

Abstract
Transcriptional regulation by the androgen receptor (AR) is critical for male sexual development and prostate cancer. In this study, we used an expression cloning strategy to identify molecules that regulate AR-driven transcription. Screening of a human cDNA library resulted in isolation of caspase-8 (Casp8), an initiator caspase that mediates death-receptor-induced apoptosis. Casp8 repressed AR-dependent gene expression independently of its apoptotic protease activity by disrupting AR amino-terminal and carboxy-terminal (N/C) interaction and inhibiting androgen-induced AR nuclear localization. Protein–protein interaction analysis revealed that three motifs in Casp8 specifically interacted with the motifs that are known to be involved in AR N/C interaction. Substitutions of the amino-acid residues critical for AR–Casp8 interactions abolished the Casp8-mediated inhibition of AR transactivation. In addition, knockdown of Casp8 by RNA interference specifically affected the androgen-dependent expression of AR-targeting genes in LNCaP cells. These results indicate that Casp8 has a novel function beyond its known role in the mediation of apoptosis.
Keywords: androgen receptor, caspase-8, cofactor, nuclear receptor, transcription
Introduction
The androgen receptor (AR) mediates androgen functions in male sex differentiation, and mutations in AR cause androgen insensitivity syndrome, which is characterized by incomplete male sexual development (Quigley et al, 1995). AR signaling plays a pivotal role in the development and maintenance of the normal prostate and is linked to prostate tumorigenesis and prostate cancer progression (Craft et al, 1999; Dehm and Tindall, 2005; So et al, 2005).
AR is a ligand-activated transcription factor and contains a carboxy-terminal ligand-binding domain (LBD), a central DNA-binding domain, and an NH2-terminal domain (Tsai and O'Malley, 1994; Mangelsdorf et al, 1995). A unique property of AR that distinguishes it from other steroid receptors is that androgen binding stabilizes AR protein, whereas other steroid receptors are downregulated by ligand binding (Langley et al, 1995; Ikonen et al, 1997; Berrevoets et al, 1998; He and Wilson, 2002). Androgen-induced AR stabilization results in part from N-terminal and C-terminal (N/C) interaction (Langley et al, 1995, 1998). AR N/C interactions are mediated by two N-terminal motifs (23FQNLF27 and 433WHTLF437) and the C-terminal AF2 (He et al, 2000, 2004). Several AR mutations that are responsible for androgen insensitivity syndrome disrupt the N/C interaction, suggesting the physiological significance of this interaction (He and Wilson, 2002; Wilson et al, 2003). Recent studies have suggested that AR N/C interaction is modulated by AR cofactors (Shenk et al, 2001; Bai et al, 2005; Hsu et al, 2005). Hence, the N/C interaction provides an additional basis for AR regulation in response to various signals.
Studies have identified various cofactors and proposed that they play important roles in the regulation of AR functions (Brinkmann et al, 1999; Janne et al, 2000; Gelmann, 2002; Heinlein and Chang, 2002). The detailed molecular mechanisms of AR cofactor action in modulation of AR-driven gene expression are still elusive. Some AR cofactors possess histone acetyltransferase activity or histone methyltransferase activity; therefore, they may activate AR transactivation as a result of their ability to remodel the chromatin structure (Xu, 2005). On the other hand, negative AR cofactors such as histone-deacetylase-containing complexes reduce AR transcriptional activity by regulating the status of histone acetylation (Baniahmad, 2005; Wang et al, 2005). Post-translational modifications of AR by cofactors such as CBP/p300 may modulate AR activity (Fu et al, 2003). Other AR cofactors lacking such enzymatic activities may function through multiple mechanisms, including control of ligand/AR binding affinity (Dedhar et al, 1994; Sato et al, 1997), AR stability (Lee and Chang, 2003a), AR subcellular localization (Poukka et al, 2000), or recruitment of basal transcription machinery (Lee and Chang, 2003b).
Caspases play critical roles in initiation and execution of programmed cell death (Shi, 2002). They constitute a family of cysteine proteases that cleave target proteins at sites next to aspartic acid residues. Caspase-8 (Casp8) is the initiator caspase for death-receptor-mediated apoptosis (Salvesen, 1999). Like other members of the caspase family, Casp8 is initially synthesized as a zymogen with two death effector domains at the N-terminus and a protease domain at the C-terminus (Boatright and Salvesen, 2003). However, increasing evidence suggests that caspases also have functions in addition to their known apoptotic roles (De Maria et al, 1999; Faouzi et al, 2001; Franchi et al, 2003; Kang et al, 2004). For examples, Casp8 activates NF-κB signaling independently of its activity as a proapoptotic protease (Chaudhary et al, 2000; Hu et al, 2000). Further genetic and biochemical analyses have shown that Casp8 functions both as a pivotal molecule for death-receptor-induced apoptosis and as a selective signal transducer for NF-κB activation by antigen receptor (Su et al, 2005). During Drosophila oogenesis, loss of function of the dcp-1 gene, which encodes a caspase (Drice), led to nurse cells defective in cytoskeletal reorganization and nuclear breakdown that normally accompany the apoptotic process (McCall and Steller, 1998). Expression of the active Drice (tdcp-1) led to widespread apoptosis and dying egg chambers similar to the normal nurse cell death that occurs in late oogenesis (Peterson et al, 2003). However, the cytoskeletal events induced by tdcp-1 were significantly different from those normally seen in nurse cells during late oogenesis. These observations suggest that the cytoskeletal events may be Drice (caspase)-dependent but caspase-enzymatic activity-independent processes.
Using a human cDNA-expressing library, we isolated Casp8 and found that it inhibits AR transcriptional activity by disrupting AR N/C interaction and inhibiting androgen-induced AR nuclear localization.
Results
Casp8 is identified as a negative AR cofactor
We used a highly sensitive quantitative reporter assay (Pomerantz et al, 2002) to identify factors capable of regulating AR-dependent transcription. A pool of cDNAs in a mammalian expression vector was transiently transfected into PC3mm2 cells with the pGL3-4 × ARE-E4-luc reporter. From four pools (total 20 000 cDNAs), six positive cDNAs were obtained. One of these positive cDNAs encoded the full-length Casp8. To confirm the effect of Casp8 on AR-dependent transcription, the pGL3-4 × ARE-E4-luc reporter was cotransfected with expression plasmids for AR and/or Casp8 into PC3mm2 cells in the absence or presence of androgen (R1881). AR activated the reporter about 34-fold in the presence of androgen, and coexpressed Casp8 inhibited this activity in a dose-dependent manner (up to 13-fold) (Figure 1A). Casp8 also strongly inhibited AR-driven transcription from the natural prostate-specific androgen (PSA) enhancer (−4354 to −858) reporter construct (Figure 1B) (Schuur et al, 1996; Cleutjens et al, 1997) but did not inhibit Gal4-p53-driven transcription from the pGL3-4 × Gal4-E4-luc reporter (Figure 1C). Expression of Casp8 in transfected cells was demonstrated by RT–PCR (Figure 1A, middle panel) and Western blot analysis (Figure 1A, bottom panel).
Figure 1.
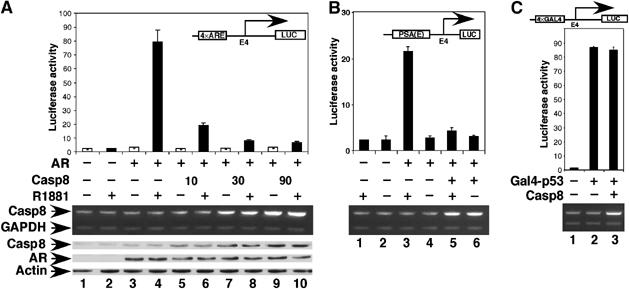
Casp8 downregulates AR transcriptional activity in a dosage-dependent manner. pGL3-4 × ARE-E4-Luc (A), pGL3-PSA(E)-E4-luc (B), or pGL3-4 × Gal4-E4-luc (C) reporter (100 ng), pcDNA-AR (30 ng) or pcDNA-Gal4-p53 (10 ng) and indicated amounts of pcDNA-Casp8 were cotransfected into PC3mm2 cells. Transfected cells were grown in the absence or presence of 10 nM R1881 for 48 h. Cells were then harvested for luciferase assay (top panel), RT–PCR (middle panel), and Western blot analysis (bottom panel). Values represent mean±s.d. (N=3).
Similar assay was performed to determine whether expression of other caspases affects AR-driven transcription. AR activated the reporter about 36-fold in the presence of androgen, and coexpressed Casp8 inhibited this activity (12-fold) (Figure 2). In contrast, expression of Casp3, Casp6, Casp7, and Casp9 did not significantly inhibit AR-driven transcription from the pGL3-4 × ARE-E4-luc reporter.
Figure 2.
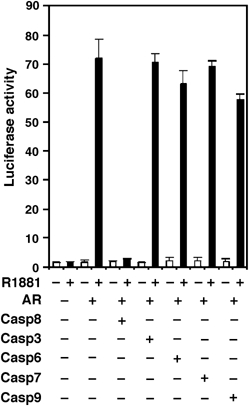
Caspases other than Casp8 do not inhibit AR-driven transcription. pGL3-4 × ARE-E4-luc reporter (100 ng), pcDNA-AR (30 ng), or indicated caspase-expressing constructs (30 ng) was cotransfected into PC3mm2 cells. Transfected cells were grown in the absence or presence of 10 nM R1881 for 48 h. Cells were then harvested and used for luciferase assay. Values represent mean±s.d. (N=3).
To investigate whether expression of Casp8 affects levels of AR protein, we performed Western blot analysis of AR. Casp8 inhibited AR-driven promoter activity by 93% (14-fold; Figure 3A, top panel), and levels of AR protein decreased by 50% in the presence of Casp8 compared with those in the absence of Casp8 (middle panel, lane 6 versus lane 5). The decrease in AR protein by 50% only slightly lowered the AR-driven reporter activity (Figure 3A, lane 4 versus lane 5). These results indicated that the decrease in AR protein expression induced by Casp8 expression did not account for loss of AR transcriptional activity mediated by Casp8.
Figure 3.
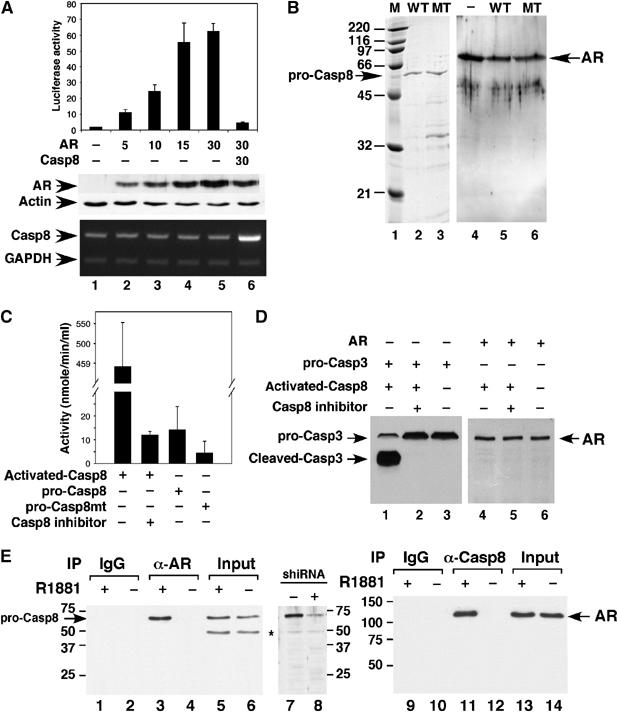
(A) Casp8-mediated inhibition of AR transcriptional activity did not result from AR degradation. pGL3-4 × ARE-E4-luc (100 ng), the indicated amounts of pcDNA-f:AR, and pcDNA-f:Casp8 were transfected into PC3mm2 cells. Transfected cells were grown in the presence of 10 nM R1881 for 48 h and then harvested for luciferase assay (upper panel), Western blot analysis (middle panel), and RT–PCR (bottom panel). Values represent mean±s.d. (N=3). (B) Casp8 did not cleave AR in vitro. FLAG epitope-tagged Casp8 (wild-type) and mutant (C345A, which lacks enzymatic activity) were expressed in PC3 cells and immunopurified using M2-agarose beads. Wild-type (WT; lane 2) and mutant (MT; lane 3) Casp8 (100 ng) were analyzed using 12% SDS–PAGE and then stained with Coomassie blue R250. Lane 1, standard molecular weight markers (kDa) (Bio-Rad). Recombinant AR (40 μg) was incubated without (lane 4) or with wild-type (lane 5) or mutant (lane 6) Casp8 (50 μg) in a buffer containing 100 mM HEPES, pH 7.5, and 10 mM DTT in a total volume of 100 μl for 20 min at 37°C. Reaction mixtures were submitted for Western blot analysis. (C) Enzymatic activity assay of pro-Casp8 and activated casp8 by using the Caspase-8 Assay Kit (Sigma-Aldrich). In a total volume of 100 μl, 1.8 pmol of activated Casp8, pro-Casp8, or pro-Casp8mt were incubated with substrate (Ac-IETD-pNA) in the presence or absence of Casp8 inhibitor (Ac-IETD-CHO) at 37°C for 1 h and enzymatic activity was determined according to the manufacturer's instructions. (D) Activated Casp8 did not cleave AR. In a total volume of 100 μl, 3 pmol of pro-Casp3 (R&D Systems Inc.) or AR was incubated with 1.8 pmol of activated Casp8 at 37°C in the presence or absence of Casp8 inhibitor for 30 min. Reaction mixtures were submitted to Western blot with anti-Casp3 (lanes 1–3) (Cell Signaling) or anti-AR (lanes 4–6) antibodies. (E) Casp8 interacted with AR in LNCaP cells. LNCaP cells were grown in the presence (lanes 1, 3, 5, 9, 11, and 13) or absence (lanes 2, 4, 6, 10, 12, and 14) of 10 nM R1881 for 48 h. Whole-cell lysates were incubated with immobilized anti-AR (lanes 3 and 4) or anti-Casp8 (lanes 11 and 12) antibodies for 2 h at 4°C. After being washed, bound proteins were eluted with 0.1 M glycine, pH 2.5, and the eluates were neutralized with 1.5 M Tris–HCl, pH 8.8, and submitted to Western blot analysis with an anti-Casp8 or anti-AR antibody. Western blotting was also performed using whole-cell lysates derived from control (lane 7) and Casp8 shRNA-expressing LNCaP cells (lane 8) with the anti-Casp8 antibody to validate the specificity of anti-Casp8 antibody.
As Casp8 is a cysteine protease, we next investigated whether Casp8 could degrade AR directly. The wild-type and enzymatic active site mutant (C345A) of Casp8 were expressed in and purified from PC3 cells (Figure 3B, lanes 2 and 3). Incubation of recombinant AR with wild-type (lane 5) or mutant (lane 6) Casp8 showed that caspase did not directly degrade AR. Enzymatic activity assay indicated that pro-Casp8 had very low (30-fold) proteinase activity than activated Casp8 (Figure 3C). Activated Casp8 cleaved recombinant pro-Casp3 (Figure 3D, lane 1), a well-known substrate of Casp8 but not AR (lane 4).
To further confirm the interaction of Casp8 with AR in vivo, we performed co-immunoprecipitation in LNCaP cells. Casp8 was co-immunoprecipitated with AR and AR was co-immunoprecipitated with Casp8 from the whole-cell extract made from LNCaP cells grown in the presence of androgen (Figure 3E, lanes 3 and 11). In the absence of ligand, no Casp8–AR interaction was detected (lanes 4 and 12). These results suggested that Casp8 interacted with AR in LNCaP cells in a ligand-dependent manner.
Additional analyses indicated that Casp8-mediated inhibition of AR transcriptional activity is not through a caspase-dependent apoptotic pathway, an Akt–Mdm2 pathway, or HDAC-containing corepressor complexes (see Supplementary data).
Casp8 disrupts AR N/C interaction
A unique property of AR is the androgen-induced intramolecular (N/C) interaction. The N/C interaction is required for AR function in vivo and androgen-induced AR protein stabilization. The mammalian two-hybrid system was used to evaluate the effect of Casp8 on AR N/C interaction. We observed a 21-fold activation of the reporter when cells were co-transferred with pACT-AR/N and pBIND-AR/C, indicating a strong AR N/C interaction (Figure 4A). Coexpression of Casp8 with pACT-AR/C and pBIND/N dramatically decreased the reporter activity, indicating that Casp8 inhibited AR N/C interaction. As a positive control, coexpression of pACT-ID with pBIND-MyoD resulted in activation (128-fold) of the reporter, which was not affected by co-transfection of pcDNA-Casp8. As negative controls, expression of neither pACT-AR/N nor pBIND-AR/C significantly activated the reporter activity. The androgen-induced AR N/C interaction stabilized AR (Langley et al, 1995, 1998); inhibition of this interaction by Casp8 might result in AR degradation (Figure 3A).
Figure 4.
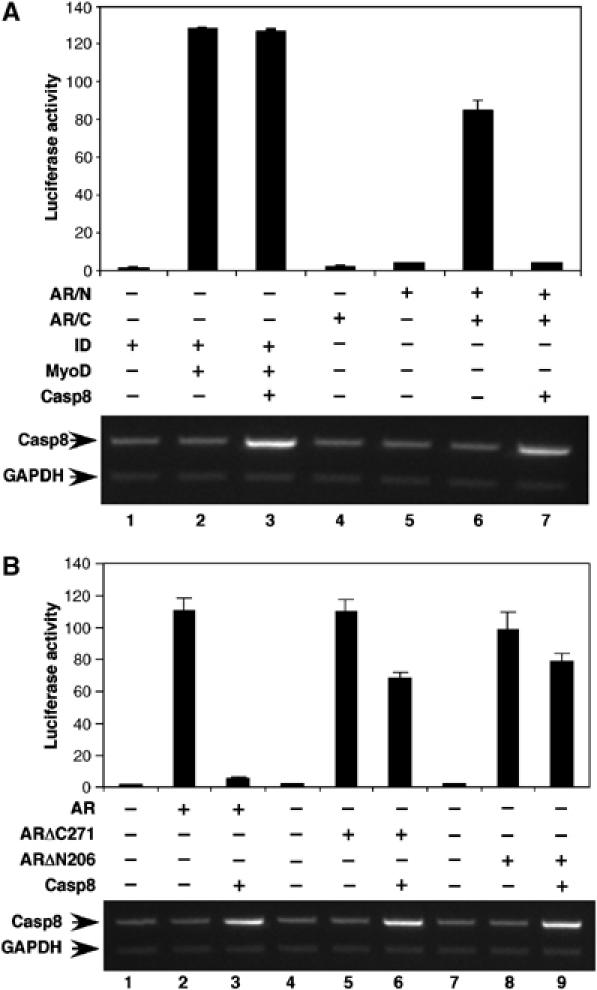
Casp8 disrupts the AR N/C interaction. (A) The mammalian two-hybrid assay was performed in PC3mm2 cells. Cells were transfected with pACT-AR/N, pBIND-AR/C, or both. Cells were transfected with pACT-ID or pBIND-MyoD as control. Transfected cells were grown in the presence of 10 nM R1881 for 48 h and then harvested for luciferase assay. Casp8 Expression was detected by RT–PCR (bottom panel). (B) An intact AR is required for Casp8-mediated inhibition of AR transactivation. pGL3-4 × ARE-E4-luc, pcDNA-AR, pcDNA-ARΔC271, or pcDNA-ARΔN206 and pcDNA-Casp8 were cotransfected into PC3mm2 cells. Cells were grown in the presence of 10 nM R1881 for 48 h and then harvested for luciferase assay. Values represent mean±s.d. (N=3).
Furthermore, we performed transient transfection assays using different AR truncations to determine if intact AR is necessary for Casp8-mediated transcriptional inhibition. ARΔC271 (with the C-terminal 271 amino-acid residues deleted) and ARΔN206 (with the N-terminal 206 amino-acid residues deleted) still had transactivation activities but no N/C interaction. ARΔC271 and ARΔN206 activated luciferase reporter activity (58- and 56-fold, respectively) to an extent similar to that of the full-length AR (71-fold) (Figure 4B). In contrast to the full-length AR, transactivations by the ARΔC271 and ARΔN206 truncations were only slightly inhibited by Casp8.
Casp8 physically interacts with AR
Three motifs (23FQNLF27, 433WHTLF437, and the C-terminal AF2) have been found to mediate AR N/C interaction (He et al, 2000; He and Wilson, 2002). Our hypothesis was that Casp8 physically interacts with these motifs, thereby disrupting AR N/C interaction. To investigate this possibility, we performed in vitro protein–protein pull-down assays. Figure 5A shows that Casp8 binds to AR (1–36) (lane 2), AR (334–566) (lane 3), and AR (662–919) (lane 4) but not glutathione S-transferase (GST) (lane 1). To identify domains in Casp8 that interact with AR, the various truncations of Casp8 were expressed as S35-labeled proteins and incubated with GST (Figure 5B, lanes 1, 4, 7, and 10; Figure 5C, lanes 1, 4, 7, and 10; Figure 5D, lanes 1, 4, and 7), GST-AR (1–36) (Figure 5B, lanes 2, 5, 8, and 11), GST-AR (334–566) (Figure 5C, lanes 2, 5, 8, and 11), or GST-AR (662–919) (Figure 5D, lanes 2, 5, and 8) fusion proteins immobilized on glutathione-agarose beads. As shown in Figure 5B, the N-terminal part (amino-acid residues 1–49) of Casp8 strongly bound to GST-AR (1–36) (lane 5) but not GST alone (lane 4), whereas further deletion of six residues (44LFQRLQ49) resulted in the complete loss of this interaction (lane 2). Mutation of F45A on Casp8 (1–49) completely abolished this interaction (Figure 5B, lane 8). Furthermore, mutation of F26A/F27A in AR (1–36) also lost this interaction (Figure 5B, lane 11). These results indicate that the motif 44LFQRLQ49 in Casp8 interacts with the AR motif 23FQNLF27, which has been shown to be involved in AR N/C interaction. Similar analyses (Figure 5C and D) revealed that amino-acid residues 291–300 (291NMDCFICCIL300) of Casp8 weakly interacted with the second motif (433WHTLF437) in AR that is involved in AR N/C interaction, and amino-acid residues 171–180 (171LLKIINDYEE180) of Casp8 interacted with the LBD of AR (probably the AF2). Motifs involved in Casp8–AR interactions are diagrammed in Figure 5E.
Figure 5.
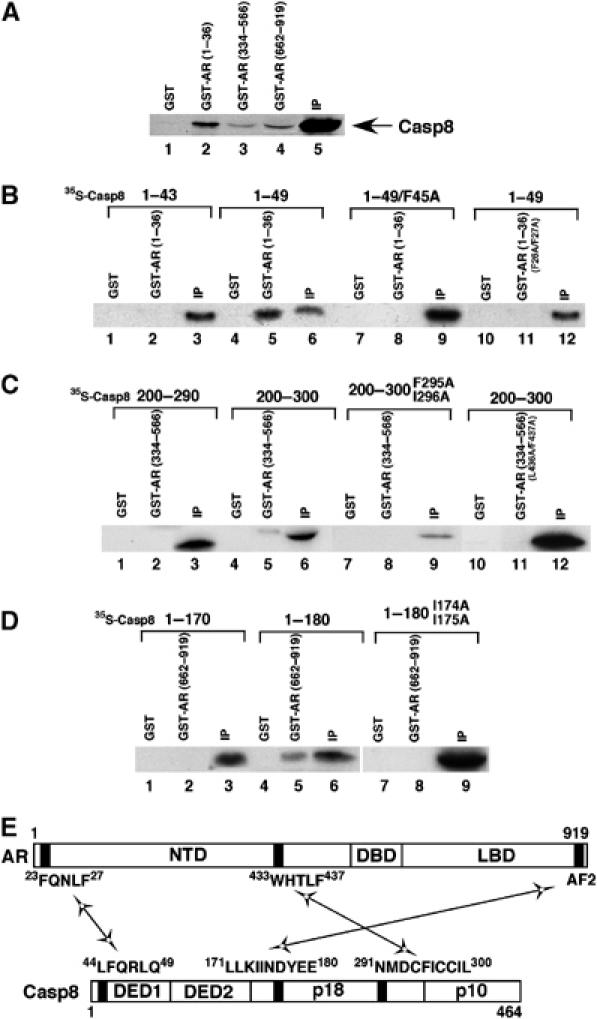
Casp8 physically interacts with AR. (A) Casp8 interacts with multiple regions in AR. GST, GST-AR (1–36), GST-AR (334–566), or GST-AR (662–919) was immobilized on glutathione-Sepharose beads and incubated with 35S-labeled Casp8. Proteins retained on the beads were separated by SDS–PAGE and the gel was analyzed by autoradiography. Five percent of input (IP) for the interaction assay was loaded on the same gel. (B–D) Identification of motifs that mediate Casp8–AR interactions. GST, GST-AR (1–36), GST-AR (1–36/F26A/F27A), GST-AR (334–566), GST-AR (334–566/L436A/F437A), or GST-AR (662–919) was immobilized on glutathione-Sepharose beads and incubated with 35S-labeled Casp8 (1–43), Casp8 (1–49), Casp8 (1–49/F45A), Casp8 (200–290), Casp8 (200–300), Casp8 (200–300/F295A/I296A), Casp8 (1–170), Casp8 (1–180), or Casp8 (1–180/I174A/I175A) as indicated. After being washed, beads were submitted to SDS–PAGE and the gel was analyzed by autoradiography. The input (IP) (5%) for the interaction assay was loaded onto the same gel. (E) Diagram of motifs in AR and Casp8 that are involved in Casp8–AR interactions. Black boxes represent motifs that mediate Casp8–AR interactions.
Interacting motifs are required for Casp8-mediated inhibition of AR-driven transcription
To further confirm the function of the motifs in Casp8, we generated point mutations in Casp8, which selectively abolished the interactions with the motifs in AR (Figure 5B–D, lane 8). Consistent with protein–protein pull-down results, the mutations F45A and I174A/I175A in Casp8 dramatically abolished Casp8-mediated inhibition of AR transcriptional activity, and the double mutation F295A/I296A partially affected this activity (Figure 6A). These results suggest that three AR-binding motifs in Casp8 are essential for the observed strong inhibition of AR-driven transcription by Casp8.
Figure 6.
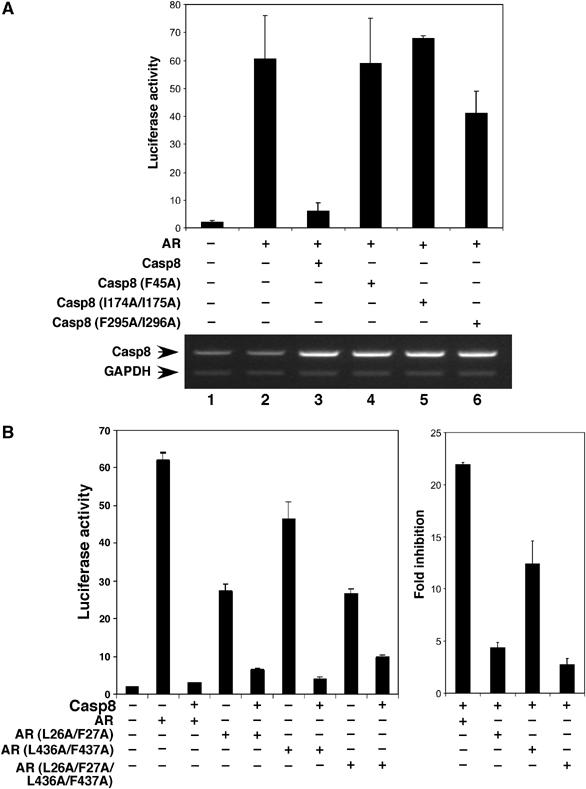
Mutations in Casp8–AR interaction motifs abolish Casp8-mediated inhibition of AR transactivation. (A) Casp8 with mutations in interaction motifs lost its ability to inhibit AR-driven transcription. PC3mm2 cells were transfected with pGL3-4 × ARE-E4-luc, pcDNA-AR, pcDNA-Casp8 (wild-type), or pcDNA-Casp8 (F45A, I174A/I175A, or F295A/I296A) mutants. Transfected cells were grown in the presence of 10 nM R1881 for 48 h and then harvested for luciferase assay (top panel) and RT–PCR. (B) Mutations in interaction motifs in AR abolish Casp8-mediated inhibition of AR transcriptional activity. PC3mm2 cells were transfected with pGL3-4 × ARE-E4-luc, pcDNA-Casp8, pcDNA-AR (wild-type), or pcDNA-AR (L26A/F27A, L436A/F437A, or L26A/F27A/L436A/F437A) mutants. The transfected cells were grown in the presence of 10 nM R1881 for 48 h and then harvested for luciferase assay (left panel). Fold inhibition of wild-type and mutant AR by Casp8 is represented on the right. Values represent mean±s.d. (N=3).
Furthermore, three AR mutants (L26A/L27A, L436A/F437A, and L26A/L27A/L436A/F437A) that have been shown to be defective in the N/C interaction (He et al, 2000) were used in the luciferase assays. As shown in Figure 6B, AR mutants L26A/L27A and L26A/L27A/L436A/F437A lost inhibitory effects of Casp8 to a large extent, whereas AR mutant L436A/F437A had a mild effect. This might reflect the weak interaction between motif 433WHTLF437 in AR and motif 291NMDCFICCIL300 in Casp8 (Figure 5C, lane 5). Thus, these results further confirmed that physical interactions between the motifs in Casp8 and AR are essential for Casp8-mediated inhibition of AR-dependent transcription.
An intramolecular interaction exists in Casp8
When studying Casp8–AR interactions, we observed that GST-AR (1–36) strongly interacted with 35S-labeled Casp8 (1–100) and Casp8 (1–150) (Figure 7A, lanes 2 and 5) but not with Casp8 (1–200) (lane 8). These observations led us to hypothesize that there is an intramolecular interaction in Casp8 molecule that inhibits intermolecular interaction between AR (1–36) and Casp8 (1–200). The in vitro-translated, 35S-labeled Casp8 (150–200) (Figure 7A, lane 12) bound to GST-Casp8 (1–49) (lane 11) and not to GST (lane 10) beads. This result confirms that there is an intramolecular interaction in the Casp8 molecule. The mutant (F45A) of Casp8 (1–49) completely abolished this intermolecular interaction (Figure 7A, lane 14), indicating that the intramolecular interacting motif overlaps with the motif (44LFQRLQ49) involved in intermolecular interaction with AR.
Figure 7.
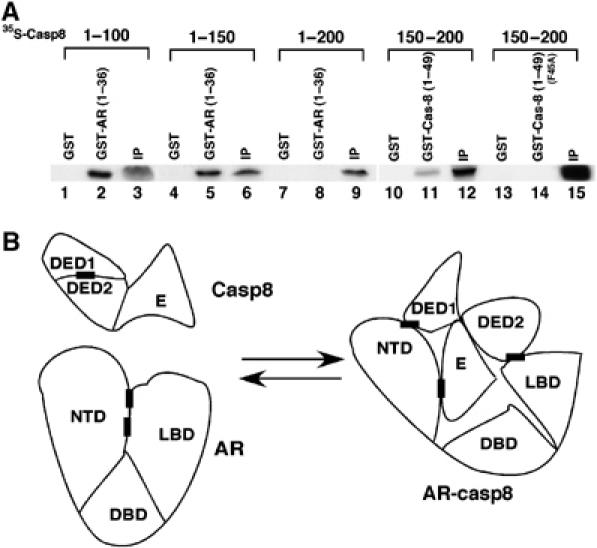
(A) An intramolecular interaction exists in Casp8 molecule. GST, GST-AR (1–36), GST-Casp8 (1–49), or GST-Casp8 (1–49/F45A) mutant was immobilized on glutathione-Sepharose beads and incubated with 35S-labeled Casp8 (1–100), Casp8 (1–150), Casp8 (1–200), or Casp8 (150–200) as indicated. After being washed, the beads were submitted to SDS–PAGE and the gel was analyzed by autoradiography. The input (IP) (5%) for the interaction assay was loaded onto the same gel. (B) Diagram of Casp8–AR interactions. Black boxes represent motifs that mediate Casp8–AR interactions.
A model of the interactions between Casp8 and AR is shown in Figure 7B. The intramolecular interactions in Casp8 involve the motif (amino-acid residues 44–49) and a motif located within amino-acid residues 150–200 (not mapped in detail). Our studies suggested that three motifs in Casp8 are involved in the intermolecular interactions with AR. These are as follows: motif 44LFQRLQ49, which mediated a strong interaction (Figure 5B, lane 5; 17% input was pulled down) with AR motif 23FQNLF27; motif 171LLKIINDYEE180, which mediated an intermediate interaction (Figure 5D, lane 5; 4% input was pulled down) with AR LBD; and motif 291NMDCFICCIL300, which mediated a weak interaction (Figure 5C, lane 5; 2% input was pulled down) with AR motif 433WHTLF437.
RNA interference of Casp8 leads to altered expression of AR target genes
To further confirm a role for Casp8 in the regulation of AR target genes in vivo, we performed RNA interference assay. Infection of LNCaP with a retrovirus expressing shCasp8D-1 (lane 2) and shCasp8D-3 (lane 4) reduced the level of Casp8 protein by 50 and 70%, respectively (Figure 8A). However, infection of LNCaP cells with a retrovirus expressing shCasp8D-2 (Figure 8A, lane 3) only slightly decreased the level of Casp8 protein. The levels of AR protein or β-actin were not affected by these shRNAs (Figure 8A, middle and bottom panels). Northern blot analysis demonstrated that the decrease in the levels of Casp8 induced by shRNAs resulted in a dramatic increase (up to 5.6-fold) in the levels of PSA gene expression in the presence of androgen (Figure 8B, lanes 2 and 4 versus lane 1). These shRNAs did not significantly affect the androgen-independent expression of PSA (Figure 8B, lanes 6–8 versus lane 5) or β-actin (bottom panel) gene.
Figure 8.
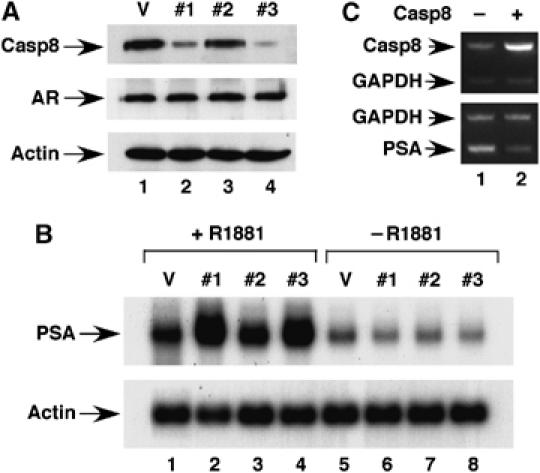
Casp8 regulates AR transactivation in vivo. (A) Knockdown of Casp8 expression by shRNAs in LNCaP cells. LNCaP cells were infected with vector control retrovirus (lane 1) or Casp8 shRNA-expressing virus (lanes 2–4). Infected cells were analyzed by Western blot. (B) Downregulation of Casp8 expression affected expression of AR target gene. Retrovirus-infected LNCaP cells were grown in the presence (lanes 1–4) or absence (lanes 5–8) of 10 nM R1881 for 24 h. Gene expression was detected by Northern blot. (C) Casp8 overexpression inhibited PSA gene expression. LNCaP cells were seeded in 6-cm dishes and transfected with 2 μg of control vector pCNDA3.1 (lane 1) or pcDNA-Casp8 (lane 2). Cells were grown in the presence of 10 nM R1881 for 24 h and RNAs were prepared from transfected cells for RT–PCR analysis to detect Casp8 (top panel) and PSA (bottom panel).
We also investigated, using real-time PCR, whether decreased Casp8 expression alters expression of other known AR target genes. Casp8 downregulation enhanced expression of Nkx3.1 and KLK2 genes and inhibited expression of Maspin (Table I), consistent with previous observations that AR pathway positively regulated expression of Nkx3.1 (Prescott et al, 1998; Bhatia-Gaur et al, 1999) and KLK2 (Wang et al, 2006), but repressed Maspin expression genes (Zhang et al, 1997). However, the Casp8 downregulation did not affect expression of TFF1 and DIO1 genes, which are regulated by the estrogen and thyroid hormone receptors, respectively (see Supplementary data). These results indicated that Casp8 specifically modulates expression of AR target genes. Consistent with this conclusion, Casp8 overexpression in LNCaP cells inhibited expression of the PSA gene (Figure 8C, lane 2 versus lane 1).
Table 1.
Data of real-time PCR assay
| 2−ΔΔCt | ||||||
|---|---|---|---|---|---|---|
| LNCaP+vector | LNCaP+shRNA | Fold changea | ||||
| Average | s.d. | Average | s.d. | Average | s.d. | |
| NKX3.1 | 10.96 | 0.51 | 19.77 | 3.17 | 1.80 | 0.20 |
| KLK2 | 10.89 | 0.76 | 16.81 | 0.76 | 1.69 | 0.55 |
| Maspin | 5.15 | 0.54 | 1.46 | 0.01 | −3.53 | 0.36 |
| aFor NKX3.1 and KLK2, fold change=relative quantification of the Casp8 knockdown group/relative quantification of the control group; for Maspin, fold change=−relative quantification of the control group/relative quantification of the Casp8 knockdown group. | ||||||
Casp8 inhibits the androgen-driven AR nuclear localization
We next investigated whether Casp8 affects the interaction of AR with DNA in LNCaP cells. Nuclear extracts (NE) were prepared from control, Casp8-expressing, and Casp8 shRNA-expressing LNCaP cells and submitted to electrophoretic mobility shift assays with an ARE-containing probe. While a strong signal of AR–DNA complex was observed in LNCaP NE (Figure 9A, lanes 3 and 5), this signal was decreased six-fold by Casp8 overexpression (lane 4) and increased 1.5-fold by Casp8 downregulation via shRNA expression (lane 6). Mutations of the conserved nucleotides in ARE (lanes 7–12) or addition of the cold probe (lanes 2 and 8) specifically abolished AR–DNA complex formation, indicating specificity of the AR–DNA complex. These results suggested that inhibition of AR-driven transcription by Casp8 might be through decreased AR–DNA interaction in the presence of Casp8.
Figure 9.
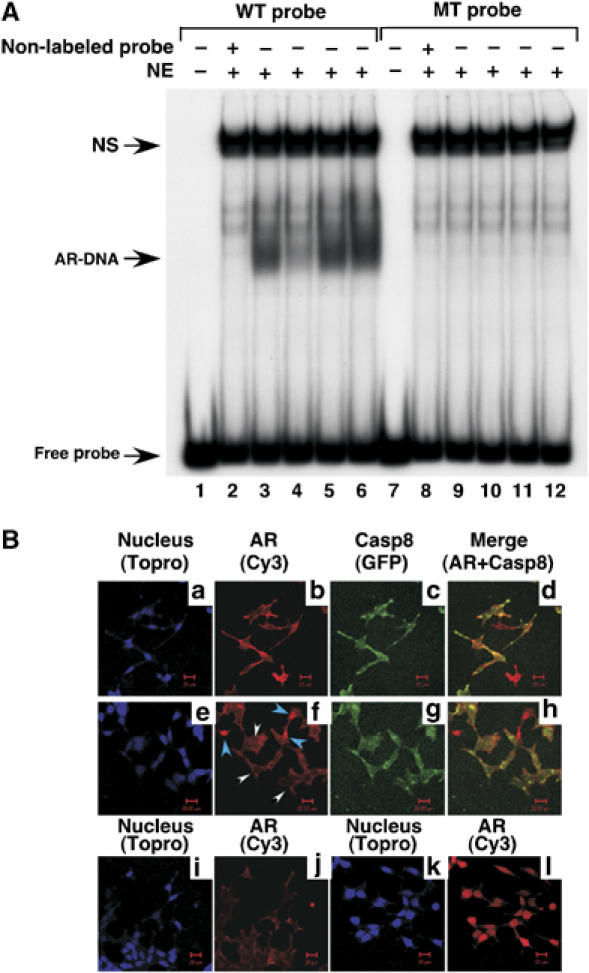
(A) Casp8 inhibits AR interaction with DNA in LNCaP cells. NE were prepared from LNCaP (lanes 2, 3, 8 and 9) or LNCaP trasfected with pcDNA-Casp8 (lanes 4 and 10), retrovirus vector (lanes 5 and 11), or Casp8 shRNA (#3) (lanes 6 and 12). (B) Casp8 expression inhibited the androgen-driven AR nuclear localization. LNCaP cells or LNCaP cells transfected with pcDNA-EGFP-Casp8 were grown in the absence (panels a–d, i, j) or presence (panels e–h, k, l) of 10 nM R1881. Signals were detected for nucleus (blue) (panels a, e, i, k), AR (red) (panels b, f, j, l), and EGFP-Casp8 (Green) (panels c, g) under a confocal microscope. In panel f, EGFP-Casp8-positive and -negative cells are indicated by white and blue arrows, respectively. Panels d and h are merged images of panels b and c and f and g, respectively.
To investigate whether Casp8 affects androgen-driven AR nuclear localization, the EGFP-Casp8 fusion protein was expressed in LNCaP cells by transient transfection. The EGFP-Casp8 protein inhibited AR-driven transcription and interacted with AR similarly to the non-fused Casp8 (data not shown). Control and EGFP-Casp8-transfected LNCaP cells were grown in the absence or presence of androgen for 48 h and submitted to immunostaining for AR. The androgen-driven nuclear translocation of AR was clearly demonstrated in control LNCaP cells (Figure 9B, panel l versus panel j). However, this translocation was inhibited in EGFP-Casp8-expressing LNCaP cells (panel f, indicated by white arrows) but not in cells lacking EGFP-Casp8 expression (indicated by blue arrows). These observations indicate that Casp8 inhibited the androgen-dependent AR nuclear translocation.
Discussion
In an effort to better understand the disparate mechanisms of AR activation and repression, we used an expression cloning approach to find new factors, new mechanistic insights, and new pathways. In the present study, we found that Casp8 physically and functionally interacted with AR and inhibited AR-driven transcription by disrupting AR N/C interaction and inhibiting the androgen-driven AR nuclear translocation. Our results suggest the existence of a functional link between Casp8 and AR signalings and indicate that Casp8 has a novel role beyond its activity as a cell death protease.
Identification of Casp8 as a negative AR cofactor
Transcriptional corepressors mediate repression by various nuclear receptors (Baniahmad, 2005; Wang et al, 2005). Some nuclear receptors (including retinoid receptor, thyroid hormone receptor, vitamin D receptor, and certain orphan receptors) that are not associated with heat-shock proteins in their unliganded state repress transcription by recruitment of corepressor complexes (Tsai and O'Malley, 1994; Heinlein and Chang, 2002). In contrast, unliganded steroid receptors (including AR) generally associate with heat-shock proteins and, upon ligand binding, dissociate from the heat-shock proteins, translocate to the nucleus, and associate with coactivators or corepressors to activate or repress target genes (Brinkmann et al, 1999; Janne et al, 2000; Gelmann, 2002; Haendler, 2002; Heinlein and Chang, 2002). The molecular mechanisms that underlie the selectivity of coactivators versus corepressors by liganded AR have remained elusive. It has been shown that some proteins repress AR-dependent transcription by physically interacting with AR to affect subcellular localization of the latter (Poukka et al, 2000) or AR transactivation (Baniahmad, 2005; Wang et al, 2005). Similarly, Casp8 negatively regulates AR-driven transcription. First, physical Casp8–AR interactions are supported by co-immunoprecipitation and GST-fusion protein pull-down data. Second, Casp8 strongly inhibited androgen-dependent reporters in the transient transfection assay, independently of its protease activity. Third, two shRNAs that specifically targeted Casp8 mRNA decreased the levels of Casp8 protein and hence affected androgen-driven gene expression in LNCaP cells. Together, these results clearly show that Casp8 has a pathway-specific and factor-specific role in the inhibition of AR signaling.
Casp8 regulates AR N/C interaction
The existence of AR N/C interaction provides an additional regulation point for AR-driven transcription. Indeed, MAGE-11 was identified as a positive AR cofactor that specifically bound the AR N-terminal 23FQNLF27 motif (He and Wilson, 2002). This interaction increased exposure of AF2 for the recruitment of SRC/p160 coactivators. It was reported that various cofactors differentially influenced AR N/C interaction, although the relevance of the effects of their cofactor functions on AR-driven transcription was not firmly established by detailed deletions and site-specific point mutations (Bai et al, 2005). Similarly, p53 was shown to repress androgen-induced transcription of PSA gene (Shenk et al, 2001). This inhibitory effect coincided with disruption of AR N/C interaction and inhibition of AR interaction with DNA.
The results presented here clearly link Casp8-mediated inhibition of AR-driven transcription to its ability to disrupt AR N/C interaction. We found that three motifs in Casp8 mediated interactions with AR and that these motifs were similar to the LXXLL motif, known to mediate nuclear receptor cofactor interaction. More importantly, we found that these motifs specifically interacted with AR motifs that mediate AR N/C interaction. The interactions between the motifs in Casp8 and the motifs in AR disrupted AR N/C interaction and consequently inhibited AR-driven transcription. These studies were well controlled by using site-specific point mutations on both Casp8 and AR.
Casp8 overexpression inhibited the androgen-driven AR nuclear translocation. Our preliminary observation indicated that recombinant Casp8 directly inhibited the AR–ARE interaction (data not shown). These results suggest that Casp8 inhibited AR-driven transcription by dual mechanisms, inhibiting the androgen-driven AR nuclear translocation and AR–ARE interaction.
The AR signaling pathway has multiple functions in male sexual development and maintenance through regulation of many genes whose functions are required for cell proliferation and differentiation. Identification and characterization of cofactors that are able to regulate AR-dependent gene expression are critical to understanding the molecular mechanisms of AR action in these processes. Our findings reveal a novel function of Casp8 in the regulation of AR function and suggest a possible linkage between the AR signaling pathway and the Casp8-mediated apoptotic process.
Materials and methods
Cell culture, chemicals, and antibodies
LNCaP and PC3mm2 cells were cultured in RPMI 1640 medium (Cellgro) with 5% (v/v) fetal bovine serum (HyClone). Androgen-free medium was composed of phenol red-free RPMI 1640 medium (Cellgro) and 5% (v/v) dextran/charcoal stripped fetal bovine serum. The synthetic androgen R1881 and transfection reagents (Lipofectamine and Lipofectamine 2000) were obtained from New England Nuclear and Invitrogen, respectively. Mouse anti-human Casp8, Casp3, and monoclonal anti-actin antibodies were purchased from Cell Signaling and Sigma, respectively. The rabbit anti-AR antibody was described previously (Yu et al, 2001). The cDNA fragment of human Casp8 encoding amino-acid residues 185–447 was subcloned into the vector pET15d (Novagene), expressed as a 6His-tagged protein in Escherichia coli BL21, and purified using NTA Ni2+ agarose. The SDS–PAGE-purified recombinant protein (10 mg) was sent to Sigma Genosys for polyclonal antibody production. The antibody was purified through a 6His-Casp8 (185–447) affinity column.
Library screening
E. coli (DH10B) was transfected with DNA from four wells of an arrayed human colon cDNA library (Origene) and plated to obtain about 100 colonies per plate (100 mm). Colonies were scraped onto two plates, and the plates were grown at 37°C overnight. One plate was stored as a stock at 4°C and the other plate was used to make plasmid DNA. PC3mm2 cells (5 × 104) were plated in each well of a 24-well plate 24 h before transfection. A total of 700 ng of DNA was transfected using Lipofectamine, including pRL-CMV (5 ng), pGL3-4 × ARE-E4-luc (100 ng), pcDNA-AR (30 ng), and pooled DNA (565 ng). Control transfection contained pcDNA3.1 (565 ng) instead of pooled cDNA. Transfected cells were grown in the presence of 10 nM R1881 for 48 h and then harvested for luciferase assay. Fold stimulation was calculated for each sample by dividing the value for luciferase activity by the value of control sample. The pool was considered positive if it stimulated or inhibited the luciferase reporter more than two-fold. Colonies on the stock plate of a positive pool were scraped onto 10 duplicated plates (10 colonies per plate) and plates were grown at 37°C overnight. Ten plates were stored as stocks at 4°C and another 10 plates were used to make plasmid DNA. DNA from each plate was assayed. Individual colonies on the positive stock plate (10 colonies per plate) were grown, and plasmid DNA was isolated for the assay. Positive clones were identified by DNA sequencing analysis.
Construction of plasmids
Plasmids pGL3-4 × ARE-E4-Luc, pGL3-PSA(E)-E4-luc, pcDNA-AR, pcDNA-ARΔN206 (with 206 amino-acid residues from the N-terminus of AR deleted), and pcDNA-ARΔC271 (with 271 amino-acid residues from the C-terminus of AR deleted) were described previously (Liu et al, 2003; Gao et al, 2004). Plasmids pCMVhAR L26A/F27A, L436A/F437A, and L26A/F27A/L436A/F437A were kindly provided by Dr Elizabeth M Wilson (The University of North Carolina) (He et al, 2000). All Casp8 cDNA fragments were amplified using PCR with specific oligonucleotides and cloned into the vectors pET15d, pGEX-2TL (Amersham Biosciences), and pcDNA3.1 (Invitrogen). Fragments were expressed as 6His-tagged (via pET15d) or GST-fusion (via pGEX-2TL) proteins in E. coli BL21 and purified through NTA Ni2+ agarose or glutathione-Sepharose columns, respectively. DNA fragments encoding AR N-terminus (amino-acid residues 1–661) and C-terminus (amino-acid residues 662–919) were amplified by PCR and subcloned into the pACT and pBIND vectors (Promega) to generate pACT-AR/N and pBIND-AR/C, respectively. The Casp8 site-specific point mutations were prepared using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) and confirmed by DNA sequencing.
Cell transfection and luciferase assay
All transfection assays were performed according to the manufacturer's protocol for Lipofectamine or Lipofectamine 2000 (Invitrogen). Briefly, cells were seeded in 6-well plates (4 × 105 LNCaP, 3 × 105 PC3mm2) or 24-well plates (5 × 104 PC3mm2 cells) 24 h before transfection. The total amounts of DNA for each transfection reaction were adjusted to 1500 ng (six-well plate) and 300 ng (24-well plate) by the addition of pcDNA3.1 plasmid. The transfected cells were incubated in the absence or presence of 10 nM R1881 for 36–48 h and assayed for luciferase activity.
RT–PCR analysis
RNA was isolated from cells using the TRIzol Reagent (Invitrogen), and cDNA synthesis was performed using SuperScript III First-Strand Synthesis System for RT–PCR (Invitrogen). Amplification of Casp8 and GAPDH cDNA was performed in one reaction (see Supplementary data for primer sequences). PCR conditions were as follows: 94°C for 2 min followed by 23 cycles at 94°C for 30 s, 53°C (for Casp8) or 57°C (for PSA) for 30 s, and 72°C for 30 s. Plasmids with Casp8 or PSA cDNA inserts were used as positive controls, and water was used as a negative template control. PCR products were separated by 1.5% agarose gel electrophoresis and quantified using a Gel Doc 1000 apparatus (Bio-Rad).
Real-time PCR
Total RNAs were isolated from cultured cells using the TRIzol Reagent and reverse transcribed using the Reaction Ready First-Strand cDNA Synthesis Kit (SupperArray Bioscience Corp.). The cDNA products were PCR-amplified (40 cycles of 30 s at 94°C; 20 s at 55°C; 30 s at 72°C) with the RT2 real-time SYBR green PCR master mix and the gene-specific primer sets for human PSA (PPH01002A), NKX3.1 (PPH02267A), KLK2 (PPH01003A), Maspin (PPH00695A), and β-actin (PPH00073A) genes (SupperArray Bioscience Corp.) by SmartCycler II (Cepheid). Raw data processing and quantification were performed with the SmartCycler software (version 2.0C). The 2−ΔΔCt method was used to determine the relative quantification (in the presence of ligand versus in the absence of ligand) of target gene expression (Livak and Schmittgen, 2001). For NKX3.1 and KLK2, the equation fold change=relative quantification of the Casp8 knockdown group/relative quantification of the control group. For Maspin, the equation fold change=−relative quantification of the control group/relative quantification of the Casp8 knockdown group.
Mammalian two-hybrid analysis
Mammalian two-hybrid analysis was performed using the CheckMate Mammalian Two-Hybrid System (Promega). PC3mm2 cells were transfected with the pG5-luc reporter and pACT-AR/N, pBIND-AR/C, individually or together. The pRL-CMV plasmid was included as an internal control. Transfected cells were incubated for 36–48 h with medium containing 10 nM R1881 and then harvested for luciferase assay. The plasmids pACT-ID and pBIND-MyoD provided with the kit were used as controls.
GST affinity matrix-binding assay
The GST and GST-fusion proteins were expressed using the vector pGEX-2TL (+) in bacteria and immobilized on glutathione-Sepharose-4B beads (Amersham Biosciences). Then, 10 μl of beads (with 1 μg of immobilized protein) was incubated with 5 μl of rabbit reticulocyte lysate containing 35S-labeled proteins in a final volume of 200 μl in a buffer containing 20 mM HEPES (pH 7.9), 0.2 mM EDTA, 20% glycerol, 2 mM DTT, 150 mM KCl, 0.1% NP-40, and 0.5 mg/ml bovine serum albumin for 2 h at 4°C. The beads were washed five times (1 ml each) with the incubation buffer, boiled in 10 μl of 2 × SDS gel sample buffer, and submitted to SDS–PAGE. The gel was analyzed using autoradiography.
Western blot analysis
Proteins (20 μg) were separated by 10% (for AR) or 12% (for Casp8) SDS–PAGE and transferred to a nitrocellulose membrane. The membrane was blotted with the primary antibody (anti-FLAG M2 monoclonal, 1:2000; anti-Casp8, 1:1000; anti-Casp3, 1:1000; or anti-AR polyclonal, 1:5000). After washing, the membrane was incubated with an HRP-conjugated secondary antibody (anti-rabbit or anti-mouse IgG) for 2 h. Immunoreactive bands were visualized with the ECL Advance Western Blotting Detection system (Amersham Biosciences).
RNA interference
Three sets of oligos (Casp8D-1, Casp8D-2, and Casp8D-3) (see Supplementary data for the sequences) were synthesized, annealed, and subcloned into BglII/HindIII sites of pSuperior.retro.puro (OligoEngine) to generate pCasp8D-1, pCasp8D-2, and pCasp8D-3, respectively. Similarly, a set of nonspecific control oligos was synthesized and subcloned into pSuperior.retro.puro. Retroviral production and infection of LNCaP cells were performed as reported previously (Gao et al, 2005). Whole-cell lysates (20 μg of protein) made from infected cells were submitted to Western blot analysis with anti-Casp8, anti-AR, and anti-actin antibodies. The mRNAs isolated from the infected cells were submitted to Northern blot and RT–PCR as described previously (Gao et al, 2005).
Electrophoretic mobility shift assay
The ARE-containing wild-type and mutant probes derived from the PSA gene (−152 to −174) (Liu et al, 2003) were labeled with [α-32P]dCTP using a fill-in reaction with the Klenow enzyme. The NE were made from cultured cells, and electrophoretic mobility shift assays were performed as described previously (Liu et al, 2003).
Immunohistochemistry staining
LNCaP cells were fixed and permeabilized with cold acetone, blocked with 4% fish gel, and incubated with the rabbit anti-AR antibody (1:1000 dilution) at 4°C for 12 h. Cells were incubated with Cy3-conjugated goat anti-rabbit IgG (1:500 dilution; Jackson ImmunoResearch Laboratories Inc.) at 25°C for 1 h. The nuclei were counterstained with Topro (1:10 000 dilution; Jackson ImmunoResearch Laboratories Inc.). The slides were viewed under a confocal microscope.
Supplementary Material
Supplementary data
Acknowledgments
We thank Don Norwood and Michael Worley for critical editorial review. This work was supported in part by a grant (1R01 DK065156 01) to ZW from the National Institute of Diabetes and Digestive and Kidney Diseases and the National Institutes of Health and by a Cancer Center Support Core grant (CA16672) to MD Anderson Cancer Center from the National Cancer Institute, National Institutes of Health.
References
- Bai S, He B, Wilson EM (2005) Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol Cell Biol 25: 1238–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baniahmad A (2005) Nuclear hormone receptor co-repressors. J Steroid Biochem Mol Biol 93: 89–97 [DOI] [PubMed] [Google Scholar]
- Berrevoets CA, Doesburg P, Steketee K, Trapman J, Brinkmann AO (1998) Functional interactions of the AF-2 activation domain core region of the human androgen receptor with the amino-terminal domain and with the transcriptional coactivator TIF2 (transcriptional intermediary factor2). Mol Endocrinol 12: 1172–1183 [DOI] [PubMed] [Google Scholar]
- Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, Kim M, Desai N, Young P, Norton CR, Gridley T, Cardiff RD, Cunha GR, Abate-Shen C, Shen MM (1999) Roles for Nkx3.1 in prostate development and cancer. Genes Dev 13: 966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatright KM, Salvesen GS (2003) Mechanisms of caspase activation. Curr Opin Cell Biol 15: 725–731 [DOI] [PubMed] [Google Scholar]
- Brinkmann AO, Blok LJ, de Ruiter PE, Doesburg P, Steketee K, Berrevoets CA, Trapman J (1999) Mechanisms of androgen receptor activation and function. J Steroid Biochem Mol Biol 69: 307–313 [DOI] [PubMed] [Google Scholar]
- Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, Hood L (2000) Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene 19: 4451–4460 [DOI] [PubMed] [Google Scholar]
- Cleutjens KB, van der Korput HA, van Eekelen CC, van Rooij HC, Faber PW, Trapman J (1997) An androgen response element in a far upstream enhancer region is essential for high, androgen-regulated activity of the prostate-specific antigen promoter. Mol Endocrinol 11: 148–161 [DOI] [PubMed] [Google Scholar]
- Craft N, Shostak Y, Carey M, Sawyers CL (1999) A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med 5: 280–285 [DOI] [PubMed] [Google Scholar]
- De Maria R, Zeuner A, Eramo A, Domenichelli C, Bonci D, Grignani F, Srinivasula SM, Alnemri ES, Testa U, Peschle C (1999) Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 401: 489–493 [DOI] [PubMed] [Google Scholar]
- Dedhar S, Rennie PS, Shago M, Hagesteijn CY, Yang H, Filmus J, Hawley RG, Bruchovsky N, Cheng H, Matusik RJ, Giguere V (1994) Inhibition of nuclear hormone receptor activity by calreticulin. Nature 367: 480–483 [DOI] [PubMed] [Google Scholar]
- Dehm SM, Tindall DJ (2005) Regulation of androgen receptor signaling in prostate cancer. Expert Rev Anticancer Ther 5: 63–74 [DOI] [PubMed] [Google Scholar]
- Faouzi S, Burckhardt BE, Hanson JC, Campe CB, Schrum LW, Rippe RA, Maher JJ (2001) Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-kappa B-independent, caspase-3-dependent pathway. J Biol Chem 276: 49077–49082 [DOI] [PubMed] [Google Scholar]
- Franchi L, Condo I, Tomassini B, Nicolo C, Testi R (2003) A caspaselike activity is triggered by LPS and is required for survival of human dendritic cells. Blood 102: 2910–2915 [DOI] [PubMed] [Google Scholar]
- Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D, Zhang X, Albanese C, Balk S, Chang C, Fan S, Rosen E, Palvimo JJ, Janne OA, Muratoglu S, Avantaggiati ML, Pestell RG (2003) Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol 23: 8563–8575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Lee P, Wang H, Gerald W, Adler M, Zhang L, Wang YF, Wang Z (2005) The androgen receptor directly targets the cellular Fas/FasL-associated death domain protein-like inhibitory protein gene to promote the androgen-independent growth of prostate cancer cells. Mol Endocrinol 19: 1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Liu GZ, Wang Z (2004) Modulation of androgen receptor-dependent transcription by resveratrol and genistein in prostate cancer cells. Prostate 59: 214–225 [DOI] [PubMed] [Google Scholar]
- Gelmann EP (2002) Molecular biology of the androgen receptor. J Clin Oncol 20: 3001–3015 [DOI] [PubMed] [Google Scholar]
- Haendler B (2002) Androgen-selective gene regulation in the prostate. Biomed Pharmacother 56: 78–83 [DOI] [PubMed] [Google Scholar]
- He B, Gampe RT Jr, Kole AJ, Hnat AT, Stanley TB, An G, Stewart EL, Kalman RI, Minges JT, Wilson EM (2004) Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol Cell 16: 425–438 [DOI] [PubMed] [Google Scholar]
- He B, Kemppainen JA, Wilson EM (2000) FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem 275: 22986–22994 [DOI] [PubMed] [Google Scholar]
- He B, Wilson EM (2002) The NH(2)-terminal and carboxyl-terminal interaction in the human androgen receptor. Mol Genet Metab 75: 293–298 [DOI] [PubMed] [Google Scholar]
- Heinlein CA, Chang C (2002) Androgen receptor (AR) coregulators: an overview. Endocr Rev 23: 175–200 [DOI] [PubMed] [Google Scholar]
- Hsu CL, Chen YL, Ting HJ, Lin WJ, Yang Z, Zhang Y, Wang L, Wu CT, Chang HC, Yeh S, Pimplikar SW, Chang C (2005) Androgen receptor (AR) NH2- and COOH-terminal interactions result in the differential influences on the AR-mediated transactivation and cell growth. Mol Endocrinol 19: 350–361 [DOI] [PubMed] [Google Scholar]
- Hu WH, Johnson H, Shu HB (2000) Activation of NF-kappaB by FADD, Casper, and caspase-8. J Biol Chem 275: 10838–10844 [DOI] [PubMed] [Google Scholar]
- Ikonen T, Palvimo JJ, Janne OA (1997) Interaction between the amino- and carboxyl-terminal regions of the rat androgen receptor modulates transcriptional activity and is influenced by nuclear receptor coactivators. J Biol Chem 272: 29821–29828 [DOI] [PubMed] [Google Scholar]
- Janne OA, Moilanen AM, Poukka H, Rouleau N, Karvonen U, Kotaja N, Hakli M, Palvimo JJ (2000) Androgen-receptor-interacting nuclear proteins. Biochem Soc Trans 28: 401–405 [PubMed] [Google Scholar]
- Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, Ramakrishnan P, Lapidot T, Wallach D (2004) Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol 173: 2976–2984 [DOI] [PubMed] [Google Scholar]
- Langley E, Kemppainen JA, Wilson EM (1998) Intermolecular NH2-/carboxyl-terminal interactions in androgen receptor dimerization revealed by mutations that cause androgen insensitivity. J Biol Chem 273: 92–101 [DOI] [PubMed] [Google Scholar]
- Langley E, Zhou ZX, Wilson EM (1995) Evidence for an anti-parallel orientation of the ligand-activated human androgen receptor dimer. J Biol Chem 270: 29983–29990 [DOI] [PubMed] [Google Scholar]
- Lee DK, Chang C (2003a) Endocrine mechanisms of disease: expression and degradation of androgen receptor: mechanism and clinical implication. J Clin Endocrinol Metab 88: 4043–4054 [DOI] [PubMed] [Google Scholar]
- Lee DK, Chang C (2003b) Molecular communication between androgen receptor and general transcription machinery. J Steroid Biochem Mol Biol 84: 41–49 [DOI] [PubMed] [Google Scholar]
- Liu GZ, Wang H, Wang Z (2003) Identification of a highly conserved domain in the androgen receptor that suppresses the DNA-binding domain–DNA interactions. J Biol Chem 278: 14956–14960 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM (1995) The nuclear receptor superfamily: the second decade. Cell 83: 835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall K, Steller H (1998) Requirement for DCP-1 caspase during Drosophila oogenesis. Science 279: 230–234 [DOI] [PubMed] [Google Scholar]
- Peterson JS, Barkett M, McCall K (2003) Stage-specific regulation of caspase activity in Drosophila oogenesis. Dev Biol 260: 113–123 [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Denny EM, Baltimore D (2002) CARD11 mediates factor-specific activation of NF-kappaB by the T cell receptor complex. EMBO J 21: 5184–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poukka H, Karvonen U, Yoshikawa N, Tanaka H, Palvimo JJ, Janne OA (2000) The RING finger protein SNURF modulates nuclear trafficking of the androgen receptor. J Cell Sci 113 (Part 17): 2991–3001 [DOI] [PubMed] [Google Scholar]
- Prescott JL, Blok L, Tindall DJ (1998) Isolation and androgen regulation of the human homeobox cDNA, NKX3.1. Prostate 35: 71–80 [DOI] [PubMed] [Google Scholar]
- Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS (1995) Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr Rev 16: 271–321 [DOI] [PubMed] [Google Scholar]
- Salvesen GS (1999) Caspase 8: igniting the death machine. Struct Fold Des 7: R225–R229 [DOI] [PubMed] [Google Scholar]
- Sato N, Sadar MD, Bruchovsky N, Saatcioglu F, Rennie PS, Sato S, Lange PH, Gleave ME (1997) Androgenic induction of prostate-specific antigen gene is repressed by protein–protein interaction between the androgen receptor and AP-1/c-Jun in the human prostate cancer cell line LNCaP. J Biol Chem 272: 17485–17494 [DOI] [PubMed] [Google Scholar]
- Schuur ER, Henderson GA, Kmetec LA, Miller JD, Lamparski HG, Henderson DR (1996) Prostate-specific antigen expression is regulated by an upstream enhancer. J Biol Chem 271: 7043–7051 [DOI] [PubMed] [Google Scholar]
- Shenk JL, Fisher CJ, Chen SY, Zhou XF, Tillman K, Shemshedini L (2001) p53 represses androgen-induced transactivation of prostate-specific antigen by disrupting hAR amino- to carboxyl-terminal interaction. J Biol Chem 276: 38472–38479 [DOI] [PubMed] [Google Scholar]
- Shi Y (2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 9: 459–470 [DOI] [PubMed] [Google Scholar]
- So A, Gleave M, Hurtado-Col A, Nelson C (2005) Mechanisms of the development of androgen independence in prostate cancer. World J Urol 23: 1–9 [DOI] [PubMed] [Google Scholar]
- Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J, Salmena L, Hakem R, Straus S, Lenardo M (2005) Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science 307: 1465–1468 [DOI] [PubMed] [Google Scholar]
- Tsai MJ, O'Malley BW (1994) Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 63: 451–486 [DOI] [PubMed] [Google Scholar]
- Wang G, Jones SJ, Marra MA, Sadar MD (2006) Identification of genes targeted by the androgen and PKA signaling pathways in prostate cancer cells. Oncogene 25: 7311–7323 [DOI] [PubMed] [Google Scholar]
- Wang L, Hsu CL, Chang C (2005) Androgen receptor corepressors: an overview. Prostate 63: 117–130 [DOI] [PubMed] [Google Scholar]
- Wilson EM, He B, Langley E (2003) Methods for detecting domain interactions in nuclear receptors. Methods Enzymol 364: 142–152 [DOI] [PubMed] [Google Scholar]
- Xu W (2005) Nuclear receptor coactivators: the key to unlock chromatin. Biochem Cell Biol 83: 418–428 [DOI] [PubMed] [Google Scholar]
- Yu X, Li P, Roeder RG, Wang Z (2001) Inhibition of androgen receptor-mediated transcription by amino-terminal enhancer of split. Mol Cell Biol 21: 4614–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Magit D, Sager R (1997) Expression of maspin in prostate cells is regulated by a positive ets element and a negative hormonal responsive element site recognized by androgen receptor. Proc Natl Acad Sci USA 94: 5673–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data