

Abstract
To determine the minimum requirements for substrate recognition and processing by proteasomes, the functional elements of a ubiquitin-independent degradation tag were dissected. The 37-residue C-terminus of ornithine decarboxylase (cODC) is a native degron, which also functions when appended to diverse proteins. Mutating the cysteine 441 residue within cODC impaired its proteasome association in the context of ornithine decarboxylase and prevented the turnover of GFP-cODC in yeast cells. Degradation of GFP-cODC with C441 mutations was restored by providing an alternate proteasome association element via fusion to the Rpn10 proteasome subunit. However, Rpn10-GFP was stable, unless extended by cODC or other peptides of similar size. In vitro reconstitution experiments confirmed the requirement for both proteasome tethering and a loosely structured region. Therefore, cODC and degradation tags in general must serve two functions: proteasome association and a site, consisting of an extended peptide region, used for initiating insertion into the protease.
Keywords: degron, ornithine decarboxylase, proteasome, proteolysis, ubiquitin
Introduction
Proteasomes are molecular machines that destroy proteins (Glickman and Ciechanover, 2002). This irreversible process requires exquisite specificity, and cells have elaborated complex means to avoid false positives and negatives. What are the minimum properties needed to specify degradation? Two contrasting models have been proposed to explain the function of a degradation signal or degron. A degron may simply increase the local concentration of a substrate protein at the proteasome (Janse et al, 2004). According to this prospective, any means of proteasome localization, including non-physiologic modes of tethering, will suffice for degradation. In an alternate view, the degron provides a means of proteasome association, usually via a ubiquitin chain, but substrates additionally require the presence of an unstructured domain from which unfolding or degradation initiates (Prakash et al, 2004). The degradation tag of the enzyme ornithine decarboxylase (ODC) provides a favorable test bed to resolve this question. Most substrates of the 26S proteasome are recognized via conjugation to a polyubiquitin chain (Hochstrasser, 1996). However, a restricted subset of substrates is recognized and degraded without ubiquitylation (Verma and Deshaies, 2000; Hoyt and Coffino, 2003). One example is ODC, the first and rate-limiting enzyme of polyamine biosynthesis (Pegg, 1986; Murakami et al, 1992). Its carboxy-terminal 37 amino acids, here termed C-terminus of ornithine decarboxylase (cODC), constitute a degron necessary and sufficient to destabilize mammalian ODC. Deleting cODC stabilizes ODC, whereas appending cODC to diverse stable proteins makes them short-lived (Ghoda et al, 1989; Loetscher et al, 1991; Hoyt et al, 2003). Degradation driven by adding the cODC tag retains the attributes of native ODC degradation—26S proteasome dependence, but ubiquitin independence.
cODC requires no post-translational modification to become active; so structural studies can readily be interpreted without regard to potential secondary effects of mutagenesis on post-translational conjugation or deconjugation. Biochemical competition experiments have shown that cODC is a molecular mimic of a polyubiquitin chain and cross-competes with ubiquitin chains for recognition by the proteasome (Zhang et al, 2003). Therefore, conclusions inferred from this degron are likely to carry over to the broader class of proteasome substrates. Here we show that cODC acts as a proteasome association element, but that this is insufficient. It provides as well an unstructured or loosely structured region from which degradation can initiate. The association function of the cODC degron is dispensable if an alternative association element is provided, but this bypass mode is incapable of promoting degradation, unless supplemented by an additional region that is not tightly folded. We thus conclude that proteasome association alone is insufficient to provide a degradation signal, that an unstructured domain is also required, and that an effective substrate must supply both.
Results
C441 of the cODC degradation tag is required for recognition by the 26S proteasome
Degradation by the 26S proteasome (or other ATP-dependent proteases) is a multi-step process that requires substrate association, unfolding and insertion into a narrow portal leading to the catalytic core (Pickart and Cohen, 2004). Within the proteasome degradation tag cODC, a specific cysteine, present in native ODC as residue 441 (C441), is intolerant of mutation (Miyazaki et al, 1993). Our previous studies showed that mutating C441A within mouse ODC prolongs its degradation in both animal and yeast cells. Biochemical studies have shown that proteasomes can trap wild-type ODC, but not ODC with a C441 mutation (Murakami et al, 1999), but do not distinguish the proteasome processing step affected by the mutation. To examine the role of C441, we performed an in vitro competition assay, using purified rat 26S proteasomes, comparing wild-type ODC and C441 mutants. This assay provides a measure of proteasome recognition (Zhang et al, 2003). As shown in Figure 1A, wild-type ODC actively competes for the recognition of 26S proteasomes and inhibits the degradation of ODC. In contrast, ODCΔcODC, which lacks the degradation tag, cannot interact with the 26S proteasome and does not inhibit the degradation of wild-type ODC. Next, we examined ODC C441A and C441S mutants. Neither of these was active as a competitor. Their competition profiles resembled that of the ODCΔcODC negative control. In this assay, removing the C441 thiol (C441A) or replacing it with a hydroxyl (C441S) is as effective as removing the entire recognition tag. These results strongly suggest that the C441 thiol is required for association with the 26S proteasome, the earliest defined step in proteolysis.
Figure 1.
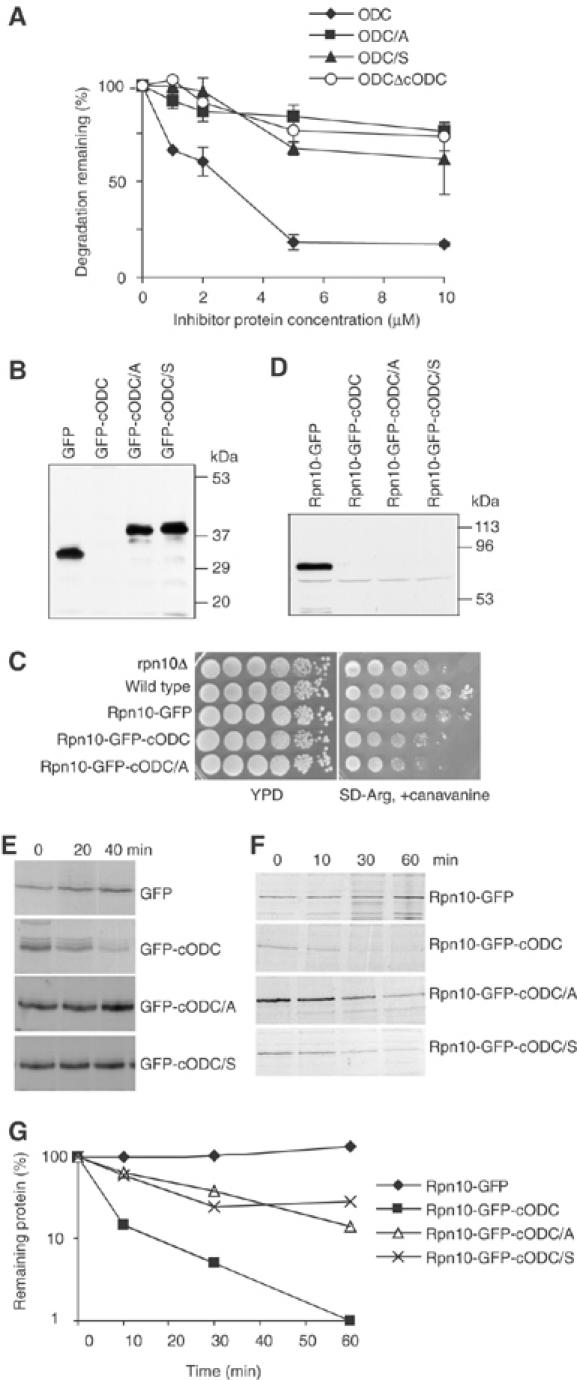
Proteasome association is required for degradation. (A) Role of C441 analyzed by competitive inhibition of ODC degradation. The effect on competitive activity of deleting or mutating cODC was analyzed. Here and in subsequent figures, cODC/S and cODC/A designate degradation tags with C441 Ser or Ala mutations. Data are normalized to ODC degradation observed in the absence of competing inhibitor protein and are expressed as percent residual degradation. Incubations with purified rat 26S proteasomes were for 1 h and contained various concentrations of inhibitors, as indicated. The extent of ODC degradation in the absence of inhibitors was 12–13%. Each plot represents the mean of two experiments; error bars indicate standard deviation. (B) Extract was prepared from wild-type cells expressing the indicated proteins. Western blots were developed with anti-GFP. (C) Yeast cells were grown to late logarithmic phase, serially diluted 10-fold and spotted on a non-selective YPD plate or on a plate selective for canavanine resistance. (D) as in (B), but with rpn10Δ cells. (E, F) Pulse–chase analysis of indicated fusion proteins was performed in wild-type (E) or rpn10Δ (F) cells. (G) The data of (F) were scanned and quantitated.
cODC provides more than an association element for the proteasome
The competition experiment shown above leaves open the question as to whether cODC is also needed for a step subsequent to proteasome association. If cODC is needed only for association, then supplying an alternative docking mechanism should bypass the effect of mutating its C441 residue or of removing the entire tag. To examine this, we used GFP and GFP-cODC as substrates in yeast cells. Western blot analysis with antibodies to GFP (Figure 1B) showed that GFP or GFP fused to a C-terminal copy of cODC with C441 mutated (GFP-cODC[C441A] and GFP-cODC[C441S]) is readily visualized, but that GFP-cODC with a wild-type copy of the tag, when expressed under identical conditions, is present at a level below the threshold of detection. This observation is consistent with previous results showing that the fusion protein formed by adding cODC to the C-terminus of GFP (GFP-cODC) is short-lived in yeast cells, and that mutating C441 is stabilizing (Hoyt et al, 2003). We expected that if C441 only works to mediate proteasome association, then providing an alternate binding pathway for GFP-cODC[C441A/S] would suppress its recognition defect and restore degradation. To provide such an alternate means of substrate–proteasome association, we fused GFP to the proteasome protein Rpn10. Rpn10 is an intrinsic protein of the proteasome. It is centrally placed within its 19S regulatory complex, stabilizes association between the 19S lid and base sub-assemblies (Glickman et al, 1998) and is a ubiquitin chain-binding protein (van Nocker et al, 1996). RPN10 is not essential for yeast cell viability, but its deletion confers sensitivity to the arginine analog canavanine. As shown in Figure 1C, Rpn10-GFP functionally complements the canavanine-sensitive phenotype of rpn10Δ cells, supporting the conclusion that the fusion protein can associate with the proteasome and provide some of the functional properties of native Rpn10. However, Rpn10-GFP-cODC did not suppress the canavanine sensitivity phenotype. This is the expected result if the fusion protein bearing the cODC degradation tag is extremely short-lived, and therefore not present in sufficient amount to complement the Rpn10 deficiency. In fact, Rpn10-GFP-cODC is not detectable by Western blot analysis whereas Rpn10-GFP is readily detected (Figure 1D). Like Rpn10-GFP-cODC, Rpn10-GFP-cODC[C441A] and Rpn10-GFP-cODC[C441S] are not detectable by Western blotting (Figure 1D), and do not suppress the canavanine sensitivity phenotype (Figure 1C). These observations suggested that the Rpn10 fusion restores a defect in proteasome association caused by mutating C441, restoring rapid degradation.
To test this directly, we performed pulse–chase experiments. Consistent with previously published data, the proteins GFP, GFP-cODC[C441A] and GFP-cODC[C441S] are extremely stable; their half-life is longer than 1 h. In marked contrast, GFP-cODC is short-lived, with a half-life of about 10 min (Figure 1E). Fusing each of these proteins to Rpn10, and thus providing them with an alternate means for association with the proteasome, drastically changed their stability pattern. Tethering GFP-cODC[C441A] and GFP-cODC[C441S] via Rpn10 reduced their half-life to about 20 min, but Rpn10-GFP, like GFP, was stable (Figure 1F and G). These data are consistent with the conclusion that C441 is a proteasome association element that can be bypassed by Rpn10-mediated tethering. Importantly, proteasome tethering via Rpn10 was not sufficient to cause degradation of GFP (Figure 1F and G). Destabilization additionally required either a wild-type cODC element or one bearing the C441 mutation. These data compel a further conclusion—that a mutant form of cODC becomes effective in the context of an alternate means of proteasome tethering, that provided by fusion to Rpn10. Rpn10-GFP-cODC, bearing a wild-type copy of cODC, is degraded extremely quickly, presumably because it has two potential interaction elements. The labile fusion proteins were visualized by Western blot analysis in a pre1-1, pre2-2 mutant, but were not seen in the ubiquitin-activating enzyme mutant uba1-2 strain (Supplementary Figure 1A), findings that imply that the degradation of these fusion proteins is proteasome-dependent but ubiquitin independent.
To further test whether degradation of C441 mutant Rpn10 fusion proteins depends on their tethering to proteasomes via Rpn10, we coexpressed Flag-Rpn10 to competitively inhibit the incorporation of Rpn10-GFP into proteasomes. These experiments used cells in which the chromosomal copy of RPN10 was disrupted. Flag-Rpn10 was encoded by a vector with the strong promoter GPD1. Affinity pulldown of proteasomes showed that the competition was effective: Flag-Rpn10 overexpression eliminated Rpn10-GFP from the 26S proteasome; in contrast, Rpn10-GFP was associated with proteasomes from cells expressing no Flag-Rpn10 (Figure 2A). In a further biochemical test of whether Rpn10 fusion proteins can enter the proteasome, we performed an in vitro competition experiment. 26S proteasomes lacking Rpn10 were prepared on an affinity matrix and incubated with labeled Rpn10-GFP in the presence of either an excess of unlabeled Rpn10-GFP protein or of a nonspecific control protein. Labeled Rpn10-GFP was captured by the 26S proteasome in the control, but was less efficiently captured when unlabeled Rpn10-GFP competitor was additionally present (Figure 2B).
Figure 2.
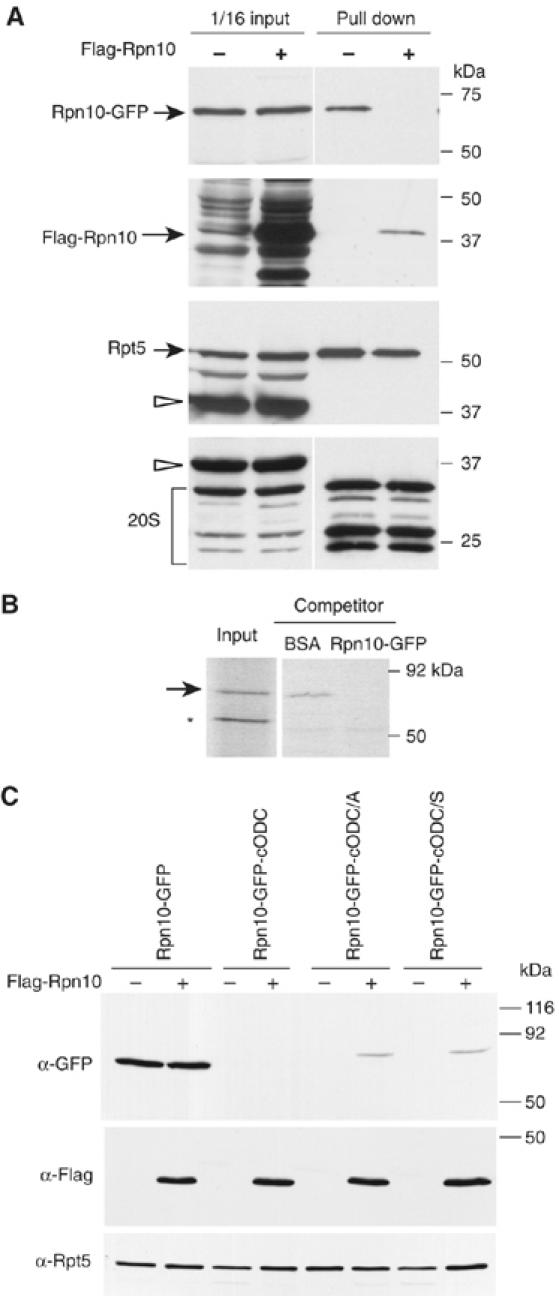
Rpn10-GFP fusion proteins are incorporated into the 26S proteasome. (A) Recovery of Rpn10-GFP with proteasomes and competition by coexpressed Flag-Rpn10. The PRE1-protein A, rpn10Δ cells expressed Rpn10-GFP and, additionally, either Flag-Rpn10 or none. 26S proteasomes were affinity purified, followed by Western blotting with antiserum to GFP, Flag, Rpt5 and the yeast 20S proteasome. Blots of extracts used for affinity purification are shown as well. Arrowheads indicate Pre1-protein A. (B) In vitro competition for proteasome association of Rpn10-GFP. Matrix-bound 26S rpn10Δ proteasomes and 35S-labeled Rpn10-GFP were co-incubated with 10 μg/ml of either BSA or unlabeled Rpn10-GFP, the matrix was washed and the bound proteins subjected to SDS–PAGE and autoradiography. The arrow indicates 35S-Rpn10-GFP, and the asterisk indicates nonspecific band. The input lane contained 1/10 of 35S-Rpn10-GFP used for incubations. (C) In vivo competition for proteasome degradation of Rpn10-GFP proteins. The indicated Rpn10-GFP fusion proteins were expressed in rpn10Δ cells either with or without excess Flag-Rpn10. Crude lysate was prepared from cells and subjected to Western blot analysis with anti-GFP and anti-Flag antibodies, and Anti-Rpt5 as is a loading control.
We then tested whether overexpression of Rpn10 could impair the capacity of Rpn10-dependent tethering to override the C441A and C441S mutations. We used the RPT5 promoter to express Rpn10-GFP fusion proteins and the stronger GPD1 promoter to express Flag-Rpn10. In the absence of competitor Rpn10 (empty vector control), Rpn10-GFP was visualized by Western blotting but Rpn10-GFP-cODC was not, because the latter is highly labile. Overexpression of Rpn10 had no effect on the signal associated with Rpn10-GFP or Rpn10-GFP-cODC. In contrast, Rpn10-GFP-cODC[C441A] and Rpn10-GFP-cODC[C441S] were visualized if Rpn10 was overexpressed, but not otherwise (Figure 2C). These results imply that Rpn10 overexpression stabilizes the Rpn10-GFP-cODC C441 mutant proteins by reducing their Rpn10-dependent proteasome tethering, but cannot impair degradation of substrates that instead utilize the intact cODC degradation tag for association. These data also support the conclusion that tethering per se is insufficient for degradation, as Rpn10-GFP is stable, but Rpn10-GFP-cODC[C441A] and Rpn10-GFP-cODC[C441S] are not. This implies that cODC provides two distinct functions required for degradation: the first function, association, depends on C441 and can be bypassed by an alternate means of association. The second function of cODC, one not dependent on C441, cannot be so bypassed and fulfills a different need, perhaps as a site at which proteasome entry is initiated.
Degradation by purified proteasomes also requires tethering plus a terminal extension
Next, we examined whether a similar pair of structural elements is also needed for substrate degradation in vitro. To support optimal tethering, 26S proteasomes were affinity purified from rpn10Δ yeast cells. Degradation of ODC or cODC-tagged proteins by affinity-purified yeast 26S proteasomes is only modestly effective (Hoyt et al, 2003; and our unpublished data), suggesting that the purification process excludes or fails to preserve an activity needed for the rapid processing of such substrates observed in vivo. Consistent with these previous observations with purified wild-type proteasomes, rpn10Δ proteasomes degraded GFP-cODC slowly (Figure 3A); GFP-cODC[C441A] and Rpn10-GFP were also slowly degraded; Rpn10 fusion was insufficient to promote degradation in the absence of a cODC or cODC[C441A] extension. In contrast, both Rpn10-GFP-cODC and Rpn10-GFP-cODC[C441A] were markedly degraded. The in vivo observation that GFP requires both a proteasome tether plus a terminal extension is thus reiterated in vitro using purified components, excluding the requirement for additional cellular constituents in substrate specification or degradation. We next tested whether Rpn10-dependent degradation observed in vitro depends on proteasome association via the Rpn10 moiety. As shown in Figure 3B, a five-fold molar excess of Rpn10 inhibited degradation of Rpn10-GFP-cODC[C441A], but GFP-cODC[C441A] did not. Thus, we conclude that the degradation can be ascribed to Rpn10-dependent association of substrate with the 26S proteasome and the presence of a terminal unstructured sequence.
Figure 3.
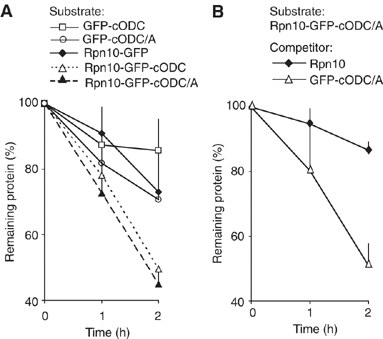
Both proteasome association and GFP extension are required for in vitro degradation. (A) The indicated Rpn10-GFP fusion proteins were incubated with rpn10Δ proteasomes and degradation measured by Western blotting with anti-GFP. (B) The degradation of Rpn10-GFP-cODC(C441A) protein by the rpn10Δ proteasome was examined as in (A) in the presence of a five-fold molar excess of either GFP-cODC(C441A) or Rpn10. Each plot represents the mean of two (A) or four (B) experiments; error bars (one sided) indicate standard deviation.
Substrates were present in these experiments at molar concentrations 30-fold in excess of proteasomes. In the case of the dually tagged substrates and rpn10Δ proteasomes, the extent of degradation over the 2-h reaction period was about 50%, implying, assuming saturation, that the proteasome requires about 8 min to fully process a substrate molecule. This rate is consistent with that seen in studies of in vitro processing of other folded substrates by purified proteasomes (Thrower et al, 2000; Zhang et al, 2003). Processing by wild-type proteasomes isolated from cells expressing endogenous Rpn10 was observed at about half this rate. The rate enhancement associated with adding a cODC extension to Rpn10-GFP was not owing to impaired folding of the GFP moiety, as the substrate recombinant proteins had very similar molar fluorescence and absorption spectra regardless of the presence of the extension (data not shown). Wild-type 26S proteasomes also degraded these substrates with similar specificity, although less efficiently than rpn10Δ proteasomes, and showed competitive inhibition with Rpn10 (Supplementary Figure 2). We speculate that wild-type proteasomes can provide significant access for Rpn10 tethering, albeit with lesser effectiveness than in the mutant case, either because Rpn10 equilibrates between a free form and one bound to a unique site of the 19S complex association, or because the 19S complex contains a secondary site for Rpn10 association.
Sequences that can furnish the post-association function of cODC
Having determined in vivo and in vitro that cODC has two distinguishable functions, we next examined the structural requirements of protein sequences that can perform its putative post-association function. As a surrogate for stability, we tested the steady-state level of proteins with different C-terminal extensions of Rpn10-GFP. We hypothesized that structural flexibility (or lack of structure) is an important determinant of cODC function as a site where proteasome entry initiates (Prakash et al, 2004), and used unstructured extensions similar in size to cODC. A truncation of the last five amino acids of cODC, to form Rpn10-GFP-cODCΔ5, did not impair turnover (Figure 4A). We next appended a part of the human DHFR, its first 34 amino acids, which does not participate in forming a tightly folded structure, and is still less likely to do so when removed from its native context. Rpn10-GFP-34DHFR was also not detected and therefore apparently unstable. We also fused a small and partially folded 35-residue alpha helix (Oakley and Kim, 1998) downstream from Rpn10-GFP. The Rpn10-GFP-helix protein was also not detectable by Western blot. We performed pulse–chase experiments to confirm that the reduced levels of Rpn10-GFP proteins with these C-terminal extensions result from their accelerated degradation. This proved to be true (Figure 4B). Thus, we conclude that a variety of terminal sequences can sustain degradation when the association process is bypassed.
Figure 4.
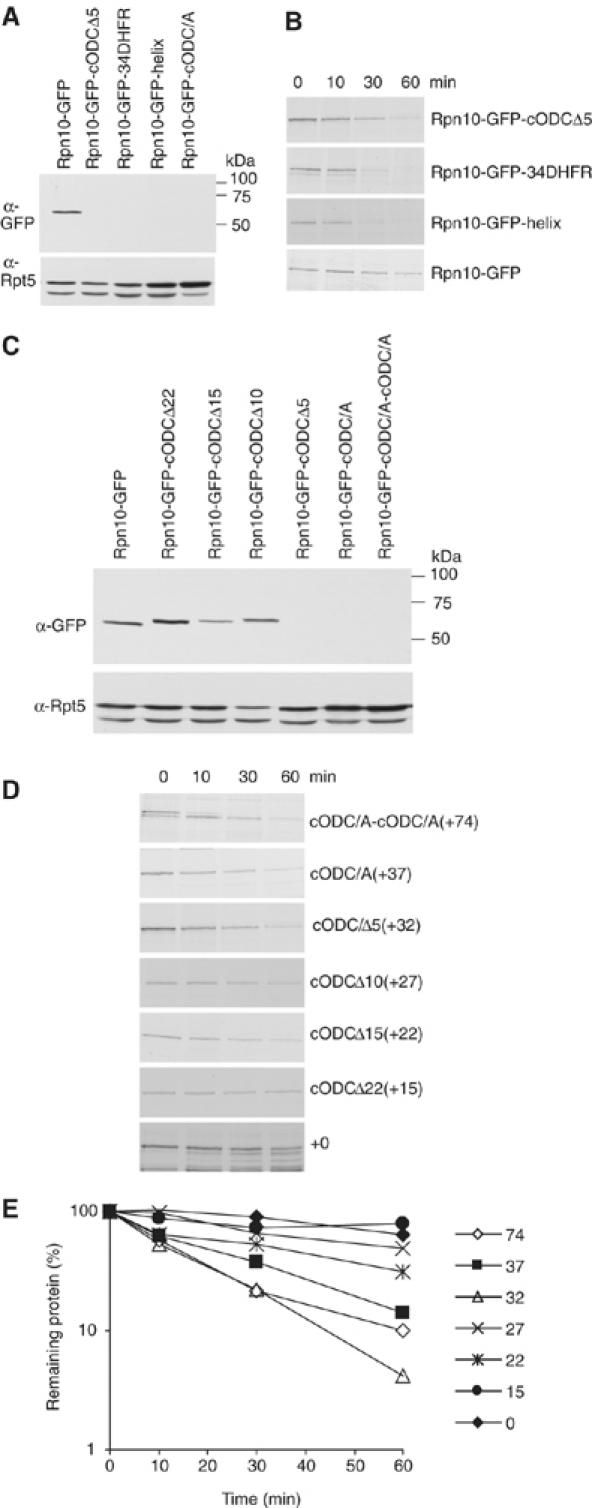
Various C-terminal extensions support degradation of Rpn10-GFP fusion proteins. (A) Rpn10-GFP proteins with the indicated C-terminal extensions were expressed and their steady-state level was analyzed by Western blot with anti-GFP. Loading was assessed with anti-Rpt5. (B) Pulse–chase analysis of proteins in (A). (C) Western blot and (D) pulse–chase analysis of Rpn10-GFP fusion proteins with deletions or a duplication of cODC. (E) Quantitation of the data of (D). For (A–D), Western blot and pulse–chase analysis utilized rpn10Δ cells.
Next, to determine the length of an unstructured sequence needed to confer degradation on tethered GFP, we progressively truncated the C-terminus of Rpn10-GFP-cODC. First, we performed Western blot analysis to see the steady-state level of proteins. As shown in Figure 4C, the construct with a +15 residue extension after GFP was stable, whereas those with +22 and +27 residue extensions were intermediate in stability compared with constructs with longer extensions +32 or +37. The construct with a +27 extension was reproducibly degraded slightly slower than that with +22. Extending GFP with two copies of cODC[C441A] to form Rpn10-GFP-cODC[2 × C441A] also resulted in destabilization, indicating that, within the limits tested, a functional extension can be longer than cODC. Pulse–chase data confirmed that the estimates of steady-state level provided a reasonably accurate assessment of stability (Figure 4D and E). These results imply that a nonspecific unstructured domain can provide the second signal needed for Rpn10-GFP degradation, a minimal extension beyond GFP of ∼20 residues is needed and one of ∼30 residues or more appears to provide enhanced degradation. In addition, there is no obvious dependence on sequence or composition.
An internal sequence can also furnish the second function of cODC
It has been reported that an internal unstructured sequence can initiate degradation (Liu et al, 2003; Prakash et al, 2004). To examine whether this is the case for our tethered protein, the same 35-residue alpha helix sequence previously used at the terminus of Rpn10-GFP (Figure 4A) was instead inserted within the 21-residue linker connecting Rpn10 and GFP in the proteins analyzed above, thus creating Rpn10-helix-GFP with an augmented linker of 56 residues. These proteins were stable in proteasome mutant pre1-1 pre2-2, but not in E1 mutant, uba1-2 (Supplementary Figure 1B), implying proteasome-dependent but ubiquitin-independent degradation. If degradation can start only from the C-terminus, Rpn10-helix-GFP like Rpn10-GFP should be stable. However, Rpn10-helix-GFP was not detectable by Western blot analysis (Figure 5A), indicating that inserting the helix destabilized the protein. A pulse–chase experiment revealed that the protein is expressed, but disappears quickly (Figure 5B and C). The fusion protein Rpn10-helix-GFP-cODC[C441A], which has a C-terminal unstructured domain, was degraded much faster than Rpn10-helix-GFP or Rpn10-GFP-cODC[C441A]. As Rpn10-GFP-cODC, proteins with either one or two [C441A] extensions are processed at similar rates, the much faster disappearance of Rpn10-helix-GFP-cODC[C441A], similar in total span to Rpn10-GFP-cODC[2 × C441A], cannot depend on the increased length conferred by introducing the helix. Its accelerated turnover suggests instead that degradation can start from either an internal or C-terminal region of Rpn10-helix-GFP-cODC[C441A]. Thus, we infer that either an internal or terminal unstructured or loosely structured region can initiate degradation.
Figure 5.
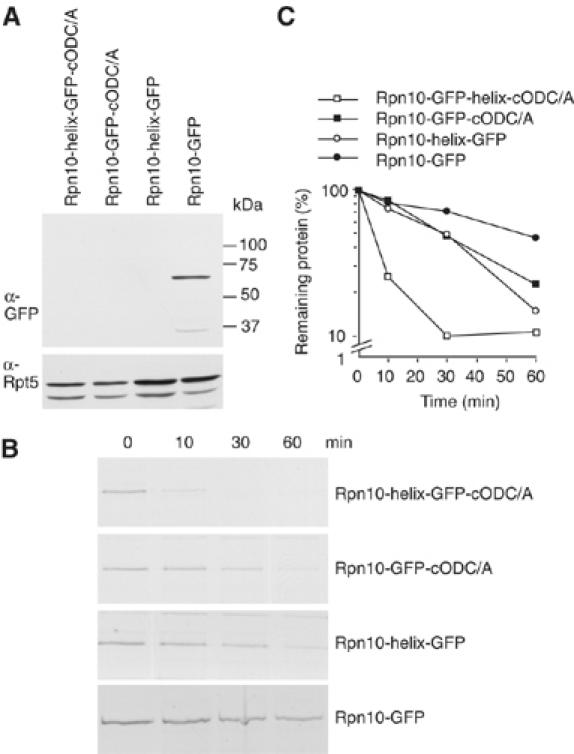
Internal sequence can also initiate degradation. (A) Rpn10-GFP proteins of the indicated structure were expressed and their steady-state level was analyzed by Western blot with anti-GFP and anti-Rpt5. (B) Pulse–chase analysis of proteins in (A). (C) Quantitation of the data of (B). For (A–C), rpn10Δ cells were used.
Discussion
What minimum functional and structural properties make a protein the target for degradation by the proteasome? Native substrates carry recognition tags that, when present, deliver proteins for degradation. The tags confer specificity, distinguishing substrates from other proteins. A ubiquitin chain is the most common tag of this kind. Does degradation depend simply on delivery to the near vicinity of a proteasome, or are there other requirements? The question was recently examined by tethering the enzyme His3 (imidazoleglycerol-phosphate dehydratase), normally a stable protein, to the proteasome (Janse et al, 2004). This was brought about by fusing Fpr1 to a proteasome protein and also attaching the Fpr1–rapamycin-binding domain of Tor1 to His3. Adding rapamycin linked Fpr1 to Tor1, thereby tethering His3 to the proteasome. Linking His3 to either Rpn10 (in the proteasome regulatory complex) or Pre10 (in the catalytic core) caused its degradation, whereas several control conditions that precluded tethering prevented degradation. Linking His3 to the proteasome in this way promoted its degradation both in intact cells and in biochemical experiments with purified components. It was thus concluded that tethering, which creates a high local concentration of His3 near the proteasome, is sufficient to convert a non-substrate to a substrate.
Matouschek and co-workers reached a different conclusion: an effective substrate requires not only a means of docking to the proteasome, but also an unstructured region (Prakash et al, 2004) that acts as a site for initiation of unfolding. Bacterial compartmented proteases seemingly operate according to similar principles, whereby substrate-targeting regions contain unstructured elements (Levchenko et al, 1997) at which unfolding begins (Kenniston et al, 2004). This region, which may lie at the terminus of the protein or constitute a surface loop, is the first portion of the substrate to enter the protease, offers the protease a purchase for unfolding the substrate, and is the first to be interiorized and hydrolyzed. According to this hypothesis, a ubiquitin chain is sufficient for proteasome docking, and folded ubiquitin-linked proteins can find their way to the proteasome, but the necessary next step, unfolding, cannot be initiated efficiently in the absence of an unstructured initiation site. How are these views and findings to be reconciled?
Here we have broadened the experimental approach to answering these questions. We used the cODC degradation tag, which is independent of ubiquitin. cODC and ubiquitin chains cross-compete for proteasome degradation, implying that the former is a molecular mimic of the latter (Zhang et al, 2003). cODC is, however, more readily subjected to manipulation of its structure than ubiquitin conjugates. Using a series of such manipulations, we show here that cODC has two functions. One is proteasome association; this is dependent on a specific cysteine thiol, that of C441. If the thiol is absent or replaced by a hydroxyl, the tag can no longer mediate association with the proteasome. If a protein bearing a form of cODC with a point mutation in C441 is provided with an alternate way to associate with the proteasome, degradation is restored. This confirms that proteasome association is a necessary function of cODC, one that requires the specific thiol, and demonstrates that the native means of association can be bypassed by a different form of tethering. However, docking in this alternate mode does not make degradation independent of cODC; it bypasses the need for the correctly positioned thiol, but mutated cODC (or a surrogate) is still needed to provide some function other than docking. Thus, association of substrates with the 26S proteasome is clearly dissected from the following process, entry of the substrate into the 26S proteasome. The requirement of loosely structured domain as an entry site is not sequence-specific, for unrelated peptides similar in size to cODC can also restore degradation of the artificially tethered protein. This suggests that a nonspecific accessible domain of adequate size and with conformational flexibility is needed. The small cODC tag provides two distinct functions through two distinguishable structural features: proteasome docking, which requires the thiol at position 441, and a terminus that is exposed, flexible and engages the proteasome to initiate unfolding, but is otherwise not strongly constrained in structure. This view is consistent with previous observations demonstrating that native ODC is unfolded and degraded starting from the native C terminal domain of ODC, the position of cODC. NMR analysis has revealed that cODC contains some highly flexible residues (D Hoffman (University of Texas, Austin) and P Coffino, unpublished). Although the degradative function of cODC requires it to be placed at the C terminus, Rpn10-tethered constructs can utilize an unstructured tag located at the terminus or internally.
The C-terminal extensions of Rpn10-GFP that support degradation are diverse in size, sequence and structure. The results are consistent with the hypothesis that degradation demands a region that is not tightly folded. None of those tested were β-sandwich domains, the protein structures that are most resistant to mechanical unfolding (Carrion-Vazquez et al, 2000). GFP is hard to unfold and consists of an 11-sheet β-barrel. The finding that extensions of GFP providing optimal degradation were the approximate length of cODC or greater suggests a priming mechanism that responds most favorably to an extended region of that length or more. It is unlikely that precise positioning of the extension or its terminus with respect to the proteasome is required. Stringent positional specificity is excluded because an extension of 74 residues is effective, as is an internal region positioned between Rpn10 and GFP. The latter finding shows as well that a free terminus is not required. One can speculate that the molecular apparatus used for tethering substrate to proteasome in the experiments of (Janse et al, 2004), the Fpr1 and Tor1 protein tags, provided a sufficiently extended region for initiation of degradation.
Native Rpn10 in cell extracts is found mainly in the proteasome-associated fraction and but also in free pools (van Nocker et al, 1996; Kominami et al, 1997; Glickman et al, 1998; Fu et al, 2001), as is Rpn10-GFP (results not shown). If free and bound pools are in equilibrium, Rpn10 must have a significant off rate. This implies that a process of proteasome ingestion and proteolysis, which begins at the C-terminus of Rpn10-GFP-cODC, will go to completion and result in extraction of the Rpn10 moiety from its position in the proteasome. The canavanine resistance phenotype of cells expressing the various Rpn10 fusions is consistent with the conclusion: cells with stable Rpn10 or Rpn10-GFP are resistant, whereas those expressing unstable Rpn10 fusions are sensitive.
cODC in its native context acts as a proteasome entry site (Zhang et al, 2004), and an analytic assumption of the foregoing has been that cODC and its surrogates act similarly in the context of Rpn10-GFP. GFP has but a few residues at its C-terminus that are not tightly structured, and this terminus plausibly requires a further extension to act as a proteasome entry site. However, the data demonstrating that extensions of Rpn10-GFP longer than 15 residues provide progressively better degradation are also consistent with an alternate interpretation: the GFP C-terminus is fully adequate to undergo insertion even without further extension, but, measured from the site of Rpn10 binding, the full span of Rpn10-GFP simply cannot reach the requisite insertion site of the proteasome. In this alternate interpretation, which applies also to the helical insert between Rpn10 and GFP, the extensions do not change the character of the C-terminus, but its reach. To test this alternate interpretation of the data, we changed the length of our standard 21-residue linker between Rpn10 and GFP, reducing it to an 8-mer. Each Rpn10-GFP construct bearing a C-terminal extension of specific length (0–37 residues) retained its individual degradative property, regardless of whether a linker of 21 or 8 residues was present (Supplementary Figure 3). This finding contradicts the prediction of the alternate model specifying total span as the determinant of degradation.
The claim that proteasome association is sufficient for degradation must be qualified, at the least, by the finding that proteins that are themselves intrinsic parts of the proteasome are stable. In addition, proteins, like Rad23 and Dsk2, which transiently associate with proteasomes, contain both UBL and UBA domains and act as substrate degradation shuttles, are not degraded. Their stability depends on the protective effect of a conserved C-terminal UBA domain (Heessen et al, 2005). The present findings raise the general question of how the working parts of the proteasome escape degradation. Two possibilities seem attractive. One is that, like UBA/UBL proteins, some bear a cis-acting inhibitory component. Most, however, may simply lack protuberant extensions that place them at risk for engulfment.
We have shown that a substrate must not only be brought to the proteasome but, once there, present an accessible region of sufficient size. This finding enhances the opportunities for experimental and therapeutic intervention in proteasome function. A better understanding of these structural requisites may offer clues as to how proteins can effectively be manipulated to provide conditional on/off switches for degradation.
Materials and methods
Strain and plasmid construction
The plasmids used in this study are tabulated in a supplement. Yeast manipulations followed standard methods (Guthrie and Fink, 1991). MHY501 (Chen et al, 1993) was used as the wild-type background strain. Other strains are isogenic to MHY501, unless noted.
MHY501: his3-D200, leu2-3,112, lys2-801, trp1-1, ura3-52, MATα
J501: rpn10Δ::LEU2
MHY91: PRE1-proteinA:TRP1, rpn10Δ::LEU2
MHY94: RPN11-proteinA:TRP1
MHY98: RPN11-proteinA:TRP1, rpn10Δ::LEU2
To create yeast expression vectors of Rpn10-GFP fusion genes, the promoter regions of RPT5 and RPN10 were PCR-amplified using Saccharomyces cerevisiae chromosome DNA as a template. The coding region plus the 5′ upstream region of GFPuv (BD Biosciences/Clontech, Palo Alto, CA), GFPuv-cODC (Hoyt et al, 2003) and its C441 mutation versions were amplified by PCR and ligated onto the SacI/EcoRI gap of a p416 vector (Mumberg et al, 1995) together with the above DNA fragments. The resulting plasmids (pJ1, 2, 3 and 33) encode a 21-amino-acid linker (-GGRSLHACRSTLEDPRVPVEK-) between the Rpn10 and GFP reading frames. Constructs encoding cognate fusion proteins with a shorter linker (-GGRVPVEK-) were constructed by restriction/religation within the multi-cloning site linking RPN10 and GFP genes. Other plasmids (listed in the table of Supplementary data) were created by PCR amplification of various extensions plus GFP-coding region, followed by ligation onto the p416 vector with RPT5 promoter and Rpn10. The helix linker used in this study is as follows, with the helix region underlined:
–GGRGG-AQLKKKLQALKKKNAQLKWKLQALKKK-LAQ-
Preparation of yeast crude extract and 26S proteasomes
For Western blot analysis of crude extracts, yeast cells in logarithmic growth at 30°C were harvested, washed and resuspended in lysis buffer A (50 mM HEPES (pH 7.5), 1% Triton X-100, 150 mM NaCl, 1 mM EDTA and 1 mM PMSF). Cells were disrupted by shaking with glass beads in a Mini-beadbeater (Biospec Products) with four cycles of 30 s pulse, followed by 30 s cooling. The supernatant was collected after centrifugation (18 000 g, 15 min) and subjected to Western blotting. Antibodies were as follows: anti-GFP polyclonal antibody (Living Colors full-length A versus polyclonal antibody) was purchased from BD Biosciences/Clontech. Anti-Rpt5 was used as a proteasome loading standard, and was purchased from Biomol (Plymouth Meeting, PA). The secondary antibody, HRP-conjugated anti-rabbit or anti-mouse IgG, and ECL detection kit were purchased from GE Health Care. The preparation and affinity purification of yeast 26S proteasomes was carried out in lysis buffer B (50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 10% glycerol, 2 mM ATP and 5 mM MgCl2). In brief, yeast crude extract was prepared as described above, but using 20 s Mini-beadbeader pulses. Crude extract from PRE1-TEV-protein A- or RPN11-TEV-protein A-tagged strains was incubated with rabbit IgG agarose resin (Sigma, St Louis, MO) for 2 h at 4°C to collect proteasomes via the proteinA tag. After washing four times with lysis buffer B containing 0.2% Triton X-100, the resin was treated with 6His-TEV protease (Invitrogen, Carlsbad, CA) for 1 h at 30°C to release proteasomes. After removing TEV protease with Ni-NTA resin, the supernatant was used as the 26S proteasome. For Western blotting of affinity-purified proteasomes, human IgG agarose resin (ICN Biomedicals, Aurora, OH) was used to captured protein A-tagged proteasomes. These were released by TEV protease treatment after washing with lysis buffer B. The rabbit IgG TrueBlot (eBioscience) was used as a secondary antibody in the GFP, Rpt5 and the 20S proteasome immunoblots. Anti-20S proteasome antibody was a gift from T Tamura (AIST, Japan).
Pulse–chase analysis
Labeling and sample treatment was as described in Hoyt et al (2003). Growing cells were washed with SD media, resuspended in SD-Met containing [35S]methionine/cysteine (NEN), 50 μCi of isotope per time period and labeled for 5 min at 30°C. Cells were harvested, resuspended in SD-Ura, 10 mM methionine/cysteine, 0.5 mg/ml cycloheximide and further incubated at 30°C. Incubations were halted by resuspension in lysis buffer A. Cells were disrupted as described, followed by centrifugation at 18 000 g for 10 min. The volume of sample for immunoprecipitation was adjusted to contain equal amounts of acid-insoluble radiolabel. Anti-GFP polyclonal antibody (1:1000 dilution) and protein A-Sepharose CL4B (GE Healthcare, Upsalla, Sweden) were added to cell lysate, followed by 2 h of incubation at 4°C. The resin was then washed four times with lysis buffer A containing 0.1% of SDS and boiled with SDS–PAGE sample buffer.
Preparation of recombinant proteins
GFP-cODC and GFP-cODC[C441A] were cloned into the pQE30 vector (Qiagen) and expressed in Escherichia coli XL1-Blue strain and affinity purified by Talon metal affinity resin (Clontech) according to the manufacturer's instructions. The coding regions of RPN10-GFP, RPN10-GFP-cODC and RPN10-GFP-cODC(C441A) in pJ1, pJ2 and pJ3 were amplified by PCR and inserted following the 6His tag of the pET28b vector, and respectively designated as pJ7, pJ184 and pJ185. Proteins were produced in BL21(DE3) strain and purified as above.35S-labeled Rpn10-GFP was prepared following the manufacturer's instructions with the Promega TNT-coupled reticulocyte lysate system. Template DNA for in vitro transcription/translation was prepared by PCR amplification using the pJ7 plasmid as a template and the following pair of primers: T7-Rpn10/JT65 5′-gtaatacgac tcactatagg gcATGGTATT GGAAGCTACA GTG- (sequences corresponding to the ORF of RPN10 are in uppercase) and GFPuv AS/JT4 5′-gggaattcat tatttgtaga gc-.
In vitro proteasome degradation and competition assays
Degradation of 35S-labeled ODC by purified rat 26S proteasomes and assay of competitive inhibition by unlabeled recombinant wild-type or mutant ODCs were performed and analyzed as described in Zhang et al (2003), and constructs for expression of recombinant proteins were as in Chen et al (2002). 35S-labeled wild-type ODC was incubated with purified rat 26S proteasomes for 1 h at 37°C and acid soluble counts were measured. Yeast proteasomes were purified from yeast strain MHY94 or MH98 as described above. Reactions contained 50 mM Tris (pH 7.5), 2 mM ATP, 1 mM DTT, 5 mM MgCl2, 0.3 mg/ml BSA, 10 nM 26S proteasome and 300 nM of substrates. Remaining substrate protein undegraded was analyzed by Western blotting using anti-GFP antibody, followed by scanning and quantification.
Determination of canavanine sensitivity
Yeast cells were grown to late logarithmic phase, diluted and spotted on SD-Arg with 1.5 μg/ml of L-canavanine or on a YPD plate and incubated at 30°C for 5 or 2 days, respectively. Cells were spotted at 3 × 107 cells/spot and 10 × serial dilutions thereof.
Quantitation of Western blot and autoradiographic band intensity
Blots were scanned and quantitatively analyzed by Totallab software (Nonlinear Dynamics, UK).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplement to Methods
Acknowledgments
JT was supported by a fellowship from the Uehara Memorial Foundation. This work was supported by NIH grants GM45335 and GM074760 to PC. We thank Tomohiro Tamura for providing anti-yeast 20S proteasome antibody.
References
- Carrion-Vazquez M, Oberhauser AF, Fisher TE, Marszalek PE, Li H, Fernandez JM (2000) Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog Biophys Mol Biol 74: 63–91 [DOI] [PubMed] [Google Scholar]
- Chen H, MacDonald A, Coffino P (2002) Structural elements of antizymes 1 and 2 required for proteasomal degradation of ornithine decarboxylase. J Biol Chem 277: 45957–45961 [DOI] [PubMed] [Google Scholar]
- Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M (1993) Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MATà2 repressor. Cell 74: 357–369 [DOI] [PubMed] [Google Scholar]
- Fu H, Reis N, Lee Y, Glickman MH, Vierstra RD (2001) Subunit interaction maps for the regulatory particle of the 26S proteasome and the COP9 signalosome. EMBO J 20: 7096–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoda L, van Daalen Wetters T, Macrae M, Ascherman D, Coffino P (1989) Prevention of rapid intracellular degradation of ODC by a carboxyl-terminal truncation. Science 243: 1493–1495 [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A (2002) The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82: 373–428 [DOI] [PubMed] [Google Scholar]
- Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D (1998) A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 94: 615–623 [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR (eds) (1991) Guide to Yeast Genetics and Molecular Biology. San Diego: Academic Press [Google Scholar]
- Heessen S, Masucci MG, Dantuma NP (2005) The UBA2 domain functions as an intrinsic stabilization signal that protects Rad23 from proteasomal degradation. Mol Cell 18: 225–235 [DOI] [PubMed] [Google Scholar]
- Hochstrasser M (1996) Ubiquitin-dependent protein degradation. Annu Rev Genet 30: 405–439 [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Coffino P (2003) A ubiquitin-free pathway into the proteasome. Cellular and Molecular Life Sciences 61: 1596–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, Zhang M, Coffino P (2003) Ubiquitin-independent mechanisms of mouse ornithine decarboxylase degradation are conserved between mammalian and fungal cells. J Biol Chem 278: 12135–12143 [DOI] [PubMed] [Google Scholar]
- Janse DM, Crosas B, Finley D, Church GM (2004) Localization to the proteasome is sufficient for degradation. J Biol Chem 279: 21415–21420 [DOI] [PubMed] [Google Scholar]
- Kenniston JA, Burton RE, Siddiqui SM, Baker TA, Sauer RT (2004) Effects of local protein stability and the geometric position of the substrate degradation tag on the efficiency of ClpXP denaturation and degradation. J Struct Biol 146: 130–140 [DOI] [PubMed] [Google Scholar]
- Kominami K, Okura N, Kawamura M, DeMartino GN, Slaughter CA, Shimbara N, Chung CH, Fujimuro M, Yokosawa H, Shimizu Y, Tanahashi N, Tanaka K, Toh-e A (1997) Yeast counterparts of subunits S5a and p58 (S3) of the human 26S proteasome are encoded by two multicopy suppressors of nin1-1. Mol Biol Cell 8: 171–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Smith CK, Walsh NP, Sauer RT, Baker TA (1997) PDZ-like domains mediate binding specificity in the Clp/Hsp100 family of chaperones and protease regulatory subunits. Cell 91: 939–947 [DOI] [PubMed] [Google Scholar]
- Liu CW, Corboy MJ, DeMartino GN, Thomas PJ (2003) Endoproteolytic activity of the proteasome. Science 299: 408–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher P, Pratt G, Rechsteiner M (1991) The C-terminus of mouse ornithine decarboxylase confers rapid degradation on dihydrofolate reductase. J Biol Chem 266: 11213–11220 [PubMed] [Google Scholar]
- Miyazaki Y, Matsufuji S, Murakami Y, Hayashi S (1993) Single amino-acid replacement is responsible for the stabilization of ornithine decarboxylase in HMOA cells. Eur J Biochem 214: 837–844 [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122 [DOI] [PubMed] [Google Scholar]
- Murakami Y, Matsufuji S, Hayashi SI, Tanahashi N, Tanaka K (1999) ATP-Dependent inactivation and sequestration of ornithine decarboxylase by the 26S proteasome are prerequisites for degradation. Mol Cell Biol 19: 7216–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Matsufuji S, Kameji T, Hayashi S, Igarashi K, Tamura T, Tanaka K, Ichihara A (1992) Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature 360: 597–599 [DOI] [PubMed] [Google Scholar]
- Oakley MG, Kim PS (1998) A buried polar interaction can direct the relative orientation of helices in a coiled coil. Biochemistry 37: 12603–12610 [DOI] [PubMed] [Google Scholar]
- Pegg AE (1986) Recent advances in the biochemistry of polyamines in eukaryotes. Biochem J 234: 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Cohen RE (2004) Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol 5: 177–187 [DOI] [PubMed] [Google Scholar]
- Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A (2004) An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol 11: 830–837 [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J 19: 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nocker S, Sadis S, Rubin DM, Glickman MH, Fu H, Coux O, Wefes I, Finley D, Vierstra RD (1996) The multiubiquitin chain binding protein Mcb1 is a component of the 26S proteasome in Saccharromyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol Cell Biol 11: 6020–6028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Deshaies RJ (2000) A proteasome howdunit: the case of the missing signal. Cell 101: 341–344 [DOI] [PubMed] [Google Scholar]
- Zhang M, MacDonald AI, Hoyt MA, Coffino P (2004) Proteasomes begin ornithine decarboxylase digestion at the carboxy terminus. J Biol Chem 279: 20959–20965 [DOI] [PubMed] [Google Scholar]
- Zhang M, Pickart CM, Coffino P (2003) Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate. EMBO J 22: 1488–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplement to Methods