

Abstract
Normal C57BL/6 mice infected with 106 colony-forming units of a highly virulent strain of Mycobacterium avium developed a progressive infection characterized by loss of T cells from the tissues and infiltration with high numbers of heavily infected macrophages. In contrast, when C57BL/6 mice were infected with 102 colony-forming units of the same strain they retained T cells and T-cell reactivity in the tissues, and granulomas evolved into large masses that, at 4 months of infection, exhibited central necrosis. The development of these necrotic lesions did not occur in nude mice, nor in mice genetically deficient in CD4, interleukin-12 (IL-12) p40, interferon-γ (IFN-γ) and CD40 and were reduced in mice deficient in CD54 or IL-6. They were less numerous but bigger in mice deficient in IL-10 or the inducible nitric oxide synthase, correlating with the increased resistance to mycobacterial proliferation of these strains as compared to control mice. The appearance of necrosis was not affected in mice deficient in CD8α, T-cell receptor δ, tumour necrosis factor receptor p55, and perforin, nor was it affected in mice over-expressing bcl2. The appearance of necrosis could be prevented by administering antibodies specific for CD4, IL-12p40, or IFN-γ from the second month of infection when organized granulomas were already found. Our results show that the immunological mediators involved in the induction of protective immunity are also major players in the immunopathology associated with mycobacteriosis.
Introduction
Infection of immunocompetent hosts with pathogenic mycobacteria such as Mycobacterium tuberculosis or M. avium complex (MAC) species leads to the development of chronic granulomatous lesions in the affected tissues. The exact function of granulomas in the control of the infection is still controversial, although some evidence is emerging of their protective role.1 The assembly of a granuloma may occur in the absence of T lymphocytes,2 probably due to the activity of natural killer (NK) cells but is much more effective in the presence of functionally active CD4+ T cells.3 The cytokines interferon-γ (IFN-γ) and tumour necrosis factor (TNF) are important for the development and maintenance of granulomas2,4 and the intercellular adhesion molecule (ICAM) -1 (CD54) is required for the initiation of granuloma formation in experimental tuberculosis infections.5
Even after effective assembly of granulomas, it is frequently the case that there is no control of mycobacterial growth. In such situations, these lesions grow and may reach macroscopic sizes. Tubercles are massive lesions typical of advanced cases of tuberculosis, which has been named after these tumoral masses.6 On macroscopic examination, the contents of these tubercular lesions have a cheesy appearance, thence the name of caseous necrosis given to the decay of the cellular material taking place inside them. Caseous necrosis is thus typical of tuberculosis6 and may also be found in lesions from patients infected with MAC.7 Caseous necrosis of experimental tuberculosis lesions can be found in guinea-pigs and rabbits infected with virulent M. tuberculosis.8,9 Unfortunately the availability of reagents to study the immune response in these species is small compared with that in mice, and there are no gene-manipulated strains that may help dissect the mechanisms underlying the occurrence of necrosis. Normal mice infected with M. tuberculosis develop lesions that seldom undergo necrosis9 as they are quite resistant to the experimentally induced tuberculosis infection. Necrosis of granulomas in mice has been observed either by using gene-disrupted mice in M. tuberculosis infections10–13 or by using normal mice infected with low doses of highly virulent M. avium[ref. 14 and our unpublished observations].
The necrosis of a tubercle represents a major problem for tuberculosis patients for several reasons. First, the necrotic debris harbours free viable bacilli that thus escape the antimicrobial activity of phagocytes and are protected against chemotherapy. Second, the lesions themselves are irreversible and cause considerable damage to the lung tissue or any other organ, e.g. they may cause the collapse of the vertebrae in Pott's disease. Third, upon rupture in the lung, these lesions liberate enormous numbers of bacilli that disseminate to other parts of the body or to the atmosphere, representing the major form of contagion between patients and new cases. It is thus important to understand the mechanisms that underlie caseation, their overlap with protective mechanisms of immunity to mycobacteriosis, and ways of preventing it without interfering with the latter. Here we dissected the cellular and molecular pathways involved in the development of tubercles and their caseation during infection of mice with low doses of a virulent M. avium strain.
Materials and methods
Mice
Female C57BL/6J, C57BL/6-CD4−/−, C57BL/6-CD8α−/−, C57BL/6-TCRδ−/−, C57BL/6-CD54−/−, C57BL/6-CD40−/−, C57BL/6-Nu/Nu, C57BL/6-Nu/Cφ, C57BL/6-IFN-γ−/−, C57BL/6-perforin−/−, and C57BL/6-IL-12p40−/− mice were purchased from the Jackson Laboratories (Bar Harbor, ME). C57BL/6-IL10−/− breeder mice were purchased from Bantin and Kingman Universal (North Hamberside, UK) and progeny was obtained in our facilities. TNF-receptor-p55-deficient mice in a C57BL/6 background (C57BL/6-TNFRp55−/−) were bred in our facilities from a breeding pair kindly provided by Dr Tak Mak (AMGEN, Ontario, Canada). Heterozygous mice (C57BL/6-TNFRp55+/−) were obtained by crossing male C57BL/6-TNFRp55+/− with female C57BL/6 mice. Interleukin-6 (IL-6) gene-knockout in a C57BL/6 background (C57BL/6-IL-6−/−) were obtained in our facilities by backcrossing an original breeding pair of 129.C57BL/6-IL6−/− mice kindly provided by Dr Manfred Kopf (Basel Institute, Switzerland) into a C57BL/6 background for seven generations. Inducible nitric oxide synthase (iNOS) -deficient mice were bred in our facilities from a breeding pair kindly provided by Drs J. D. MacMicking and C. Nathan (Cornell University, New York, NY) and J. Mudgett (Merck Research Laboratories, Rahway, NJ). The iNOS-deficent mice in a C57BL/6 background (C57BL/6-iNOS−/−) were obtained by backcrossing the original strain into a C57BL/6 background for seven generations. In all cases, backcrossing to the C57BL/6 background was monitored by performing polymerase chain reaction (PCR) using sets of primers specific for the disrupted gene on genomic DNA samples from the progeny. Transgenic mice over-expressing human bcl2 in the haematopoietic cell lineage (C57BL/6+/bcl2) were kindly supplied by J. Adams (Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia).15 Mice were kept in high-efficiency particulate air (HEPA)-filter-bearing cages and fed autoclaved chow and water. All mice were used at 6–9 weeks of age.
Bacteria
Mycobacterium avium strain 25291, exhibiting a smooth transparent morphotype, was obtained from the American Type Culture Collection (Manassas, VA). Mycobacteria were grown in Middlebrook 7H9 medium (Difco, Detroit, MI) containing 0·04% Tween-80 (Sigma, St Louis, MO) at 37° until the mid-log phase of growth. Bacteria were harvested by centrifugation and resuspended in a small volume of saline containing 0·04% Tween-80. The bacterial suspension was briefly sonicated with a Branson sonifier (Danbury, CT) to disrupt bacterial clumps, diluted and stored in aliquots at −70° until used. Before inoculation, bacterial aliquots were thawed at 37° and diluted in saline to the desired concentration.
In vivo infection
Mice were infected intravenously with 106 or 102 colony-forming units (CFU) of M. avium strain 25291 through a lateral tail vein. Infected mice were killed at different times during infection and the livers, spleens and lungs were aseptically collected and homogenized in a 0·04% Tween-80 solution in distilled water. The number of CFU of M. avium in the organs of the infected mice was determined by serial dilution and plating of the tissue homogenates into Middlebrook 7H10 agar medium (Difco, Detroit, MI) supplemented with oleic acid–albumin–dextrose–catalase (OADC). The number of bacterial colonies was counted after culture for 2 weeks at 37°. Statistical comparisons of the mycobacterial loads between deficient and control mice were performed using the Student's t-test. In one experiment, mice were treated with monoclonal antibodies specific for IFN-γ (2 mg of antibody purified from the hybridoma cell line XMG1.2, every 2 weeks), IL-12p40 (1 mg of each of the antibodies purified from the hybridoma cell lines C15.1 and C15.2, every 2 weeks) or CD4 (0·2 mg of the antibody purified from the hybridoma cell line GK1.5, every week) as previously described.16 Monoclonal antibodies or non-immune immunoglobulin G (IgG) were given intraperitoneally, starting 2 months after inducing the infection until the end of the infection period at 4 months.
In vitro stimulation of splenic cells
Single-cell suspensions from the spleens of each of the infected mice were prepared by teasing portions of the spleen with forceps in Dulbecco's modified Eagle's tissue culture medium (DMEM; Life Technologies, Paisley, UK) supplemented with 10% fetal calf serum (FCS; Life Technologies). Erythrocytes were lysed by incubation of the cell suspensions with haemolytic buffer (155 mm NH4Cl, 10 mm KHCO3, pH 7·2) for 5 min at room temperature. The cell suspensions were then thoroughly washed with Hanks' balanced salt solution (Life Technologies) and resuspended in DMEM with 10% FCS. Cells were cultivated at a density of 2×105 cells/well in a U-bottom, 96-well microtitre plate. Cells were incubated in triplicate in DMEM with 10% FCS with no further stimulus or in the presence of mycobacterial culture filtrate proteins (4 µg/ml). Supernatants from the cultures were collected after 96 hr of culture and the IFN-γ produced was quantified by a two-site sandwich enzyme-linked immunosorbent assay method using anti-IFN-γ-specific affinity-purified monoclonal antibodies (R4-6A2 as capture and biotinylated AN-18 as detecting antibody) and a standard curve was generated with known amounts of recombinant murine IFN-γ (Genzyme, Cambridge, CA). The sensitivity of the assay was 30 pg/ml. Mycobacterial culture filtrate proteins were prepared from M. avium strain 25291 grown in Sauton medium. The supernatant of the culture was concentrated by ultra-filtration, precipitated with ammonium sulphate and extensively dialysed against phosphate-buffered saline (PBS).
Flow cytometry
For the immunofluorescence staining, 106 cells were incubated in a 96-well microtitre plate with fluorescein isothiocyanate (FITC) -conjugated anti-CD4 antibody (dilution 1 : 100) and phycoerythrin (PE) -conjugated anti-CD8 antibody (dilution 1 : 100) or FITC-conjugated anti-CD3 antibody (dilution 1 : 100) and PE-conjugated anti-MAC-1 antibody (dilution 1 : 100) in PBS containing 3% FCS. All antibodies were from BD Pharmingen (San Diego, CA). The cells were washed twice with PBS containing 3% FCS and propidium iodide (Sigma) was added to the cells at a final concentration of 1 µg/ml to allow the exclusion of dead cells. The analysis of the cell populations was based on the acquisition of 10 000 events in a Becton Dickinson FACSort equipped with PCLysis II software.
Histology
Portions of the organs of the infected mice were fixed in buffered formaldehyde and embedded in paraffin. Sections were stained with haematoxylin & eosin or stained for acid-fast bacteria by the Ziehl–Neelsen method and counterstained with methylene blue.
Results
Low-dose infection by virulent M. avium leads to the development of necrotic lesions
Preliminary observations on C57BL/6 mice infected with low doses, i.e. 100–200 CFU given intravenously, of highly virulent M. avium (strain ATCC 25291) showed that during prolonged periods of infection extending up to 4 months, lesions resembling classical tubercles developed in the organs targeted by the infection: livers, spleens and lungs. This contrasted with what had been observed in models of M. avium infection utilizing higher doses of mycobacterial inocula. We thus studied the course of infection initiated by intravenously infecting C57BL/6 mice with either a low dose of M. avium 25291 (102 CFU) or a higher dose of 106 CFU. Infection was followed for up to 3 months. Figure 1 shows the mycobacterial load in the organs during infection and confirms our previous experience with this strain of M. avium since both infections were progressive with little evidence of control of the mycobacterial proliferation. The development of the lesions was followed in those organs and we present here the typical findings in the livers which illustrate the general patterns of the development of those lesions (Fig. 2). The lesions induced by the low-dose infection were very scarce by days 15 and 30 (Fig. 2a) and consisted of loose accumulations of cells with a high percentage of eosinophilic polymorphonuclear leucocytes. However, by day 60, well-structured granulomas were found with an extensive lymphoid cuff surrounding the macrophage core (Fig. 2b). By day 90, many of those granulomas were found as well as additional extensive lesions consisting of peripheral accumulations of lymphoid cells, a core containing picnotic nuclei as well as frequent infiltrations by neutrophils and variable degrees of fibrosis and neovascularization (Fig. 2c). In a few instances some lesions with central necrosis were found at day 90 of infection. On the other hand, infection by the higher dose was characterized by an early assembly of granulomas already well organized by day 15 of infection (Fig. 2d) and highly structured at day 30 (not shown). However, as the infection progressed, the lesions became depleted of lymphoid cells consisting mainly of highly infected macrophages on days 60 (Fig. 2e) and 90 (Fig. 2f) coalescing into big mantles of infected cells occupying most of the hepatic parenchyma. This differential picture of development of the lesions was associated with distinct immune responses. Mice infected with the higher dose of M. avium showed an early response to the microbe (days 15 and 30) illustrated here by the in vitro reactivity of spleen cells to mycobacterial antigens with production of IFN-γ (Fig. 3a). On day 60 this reactivity was lost and was associated with a severe loss of the T cells, including those with a CD4+ phenotype in the spleens of infected mice (Fig. 3b). On the other hand, immune reactivity to mycobacterial antigens was only detected on day 60 in the spleen cells of mice infected with the low dose of M. avium. This response persisted until day 90 and was associated with the persistence of normal numbers of T cells in the spleen (Fig. 3). Macrophages increased in number most markedly in the case of the infection with the higher dose leading to massive splenomegalies.
Figure 1.

Growth of Mycobacterium avium 25291 in the organs of C57BL/6 mice infected intravenously with 102 (•) or 106 (▪) CFU. Each time-point represents the mean of the log10 CFU per organ±one standard deviation.
Figure 2.

Histological analysis of livers from C57BL/6 mice infected with Mycobacterium avium 25291. Representative lesions in haematoxylin & eosin (H&E) stained sections at 30 days (a, 220×), 60 days (b, 220×), and 90 days (c, 28×) of infection with 102 CFU or at 15 days (d, 160×), 60 days (e, 160×), and 90 days (f, 110×) of infection with 106 CFU.
Figure 3.

Influence of infection dose on the fate of immune reactivity and survival of immune cells. (a) In vitro reactivity to Mycobacterium avium antigen of total spleen cells from mice infected with 102 (left panel) or 106 (right panel) CFU of M. avium 25291. Cells were cultured in vitro and IFN-γ levels were determined in the supernatants. No detectable IFN-γ was found in cultures that were not given antigen. (b) Numbers of spleen cell populations in mice infected with 102 (•) or 106 (▪) CFU of M. avium 25291. Data show the means and the bars represent the standard deviations of the means.
At 4 months of low-dose infection, macroscopically visible tubercles were present in every animal in the liver and spleen and sometimes in the lung. Microscopically, these lesions consisted of a central necrotic core with no cellular organization, surrounded by a fibrotic capsule (Fig. 4a). Acid-fact bacilli were most prominent under the capsule and were often absent in the necrotic area (Fig. 4b, c). Neutrophil infiltrates were found in the lesions prior to the appearance of full blown necrosis (Fig. 4c). Mice with 8 months of low-dose infection had even more extensive formation of tubercles and caseous necrosis (Fig. 4d). We next decided to dissect the immunological mechanisms of induction of necrosis. Since there was a big heterogeneity in lesion size and development, demonstrating the asynchrony in their origin, we did not quantify the area involved but rather concentrated our study on looking at the presence or absence of necrotic lesions. For that, up to 10 tissue sections were analysed when no necrosis was found.
Figure 4.
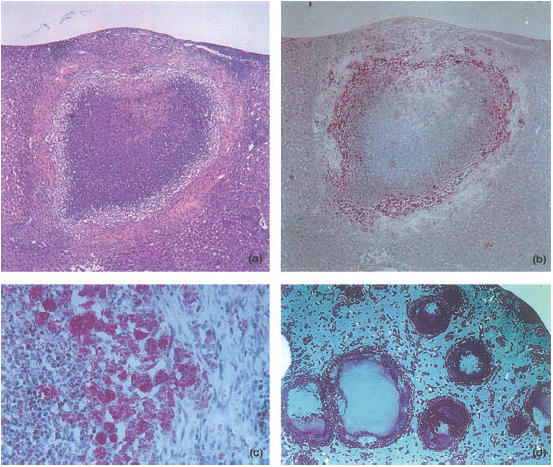
Histological analysis of necrotic lesions induced in C57BL/6 mice infected with Mycobacterium avium 25291 for 4 (a–c) or 8 (d) months. Sections were stained with H&E (a) or for acid-fast bacteria (ZN) (b–d). (a) and (b) show the same lesion magnified 28×. (c) provides a higher power view to show the layering at the periphery of the tubercles with an external area of fibrosis, and intermediate layer of heavily infected macrophages and an inner core with necrotic material and neutrophils (magnification 160×). (d) shows a low-power view of a liver of an infected mouse revealing extensive areas of caseous necrosis and a mantle of mycobacteria under the capsule (magnification 5×).
CD4+ T cells are required for the development of necrotic granulomas
The data described above suggested that T cells were required for the development of necrotic tubercles. Thus, when the inoculum was high, the depletion of the T-cell populations after the first month of infection was associated with massive infiltrates of foamy macrophages but not with necrosis. The latter was only evident when T-cell numbers were maintained, as shown by flow cytometry of the spleen cells or by microscopic evaluation of the lesions. To confirm the requirement for T cells, the phenotype of the cells involved in the induction of necrosis, and to make a preliminary assessment of the pathways involved, we infected groups of five to 11 mice with varying deficiencies with a low-dose inoculum of M. avium 25291 and studied the development of the lesions at 4 months of infection. The mycobacterial loads found in six independent experiments are depicted in Fig. 5 and a summary of the data obtained is presented in Table 1. In those experiments, a total of 44 wild-type C57BL/6 mice always developed multiple white nodules measuring from 1 to 3 mm in diameter, which were found scattered throughout the whole organ. Upon cutting with a surgical blade these nodules exhibited a soft whitish content.
Figure 5.

Mycobacterium avium loads in the livers of mice infected for 4 months with 102 CFU. Five independent experiments were performed (a–e) each studying different strains of genetically altered mice as compared to the control C57Bl/6 strain. In F, C57BL/6 mice were infected and given the indicated antibodies starting at the end of the second month of infection until 4 months when they were killed. Each symbol represents the value of CFU for one mouse and the bars represent the geometric means of the CFU.
Table 1. Summary of the data on the different mutant mice.
| Mouse strain | Growth of M. avium | Granulomas | Necrosis |
|---|---|---|---|
| C57BL/6 (B6) | Well structured | Present | |
| B6-Nu/Cφ | Well structured | Present | |
| B6-Nu/Nu | Increased* | Poorly structured† | Absent |
| B6-CD4−/− | Increased* | Poorly structured† | Absent |
| B6-CD8α−/− | Similar to control* | Well structured | Present |
| B6-TCRδ−/− | Similar to control* | Well structured | Present |
| B6-CD54−/− | Similar to control* | Well structured | Reduced‡ |
| B6-CD40−/− | Similar to control* | Poorly structured | Absent |
| B6-IL12p40−/− | Similar to control* | Incipient† | Absent |
| B6-IFNγ−/− | Increased* | Incipient | Absent |
| B6-TNFRp55+/− | Similar to control* | Well structured | Present |
| B6-IL6−/− | Similar to control* | Well structured | Reduced‡ |
| B6-IL10−/− | Reduced* | Well structured | Reduced§ |
| B6-perforin−/− | Similar to control* | Well structured | Present |
| B6-iNOS−/− | Reduced* | Well structured | Present¶ |
| B6+/bcl2 | Similar to control* | Well structured | Present |
The Student's t-test was used to analyse the statistical significance of the differences. Only statistically significant increases or decreases in viable counts are reported in this table.
Incipient granulomas were mostly formed by few heavily infected macrophages with little or no lymphoid involvement. Poorly structured granulomas had less lymphoid cuffing than granulomas of control mice.
Reduction in necrosis was observed both as a reduction in the number of lesions per organ and the occurrence of a percentage of the mice which totally lacked necrotic lesions.
The lesions showed necrosis later than control mice.
The lesions were fewer in number but bigger in size.
Neither nude mice nor CD4−/− mice exhibited such necrotic tubercles. Upon macroscopical analysis nude mice exhibited no nodules in the tissues. The CD4−/− mice exhibited few macroscopic lesions smaller in size and with no necrosis detectable by histology. Instead, histological analysis showed that the lesions were smaller and consisted of extensive accumulations of infected macrophages and few lymphoid cells (Fig. 6a). In both nude and CD4−/− mice, mycobacterial loads were significantly higher than in the respective controls (P<0·01 in both cases) (Fig. 5). In contrast, macroscopically evident nodules were present in the tissues of CD8α−/− and TCRδ−/− mice. Histologically, these nodules showed necrosis similar to that found in C57BL/6 mice (Fig. 6e, f). These latter mutations had no significant impact on the mycobacterial proliferation (Fig. 5).
Figure 6.
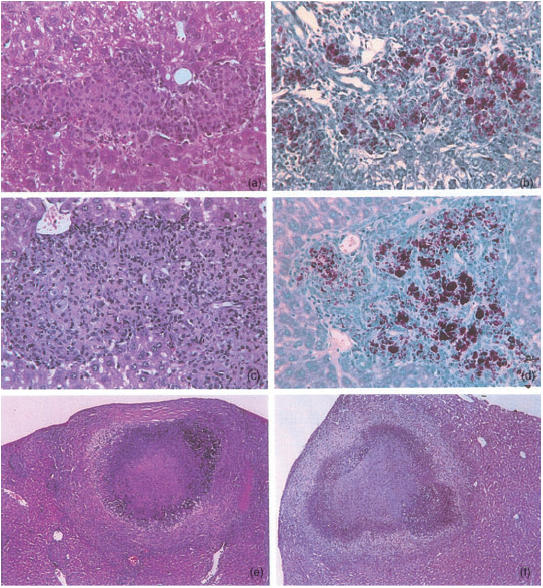
Representative lesions developing in livers of mice infected for 4 months with Mycobacterium avium 25291. HE (a and c, 80×) and ZN (b and d, 80×) stainings of the organs of nude mice (a,b) and CD4−/− mice (c,d). HE stainings of the organs of CD8α−/− (e, 55×) and TCRδ−/− mice (f, 27×).
Role of cytokines in the development of necrosis
In order to identify the molecular mediators responsible for the development of necrosis we infected mice deficient in cytokines that play a central role in the immune responses to mycobacteria, either inducing protection or, in contrast, promoting susceptibility. No necrosis was found in IL-12p40−/− and IFN-γ−/− mice. Macroscopically, there were no visible nodules in the tissues. Histologically, there was a scarcity of cellular infiltration into the liver with no granulomas but only disseminated accumulations of groups of few macrophages heavily infected with M. avium (Fig. 7a,b). Only in IFN-γ−/− mice was an enhanced mycobacterial proliferation observed (P<0·01). In contrast, TNFRp55−/− mice showed exuberant nodules with extensive necrosis (Fig. 7c). IL-6−/− mice presented smaller lesions with less evidence of necrosis (Fig. 7d) while the few macroscopically visible lesions of IL-10−/− mice were extremely large as compared to those of control mice and showed an extensive necrotic content (Fig. 7e). The organs of the latter strain had a significantly lower mycobacterial load (P<0·01), thus correlating with the lower number of lesions (Fig. 5).
Figure 7.

Representative lesions developing in livers of mice infected for 4 months (a–i, and k–p) or 8 months (j) with Mycobacterium avium 25291. HE (c–g, i–p) and ZN (a,b,h) stainings of the organs of IL-12p40−/− (a, 270×), IFN=γ−/− (b, 270×), TNFRp55−/− (c, 68×), IL-6−/− (d, 68×), IL-10−/− (e, 24×), CD54−/− (f, 50×), CD40−/− (g and h, 50×), iNOS−/− (i, 98×, and j, 24×), perforin−/− (k, 24×), and C57Bl/6+/bcl2 (l, 24×) mice, and of C57BL/6 mice treated with non-immune IgG (m, 24×), anti-CD4 (n, 68×), anti-IL-12p40 (ο, 68×), or anti-IFNγ (p, 68×) antibodies for the third and fourth months of infection.
Role of adhesion molecules or co-stimulatory molecules
CD54 has been shown to be required for the assembly of the granuloma.5 On the other hand, we have preliminary evidence suggesting that CD40 plays a role in inducing protective immunity to M. avium. We thus infected mice deficient in any of these two molecules to assess their relevance in the necrotic phenomena under study. The number of lesions in CD54−/− mice was smaller than in control animals but when they were present they exhibited a necrotic core (Fig. 7f). Although CD40−/− mice appeared macroscopically to develop lesions similar to wild-type mice, histologically they were devoid of necrosis (Fig. 7g,h).
Search for possible mediators of necrosis
Several molecules or pathways may be responsible for the induction of tissue necrosis. Among them, nitric oxide, apoptosis and T-cell cytotoxic products are possible candidates. To study them, we infected mice deficient in iNOS, mice over-expressing an anti-apoptotic molecule, Bcl2, and mice deficient in perforin. After 4 months of infection iNOS−/− mice had no evidence of necrosis (Fig. 7i). The lesions were large granulomas with an extensive cuffing by lymphoid cells. However, the livers of these animals had two orders of magnitude fewer mycobacteria than control mice (Fig. 5), a difference that was statistically significant (P<0·01). When iNOS−/− mice were killed at 8 months of infection allowing for additional mycobacterial growth, numerous fully developed necrotic tubercles were found (Fig. 7j). Mice deficient in perforin or mice overexpressing the anti-apoptotic molecule Bcl2 showed no alterations in the development of necrotic lesions (Fig. 7k and l, respectively) or in the mycobacterial growth (Fig. 5).
Necrosis can be prevented after the development of fully matured granulomas has taken place
It may be argued that the absence of necrosis in mice that had deficient IFN-γ responses may just reflect the lack of structured granulomas where the necrotic process would take place. To address this issue we infected C57BL/6 mice and at the beginning of the third month of infection, when mature granulomas were present but no necrotic lesions were yet present, a cohort of animals was treated with monoclonal antibodies causing the depletion of CD4+ cells or leading to the neutralization of IFN-γ or IL-12p40. These animals and control animals treated with non-immune IgG were killed at 4 months and the organs were analysed for the development of necrotic lesions. As shown in Fig. 7m, the control group showed many necrotic lesions. However, all of the antibody-treated groups failed to exhibit such necrotic lesions (Fig. 7n–p). The growth of the mycobacteria was not significantly affected by these treatments (Fig. 5).
Discussion
We describe here a model of mycobacterial infection featuring the development of lesions containing caseous necrotic material. The properties of the present model are as follows. First, it is associated with progressive, uncontrolled mycobacterial proliferation. The exact defect in the immune response accounting for the lack of control or, conversely, the microbial factors explaining the resistance to the immune response are as yet not identified. Second, for the assembly of the granulomas that evolve into tubercles harbouring necrotic centres to occur, T cells and an intact IL-12p40-IFNγ cytokine pathway were required. Third, the site of the lesions was determined by the initial seeding of the organs. The lesions described here were formed in the liver and in the spleen but were infrequent in the lung. That was due to the low inoculum dose used intravenously that causes negligible seeding of the lung. Thus, only at late time-points were similar lesions found in the lung, at a time of progressive mycobacterial proliferation and dissemination. This interpretation is supported by the findings of Benini et al.14 in their model of aerosol infection. Thus, when the infection is initiated by the aerogenic route, pulmonary necrotic lesions are the prominent finding.
The major player in the development of necrosis in our model is the CD4+ T-cell subset, presumably a subpopulation capable of secreting IFN-γ. In addition to IL-12p40 and IFN-γ, CD40 and IL-6, to different extents, were found to be required for the necrosis to develop. Whether CD40 is involved in the early induction of T-cell responses or is itself an effector molecule in the promotion of necrosis is not yet clear. Also, we did not examine whether CD40-deficient mice develop necrosis at later time-points. The role of IL-6 in the induction of immune responses to M. avium has already been highlighted by us17 and the participation in the necrotic process is hereby described. On the other hand, lack of IL-10 led to an improvement in the control of the infection and consequently to a reduction in the number of lesions. However, the few remaining lesions were massive and had an extensive necrotic core, consistent with a role of IL-10 in the down-regulation of pathology.
The role of nitric oxide appeared to relate more to its effects on T-lymphocyte survival and improved mycobacterial control as previously reported.18 Thus, although no necrosis was found in iNOS−/− mice at 4 months of infection, extensive necrotic tubercles were detectable at 8 months of infection. This correlated with the mycobacterial burden since the mean log10 CFU at 4 months were two orders of magnitude lower than the mean log10 CFU in control C57BL/6 mice and only at 8 months of infection, when the mycobacterial load increased, was the necrotic process fully triggered. These data strengthen the notion that both T cells and bacterial stimuli are required for necrosis to occur. It is pertinent to note at this point that Cooper et al.12 have already described the occurrence of necrosis during tuberculosis infection in iNOS−/− mice. Thus, it is possible that the caseous necrosis following tuberculosis infections in humans follows similar pathways to that described here.
The cellular and molecular basis of the necrosis has been interpreted differently by several authors. On the one hand, some favour an active damaging mechanism that has an immunological basis. It is clear that necrosis can ensue from the exposure of presensitized hosts to specific antigens in what is known as the Koch's phenomenon, although the tissue damage in this reaction differs from the central caseous necrosis that occurs in chronic infection. Also, it is known that as the immune system becomes compromised during the development of acquired immune deficiency syndrome in human immunodeficiency virus-infected patients, so the lesions become smaller and less necrotic.19 As reviewed by Dannenberg,20 this point of view regards necrosis as the result of T-cell activity, namely acting in pathways akin to delayed-type hypersensitivity reactions. An opposed view that was recently put forth postulated that necrosis occurs at the centre of lesions that became too big to allow for certain cytokines produced by T cells at the cortex of the granuloma to reach the centrally located macrophages.9 This model is based on the comparative analysis of the structure of the granulomas in different species of animals, i.e. guinea-pigs and mice, and is supported by the findings in gene-deficient mice infected with M. tuberculosis.10–13 Our data add new information by showing that necrosis required the presence of a T-cell compartment and of the cytokines forming the T helper type 1 pathway, IL-12p40 and IFN-γ. Thus, it is supportive of the former rather than the latter view. The relation to delayed-type hypersensitivity reactions is still unclear since CD54/ICAM1-deficient mice, which have their delayed-type hypersensitivity reactions abrogated5 still remain susceptible to the development of necrosis in their lesions, albeit at a lower rate.
The work of Rook and colleagues has shown that M. tuberculosis sensitizes cells, namely macrophages, to the action of TNF leading to cellular toxicity and cell death.21 However, we show here that mice deficient in the p55 TNF receptor still exhibit the development of tubercles and necrosis, as do wild-type mice. We cannot exclude the possibility that M. tuberculosis differs from M. avium in this respect but we can conclude that necrosis can occur without the participation of TNF signalling through the p55 receptor.
The actual mediators of the necrotic process remain to be identified. It is clear that the whole process is dependent on IFN-γ and the pathways that induce it (IL-12, IL-6 and CD40) but the mechanism is still elusive. We excluded a role for NO since necrosis is present in mice genetically unable to produce this molecule. We also addressed the role of apoptosis but failed to find a reduction in the development of necrotic lesions in mice over-expressing the antiapoptotic molecule, Bcl2. It is still possible, however, that apoptotic pathways insensitive to Bcl2 regulation might play a role. Finally, perforin was also found not to be involved. A trivial explanation for the lack of necrosis in mice such as those deficient in IL-12p40 or IFN-γ is that since they are unable to mount a granuloma, there are no lesions where necrosis can occur. We find this unlikely because when mice were allowed to develop mature granulomas and then treated with antibodies specific for IFN-γ, IL-12p40, or CD4, the emergence of necrosis was blocked. Thus, CD4+ T cells and the cytokine axis made of IL-12p40 and IFN-γ are not only required for the assembly of granulomas but also may play a distinct role later on in the promotion of necrosis. This latter issue also highlights the limitations in the use of gene-deficient mice in addressing the role of a specific element of the immune system early as opposed to late in infection and the need to combine such models with others which use treatments (antibodies, inhibitory drugs) which although potentially less effective may be specifically timed into the system.
During the submission of this work, Ehlers and colleagues22 dissected their model of aerogenic induction of necrosis and identified a similar dependence on αβ T cells, IL-12 and IFN-γ. As in our work, nitric oxide was not required.
The work presented here raises new clues to the study of human tuberculosis that, in our opinion, deserve further attention. If the pathogenetic basis of caseous necrosis in human tuberculosis is similar to the one described here we would speculate that the reason necrosis develops in certain clinical cases of tuberculosis infection relates to a failure to control the mycobacterial infection despite the presence of an ongoing intense immune response. It will be important to clarify if, in addition to the existence of common T-cell mechanisms leading to protective immunity and pathology (IL-12 and IFN-γ) there may be distinct ones leading to the former but not the latter.
Acknowledgments
The authors are grateful to A. I. Correia for helpful technical assistance and to T. F. Pais for help in the preparation of the antigens from M. avium. This work was supported by contracts AI-41922 from the NIH and 32629/99 from the Foundation for Science and Technology (Lisbon). M. F. received a fellowship from PRAXIS XXI.
References
- 1.Saunders BM, Frank AA, Orme IM. Granuloma formation is required to contain bacillus growth and delay mortality in mice chronically infected with Mycobacterium tuberculosis. Immunology. 1999;98:324–8. doi: 10.1046/j.1365-2567.1999.00877.x. 10.1046/j.1365-2567.1999.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith Hänsch H, Bancroft G, Ehlers S. T-cell-independent granuloma formation in response to Mycobacterium avium: role of tumour necrosis factor-α and interferon-γ. Immunology. 1997;92:413–21. doi: 10.1046/j.1365-2567.1997.00384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hänsch HCR, Smith DA, Mielke MEA, Hahn H, Bancroft GJ, Ehlers S. Mechanisms of granuloma formation in murine Mycobacterium avium infection: the contribution of CD4+ T cells. Intern Immunol. 1996;8:1299–310. doi: 10.1093/intimm/8.8.1299. [DOI] [PubMed] [Google Scholar]
- 4.Ehlers S, Benini J, Kutsch S, Endres R, Rietschel ET, Pfeffer K. Fatal granuloma necrosis without exacerbated mycobacterial growth in tumor necrosis factor receptor p55 gene-deficient mice intravenously infected with Mycobacterium avium. Infect Immun. 1999;67:3571–9. doi: 10.1128/iai.67.7.3571-3579.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson CM, Cooper AM, Frank AA, Orme IM. Adequate expression of protective immunity in the absence of granuloma formation in Mycobacterium tuberculosis-infected mice with a disruption in the intracellular adhesion molecule 1 gene. Infect Immun. 1998;66:1666–70. doi: 10.1128/iai.66.4.1666-1670.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jagirdar J, Zagzag D. Tuberculosis. Boston: Little, Brown; 1996. Pathology and insights into pathogenesis of tuberculosis; pp. 467–82. [Google Scholar]
- 7.Farhi DC, Mason UG, Horsburgh CR. Pathologic findings in disseminated Mycobacterium avium-intracellulare infection. Am J Clin Pathol. 1986;85:67–72. doi: 10.1093/ajcp/85.1.67. [DOI] [PubMed] [Google Scholar]
- 8.Lurie MB, Abramson S, Heppleston AG. On the response of genetically resistant and susceptible rabbits to the quantitative inhalation of human type tubercle bacilli and the nature of resistance to tuberculosis. J Exp Med. 1952;95:119–34. doi: 10.1084/jem.95.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orme IM. The immunopathogenesis of tuberculosis: a new working hypothesis. Trends Microbiol. 1998;6:94–7. doi: 10.1016/s0966-842x(98)01209-8. [DOI] [PubMed] [Google Scholar]
- 10.Flynn JL, Goldstein MM, Chan J, et al. Tumor necrosis factor-α is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–72. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 11.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon γ gene-disrupted mice. J Exp Med. 1993;178:2243–7. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper AM, Pearl JE, Brooks JV, Ehlers S, Orme IM. Expression of the nitric oxide synthase 2 gene is not essential for early control of Mycobacterium tuberculosis in the murine lung. Infect Immun. 2000;68:6879–82. doi: 10.1128/iai.68.12.6879-6882.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flynn JL, Goldstein MM, Triebold KJ, Koller B, Bloom BR. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc Natl Acad Sci USA. 1992;89:12013–17. doi: 10.1073/pnas.89.24.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benini J, Ehlers EM, Ehlers S. Different types of pulmonary granuloma necrosis in immunocompetent vs. TNFRp55-gene-deficient mice aerogenically infected with highly virulent Mycobacterium avium. J Pathol. 1999;189:127–37. doi: 10.1002/(SICI)1096-9896(199909)189:1<127::AID-PATH398>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 15.Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW, Adams JM. Constitutive Bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci USA. 1999;96:14943–8. doi: 10.1073/pnas.96.26.14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silva RA, Pais TF, Appelberg R. Evaluation of IL-12 in immunotherapy and vaccine design in experimental Mycobacterium avium infections. J Immunol. 1998;161:5578–85. [PubMed] [Google Scholar]
- 17.Appelberg R, Castro AG, Pedrosa J, Minóprio P. Role of interleukin 6 in the induction of protective T cells during mycobacterial infections in mice. Immunology. 1994;82:361–4. [PMC free article] [PubMed] [Google Scholar]
- 18.Gomes MS, Flórido M, Pais TF, Appelberg R. Improved clearance of Mycobacterium avium upon disruption of the iNOS gene. J Immunol. 1999;162:6734–9. [PubMed] [Google Scholar]
- 19.Doenhoff MJ. Granulomatous inflammation and the transmission of infection: schistosomiasis – and TB too? Immunol Today. 1998;19:462–7. doi: 10.1016/s0167-5699(98)01310-3. [DOI] [PubMed] [Google Scholar]
- 20.Dannenberg AM., Jr Delayed-type hypersensitivity and cell-mediated immunity in the pathogenesis of tuberculosis. Immunol Today. 1991;12:228–33. doi: 10.1016/0167-5699(91)90035-R. [DOI] [PubMed] [Google Scholar]
- 21.Filley EA, Bull HA, Dowd PM, Rook GAW. The effect of Mycobacterium tuberculosis on the susceptibility of human cells to the stimulatory and toxic effects of tumour necrosis factor. Immunology. 1992;77:505–9. [PMC free article] [PubMed] [Google Scholar]
- 22.Ehlers S, Benini J, Held HD, Roeck C, Alber G, Uhlig S. αβ T, cell receptor-positive cells and interferon-γ, but not inducible nitric oxide synthase are critical for granuloma necrosis in a mouse model of mycobacteria-induced pulmonary immunopathology. J Exp Med. 2001;194:1847–59. doi: 10.1084/jem.194.12.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]