loomis W, Junger WG. Hypertonic saline resuscitation reduces neutrophil margination by suppressing neutrophil L selectin expression. J Trauma. 1998;45:7–12. doi: 10.1097/00005373-199807000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Behrman SW, Fabian TC, Kudsk KA, Proctor KG. Microcirculatory flow changes after initial resuscitation of hemorrhagic shock with 7.5% hypertonic saline/6% dextran 70. J Trauma. 1991;31:589–98. doi: 10.1097/00005373-199105000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Diebel LN, Robinson SL, Wilson RF, Dulchavsky SA. Splanchnic mucosal perfusion effects of hypertonic versus isotonic resuscitation of hemorrhagic shock. Am Surg. 1993;59:495–9. [PubMed] [Google Scholar]
- 17.Maningas PA. Resuscitation with 7.5% NaCl in 6% dextran-70 during hemorrhagic shock in swine: effects on organ blood flow. Crit Care Med. 1987;15:1121–6. doi: 10.1097/00003246-198712000-00009. [DOI] [PubMed] [Google Scholar]
- 18.Prough DS, Whitley JM, Taylor CL, Deal DD, DeWitt DS. Small-volume resuscitation from hemorrhagic shock in dogs: effects on systemic hemodynamics and systemic blood flow. Crit Care Med. 1991;19:364–72. doi: 10.1097/00003246-199103000-00015. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez EA, Kozar RA, Suliburk JW, Weisbrodt NW, Mercer DW, Moore FA. Hypertonic saline resuscitation after mesenteric ischemia/reperfusion induces ileal apoptosis. J Trauma. 2005;59:1092–8. doi: 10.1097/01.ta.0000188935.66504.00. [DOI] [PubMed] [Google Scholar]
- 20.Bohlen HG, Gore RW. Preparation of rat intestinal muscle and mucosa for quantitative microcirculatory studies. Microvasc Res. 1976;11:103–10. doi: 10.1016/0026-2862(76)90081-9. [DOI] [PubMed] [Google Scholar]
- 21.Neutze JM, Wyler F, Rudolph AM. Changes in distribution of cardiac output after hemorrhage in rabbits. Am J Physiol. 1968;215:857–64. doi: 10.1152/ajplegacy.1968.215.4.857. [DOI] [PubMed] [Google Scholar]
- 22.Kovach AG, Rosell S, Sandor P, Koltay E, Kovach E, Tomka N. Blood flow, oxygen consumption, and free fatty acid release in subcutaneous adipose tissue during hemorrhagic shock in control and phenoxybenzamine-treated dogs. Circ Res. 1970;26:733–41. doi: 10.1161/01.res.26.6.733. [DOI] [PubMed] [Google Scholar]
- 23.Reilly PM, Wilkins KB, Fuh KC, Haglund U, Bulkley GB. The mesenteric hemodynamic response to circulatory shock: an overview. Shock. 2001;15:329–43. doi: 10.1097/00024382-200115050-00001. [DOI] [PubMed] [Google Scholar]
- 24.Deitch EA, Xu D, Franko L, Ayala A, Chaudry IH. Evidence favoring the role of the gut as a cytokine-generating organ in rats subjected to hemorrhagic shock. Shock. 1994;1:141–5. doi: 10.1097/00024382-199402000-00010. [DOI] [PubMed] [Google Scholar]
- 25.Diebel LN, Tyburski JG, Dulchavsky SA. Effect of acute hemodilution on intestinal perfusion and intramucosal pH after shock. J Trauma. 2000;49:800–5. doi: 10.1097/00005373-200011000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Mazzoni MC, Schmid-Schonbein GW. Mechanisms and consequences of cell activation in the microcirculation. Cardiovasc Res. 1996;32:709–19. [PubMed] [Google Scholar]
- 27.Mazzoni MC, Borgstrom P, Intaglietta M, Arfors KE. Lumenal narrowing and endothelial cell swelling in skeletal muscle capillaries during hemorrhagic shock. Circ Shock. 1989;29:27–39. [PubMed] [Google Scholar]
- 28.Mazzoni MC, Intaglietta M, Cragoe EJ, Jr, Arfors KE. Amiloride-sensitive Na+ pathways in capillary endothelial cell swelling during hemorrhagic shock. J Appl Physiol. 1992;73:1467–73. doi: 10.1152/jappl.1992.73.4.1467. [DOI] [PubMed] [Google Scholar]
- 29.Mazzoni MC, Borgstrom P, Warnke KC, Skalak TC, Intaglietta M, Arfors KE. Mechanisms and implications of capillary endothelial swelling and luminal narrowing in low-flow ischemias. Int J Microcirc Clin Exp. 1995;15:265–70. doi: 10.1159/000179028. [DOI] [PubMed] [Google Scholar]
- 30.Corso CO, Okamoto S, Leiderer R, Messmer K. Resuscitation with hypertonic saline dextran reduces endothelial cell swelling and improves hepatic microvascular perfusion and function after hemorrhagic shock. J Surg Res. 1998;80:210–20. doi: 10.1006/jsre.1998.5426. [DOI] [PubMed] [Google Scholar]
- 31.Mazzoni MC, Borgstrom P, Intaglietta M, Arfors KE. Capillary narrowing in hemorrhagic shock is rectified by hyperosmotic saline-dextran reinfusion. Circ Shock. 1990;31:407–18. [PubMed] [Google Scholar]
- 32.Rizoli SB, Kapus A, Fan J, Li YH, Marshall JC, Rotstein OD. Immunomodulatory effects of hypertonic resuscitation on the development of lung inflammation following hemorrhagic shock. J Immunol. 1998;161:6288–96. [PubMed] [Google Scholar]
- 33.Rizoli SB, Kapus A, Parodo J, Fan J, Rotstein OD. Hypertonic immunomodulation is reversible and accompanied by changes in CD11b expression. J Surg Res. 1999;83:130–5. doi: 10.1006/jsre.1999.5581. [DOI] [PubMed] [Google Scholar]
- 34.Rizoli SB, Kapus A, Parodo J, Rotstein OD. Hypertonicity prevents lipopolysaccharide-stimulated CD11b/CD18 expression in human neutrophils in vitro: role for p38 inhibition. J Trauma. 1999;46:794–98. doi: 10.1097/00005373-199905000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Loomis WH, Namiki S, Hoyt DB, Junger WG. Hypertonicity rescues T cells from suppression by trauma-induced anti-inflammatory mediators. Am J Physiol Cell Physiol. 2001;281:C840–8. doi: 10.1152/ajpcell.2001.281.3.C840. [DOI] [PubMed] [Google Scholar]
- 36.Oreopoulos GD, Hamilton J, Rizoli SB, Fan J, Lu Z, Li YH, et al. In vivo and in vitro modulation of intercellular adhesion molecule (ICAM)-1 expression by hypertonicity. Shock. 2000;14:409–14. doi: 10.1097/00024382-200014030-00029. [DOI] [PubMed] [Google Scholar]
- 37.Oreopoulos GD, Bradwell S, Lu Z, Fan J, Khadaroo R, Marshall JC, et al. Synergistic induction of IL-10 by hypertonic saline solution and lipopolysaccharides in murine peritoneal macrophages. Surgery. 2001;130:157–65. doi: 10.1067/msy.2001.115829. [DOI] [PubMed] [Google Scholar]
- 38.Oreopoulos GD, Wu H, Szaszi K, Fan J, Marshall JC, Khadaroo RG, et al. Hypertonic preconditioning prevents hepatocellular injury following ischemia/reperfusion in mice: a role for interleukin 10. Hepatology. 2004;40:211–20. doi: 10.1002/hep.20281. [DOI] [PubMed] [Google Scholar]
- 39.Rizoli SB, Rhind SG, Shek PN, Inaba K, Filips D, Tien H, et al. The immunomodulatory effects of hypertonic saline resuscitation in patients sustaining traumatic hemorrhagic shock: a randomized, controlled, double-blinded trial. Ann Surg. 2006;243:47–57. doi: 10.1097/01.sla.0000193608.93127.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]