

Abstract
We have investigated the effect of Flt-3 ligand (Flt-3L) on the resistance to herpes simplex virus type-1 (HSV-1) infection in BALB/c mice which are normally highly susceptible to challenge with this virus. We have confirmed data by others that in vivo treatment with Flt-3L causes an increase in dendritic cells (DC) and natural killer (NK) cells in lymphoid tissue. Increasing doses of Flt-3L caused a corresponding increase in liver and spleen CD11c+ DC which were increased up to 20-fold compared with control levels. A significant expansion of NK cells was seen in the spleen of Flt-3L-treated mice where the number of DX5+ cells was increased by up to fivefold. We subsequently tested the hypothesis that Flt-3L treatment, at the time of viral infection, might lead to enhanced immunity and protection against viral pathogenesis. Two murine models of HSV-1 (SC16) infection were used. In the first model, mice were injected with Flt-3L daily for 9 days. Control mice received mouse serum albumin (MSA). On day 7 of the Flt-3L treatment 106 plaque-forming units (PFU) of SC16 was inoculated into the ear pinna. Flt-3L treatment significantly reduced mortality following virus inoculation, with 80% survivors in this group compared with 20% survivors in the MSA-treated group. In the second model, Flt-3L-treated mice were scarified with 104 PFU of SC16. In this case there was 60% survival in the Flt-3L-treated group of mice compared with 10% survival in the MSA-treated group. Assessment by in situ hybridization for latency-associated transcripts showed that Flt-3L treatment reduced the amount of latent virus within infected neurons. These studies show that in vivo treatment with Flt-3L results in protection against challenge with live HSV-1, which may be a consequence of enhanced numbers of DC and/or NK.
Introduction
Herpes simplex virus type 1 (HSV-1) infects epidermal or epithelial cells of mucosal surfaces and undergoes multiple rounds of lytic replication. During this process HSV-1 enters the termini of local sensory neurons and retrograde axonal transport carries it to the neuronal cell bodies where it establishes latent infection. Intermittent reactivation may result in the production of infectious HSV from latently infected neurons.1,2 Although the factors that influence rates of HSV reactivation are poorly defined, there is evidence that suggests the number of viral genome copies in each neuron and the number of neurons latently infected are contributory factors.3
Protective immunity to viruses, including HSV-1, requires a co-ordinated response by both the innate and the adaptive immune system.4 The innate response is mediated through components which include natural killer (NK) cells, phagocytic cells and regulatory molecules, such as type I interferons (IFN-α/β). This system is activated within hours of infection and serves to provide protection of the host during the development of the adaptive immune response, which comprises antigen-specific T and B lymphocytes. A firmly established paradigm for the activation of antigen-specific T cells is their recognition of peptide fragments of antigen bound to presentational elements, such as major histocompatibility complex (MHC) or CD1 molecules, expressed by dendritic cells (DC).5
Two lineages of DC exist, myeloid (Langerhans' cells and interstitial DC) and lymphoid.6,7 In the human system two subsets of precursor DC have been described, pDC1 and pDC2, which have been proposed to be precursor cells of the myeloid and lymphoid lineages, respectively.8 Collectively, this group of antigen-presenting cells (APC) are regarded as the sentinels of the immune system. An accepted working hypothesis for their function is that immature, or precursor, DC located at epithelial sites encounter incoming pathogens such as viruses which are processed for antigen presentation to T cells. The encounter between DC and pathogen leads to maturation/activation of the DC and their subsequent migration to secondary lymphoid organs where they initiate antigen-specific T-cell responses.9,10 Recently, it has been shown that precursors of DC, including those exposed to HSV-1, are potent producers of IFN-α, a known activator of NK cells.11,12 This strongly suggests that these APC may also regulate NK-cell function. DC are implicated in NK-cell activation13 because of the ligand–receptor complementarity between these two cell types. For example, DC express CD80, CD86 and CD40 whilst NK cells express CD28 and CD40 ligand.14 Cross-linking of CD28 or CD40 ligand on NK cells augments their proliferation, IFN-γ production and cytotoxicity, and NK cells can kill CD40-expressing targets.15 Direct evidence for an interaction between DC and some NK cells in vitro has been the demonstration that NK1 T cells are activated by the glycolipid α-galactosylceramide (α-Gal-Ser).16 This activation is dependent upon NK1.1 T-cell recognition of α-Gal-Ser presented by CD1 molecules expressed by DC. Indirect, but strong, in vivo evidence for a functional interaction between DC and NK cells is the action of the haematopoietic cytokine Flt-3 ligand (Flt-3L). When injected in vivo, Flt-3L causes a dramatic expansion of both the myeloid and lymphoid subsets of DC, and also a significant increase in the number of NK cells.17,18 Interestingly, Flt-3L stimulates the expansion of these two cell types in similar locations, including the spleen.
Here we have investigated whether Flt-3L treatment in vivo can provide resistance to HSV-1 infection. Our studies show that Flt-3L can produce enhanced immunity that results in protection against infection with live HSV-1. This effect was accompanied by elevated numbers of DC and NK cells and supports the hypothesis that DC represent an important linkage between the innate and adaptive immune system.
Materials and methods
Mice
Female BALB/c mice, 4–6 weeks old, were obtained from Harlan, UK. All regulated procedures involving experimental animals were carried out under Project and Personal licence authority issued in accordance with The Animals (Scientific Procedures) Act 1986.
Flt-3L treatment of mice
Mice were injected subcutaneously daily for 9 consecutive days17 with 0·2 ml of human recombinant Flt-3L (a gift from Immunex Corporation, Seattle, WA) diluted in phosphate-buffered saline (PBS) containing 5 µg/ml mouse serum albumin (MSA). Control mice received PBS plus MSA alone.
Isolation of spleen and liver mononuclear cells for fluorescence-activated cell sorter (FACS) analysis
Spleen and liver were isolated from Flt-3L-treated mice, placed in FACS buffer (PBS containing 1% heat-inactivated fetal calf serum (FCS) plus 0·1% sodium azide) and processed through a nylon cell strainer (pore size 40 µm). Single cell suspensions were layered onto NycoPrep™ Animal (density 1·077 g/ml; osmolarity 265 mOsm), and centrifuged at 500 g for 15 min at room temperature.19 Mononuclear cells were recovered from the interface and washed three times in FACS buffer prior to immunofluorescence staining.
Monoclonal antibodies and immunofluorescence staining
Fluorescein isothiocyanate (FITC)-conjugated antibodies against mouse CD11c, T-cell receptor (TCR) -αβ, B220, I-Ad/I-Ed and phycoerythrin (PE)-conjugated CD11c, DX5 and CD8α, were all obtained from Pharmingen (Becton Dickinson, Cowley, Oxford, UK). FITC-conjugated anti-mouse CD4 was purchased from Sigma (Poole, Dorset, UK). FITC- or PE-conjugated isotype control antibodies [hamster immunoglobulin, rat immunoglobulin G2a (IgG2a) k, rat IgM] were also purchased from Pharmingen. Cell surface phenotype was assessed using aliquots of 1 × 106 mononuclear cells incubated with 1 µg of directly conjugated monoclonal antibody, either singly (for single-colour fluorescence) or in pairs (for two-colour fluorescence) for 20 min at 4° and washed three times with FACS buffer prior to analysis by flow cytometry using a FACSCalibur® (Becton Dickinson, San Jose, CA) gating on mononuclear cells. Ten thousand cells were analysed per sample, with dead cells excluded on the basis of forward and side light scatter. Background fluorescence was established with control isotype-specific antibodies and was routinely 1–3%.
Virus strains
The strain of HSV-1 used in these studies was SC16. This strain of virus has been extensively characterized in mice and was originally isolated from a clinical case of herpes labialis.20 Virus stocks for inoculation were prepared at low multiplicities of infection, titrated in BHK-21 cells and stored at −70°.
Virus inoculation of mice
Mice were inoculated with virus by one of two routes at 4·5 weeks of age. In the ear inoculation model, virus was inoculated into the skin of the left ear pinna21 with 10 µl of virus suspension containing between 104 and 108 plaque-forming units (PFU) SC16 in a single, discrete inoculation site. In the neck scarification model, the left side of the neck was shaved with an electric trimmer and virus was scarified onto the neck22 2 days later. Briefly, 10 µl of SC16 virus suspension containing between 102 and 105 PFU was placed on the skin in an area of 1 cm2 at the site to be scarified (left side of the ventral surface of the neck, approximately 0·5 cm lateral to the ventral mid-line) and a hypodermic needle was used to administer 8–10 superficial, light strokes in a criss-cross pattern parallel to cranio-caudal and ventro-dorsal axes to produce the scarification. Mice treated with Flt-3L were inoculated with virus on day 7 of the Flt-3L administration. In all cases, mice were observed for clinical signs including increase in ear pinna thickness, weight gain/loss and mortality. Mice which developed zosteriform lesions after neck scarification were scored according to the severity of their lesion on the scale 1=no lesion, 2=mild, 3=moderate and 4=severe lesion.
Quantification of infectious virus
Tissue samples (left and right trigeminal ganglia, and left and right CIII cervical dorsal root ganglia) were obtained at various time-points post-infection and placed in 1 ml Eagle's minimal essential medium (EMEM). Each tissue sample was minced with scissors, homogenized and sonicated on ice for 2 min.23 The samples were centrifuged at 1500 g to remove debris. Either 200 µl of neat, 10-fold diluted, or 100-fold diluted tissue sample supernatant was inoculated onto duplicate BHK-21 cell monolayers in 24-well plates and allowed to adsorb for 1 hr. Each well was subsequently overlaid with 1 ml EMEM containing 2% heat inactivated FCS plus 2% carboxymethylcellulose. Plates were stained and fixed with crystal violet following incubation at 37° in 5% CO2 for 48 hr and viral plaques were counted. All samples were coded prior to the start of the experiment and this code was not broken until all the results were recorded as PFU/tissue.
Reactivation of virus from latently infected ganglia by co-cultivation
Ganglia innervated by the ear pinna (left and right trigeminal ganglia, and left and right CIII cervical dorsal root ganglia) were obtained at > 30 days post-infection. These tissues were tested for their ability to yield virus after explantation of the ganglia and incubation at 37°, 5% CO2 for 5 days to allow reactivation to occur.24 Samples were coded and read blind.
Detection of latently infected neurons by in situ hybridization
In situ hybridization was performed on tissues to detect latency-associated transcripts (LATs) > 30 days post-infection.25 The probes used to detect LATs were generated by T7 polymerase transcription of HindIII-linearized pSLAT 2 (kindly provided by Dr Efstathiou, University of Cambridge, UK) with a digoxigenin (DIG) detection system.26 After transcription, the reaction mixtures were ethanol-precipitated and the product was resuspended in 100 µl of 10 mm Tris (pH 8)−1 mm dithiothreitol with RNase inhibitor. Tissues were fixed in periodate–lysine–paraformaldehyde at 4° for 16 hr, transferred to 50% ethanol, and then paraffin embedded. Sections (5 µm thick) were collected onto glutaraldehyde-activated, 3-aminopropyl-triethoxysilane-coated slides and were de-waxed in xylene before use. Sections were digested with 100 µg/ml proteinase K at 37° for 8 min. Overnight hybridization was carried out at 72°. Between 1 and 3 µg of DIG-labelled probe was used in each 100 µl of hybridization solution. One stringent wash in 0·1 × SSC (1 × SSC is 0·15 m NaCl plus 0·015 m sodium citrate)−30% formamide−10 mm Tris–HCl (pH 7·5) was carried out at 75° for 30 min. Bound probe was detected with alkaline phosphatase-conjugated anti-DIG Fab fragments according to the manufacturer's instructions (Boehringer Mannheim). RNase- and DNase-treated tissue sections were included in each hybridization as internal controls. Alternate sections from all tissues were counted and used to calculate the mean number of positive neurons per section. Cells whose nuclei contained a level of brown staining clearly above the background level were scored positive.
Results
Flt-3L stimulates an increase in the number of DC and NK cells
Flt-3L is a haemopoietic cytokine which promotes normal haemopoiesis and mobilization of haemopoietic stem cells.27 The administration of Flt-3L in vivo in the range of 1–10 µg per day caused a corresponding increase in spleen and liver mononuclear cell number. FACS analysis showed CD11c+ DC numbers in Flt-3L-treated mice were increased to over 20-fold in the spleen and 18-fold in the liver, compared to MSA-treated mice. DC in the spleen and liver from Flt-3L-treated mice were predominantly CD11c+ MHC class II+ (data not shown). Before and after Flt-3L treatment, spleen and liver DC were predominantly of the myeloid phenotype. However, although DC were increased in both tissues after Flt-3L treatment, a difference was seen in the relative expansion of different DC subsets within the two tissues. Spleen DC showed a greater increase in the percentage of lymphoid DC compared to myeloid DC whilst this trend was reversed with respect to the composition of liver DC (data not shown). A significant expansion of NK cells was seen in the spleen from Flt-3L-treated mice which were increased up to five-fold compared to control levels. There was some increase in lymphocyte content of the spleen and liver as the number of B and T cells in each tissue was raised by three- to four-fold following Flt-3L treatment (data not shown). Collectively, our analysis of lymphoid tissue following Flt-3L treatment confirmed similar findings reported earlier by the groups of Maraskovsky,17 Shaw18 and Brasel.28
Flt-3L protects against live HSV-1 challenge in vivo
To test the effect of increased numbers of DC and/or NK cells induced by Flt-3L on the immune response to HSV-1 it was necessary to establish the optimum dose of virus for subsequent challenge experiments. Figure 1 shows that the infection of BALB/c mice with HSV-1 SC16 resulted in mortality which was dependent upon the dose of virus and the route of inoculation. After ear pinna inoculation, mice infected with 105 PFU HSV-1 showed 80% survival, 106 and 107 PFU showed 30% survival and there were no survivors in the 108 PFU inoculated group (Fig. 1a). Mice inoculated by neck scarification showed similar responses but with lower doses of virus (Fig. 1b). Only 105 PFU HSV-1 was required to achieve 100% mortality in comparison with a dose of 108 PFU in the ear pinna model. All mice died between 5 and 7 days post-infection when inoculated by the ear pinna route and between 7 and 9 days post-infection following neck scarification.
Figure 1.

Effect of Flt-3L treatment on HSV-1-inoculated mice. Mice (n = 10) were inoculated with various doses of HSV-1 by: (a) intradermal inoculation of the left ear pinna or (b) scarification of the left side of the neck. Per cent survival was recorded daily for days 0–20 post-inoculation. Mock-infected mice were inoculated with medium. Other mice (n = 10) were injected subcutaneously daily for 9 days with human recombinant Flt-3L. On day 7 of Flt-3L treatment mice were inoculated with HSV-1 by (c) intradermal inoculation of the left ear pinna with 106 PFU HSV-1 following 3·5 µg Flt-3L per day; or (d) scarification of the left side of the neck with 104 PFU HSV-1 following 5 µg Flt-3L per day. Per cent survival was recorded from 6 days before inoculation to 20 days post-inoculation. Mock-infected mice received MSA alone.
The effect of Flt-3L on HSV-1 infection was tested using the two virus inoculation routes described above. A higher dose of Flt-3L was used in the scarification model in order to compensate for greater severity of virus effects by this route. Prophylactic treatment with 3·5 µg/day Flt-3L resulted in a significant reduction in mortality following virus inoculation by the ear pinna route (Fig. 1c). Flt-3L treatment produced 80% survivors compared with 20% survivors in the MSA-treated group. Similar results were seen using the murine neck scarification model following inoculation with 104 PFU of virus and 5 µg/day human recombinant Flt-3L. There was a 60% survival rate in mice treated with Flt-3L compared with a 10% survival rate in the MSA-treated group (Fig. 1d). There was no statistical difference in weight or clinical signs, or zosteriform lesion score, between the Flt-3L and MSA-treated groups in either of the two experimental protocols (data not shown). However, the increase in pinna thickness in the ear inoculated animals was significantly reduced (P = < 0·05) in the Flt-3L group compared with control mice on days 7–8, 14 and 16–19 post-infection (data not shown). The ear pinna represents a site of secondary viral replication in the neck scarification model, versus primary replication in the ear inoculation model.
In addition to reducing HSV-1-induced mortality, the increased number of DC and NK cells generated by Flt-3L treatment reduced the amount of latent HSV-1. Levels of latent virus were examined in mice > 30 days post-infection by conventional co-cultivation and in situ hybridization. Figure 2 shows the results of in situ hybridization to detect LAT-positive neurons in cervical dorsal root ganglia (CIII) from representative mice. Ganglionic sections from MSA-treated mice gave strong nuclear staining (Fig. 2a). Positive stain was confined to the nucleus and had a punctate distribution characteristic of the LAT probe.25 The left (ipsilateral) ganglia had more positive neurons than the right (contralateral) side. There was a decrease in the number of LAT-positive neurons in tissues from Flt-3L-treated mice (Fig. 2b). Sections of ganglia from mock-infected mice processed identically were completely negative for major LAT staining (Fig. 2c). Similar results were observed in the trigeminal ganglia (data not shown). DNase-treated, latently infected sections gave results similar to those given by the MSA-treated sections. All RNase-treated, latently infected sections were negative and a sense riboprobe also gave no positive neurons (data not shown). Table 1 shows the numerical assessment of the in situ hybridization assay. Numerous neurons stained positive for the major LAT in ganglionic sections obtained from mice which survived HSV-1 infection without Flt-3L treatment. Virtually all sections from several different experiments showed that Flt-3L-treated mice lacked LAT-positive neurons. Tissues from mock-infected animals showed background staining with no positive neurons. Table 2 shows the amount of reactivatable virus in tissues from Flt-3L-treated mice assessed by conventional co-cultivation. In MSA-treated virus-infected mice, all tissues tested were positive for virus reactivation. The left trigeminal and left CIII ganglia yielded higher titre virus than the corresponding contralateral tissues. In contrast, all Flt-3L-treated mice gave results of less than 0·5 log10 PFU/tissue which is below the level of detection for this test.
Figure 2.
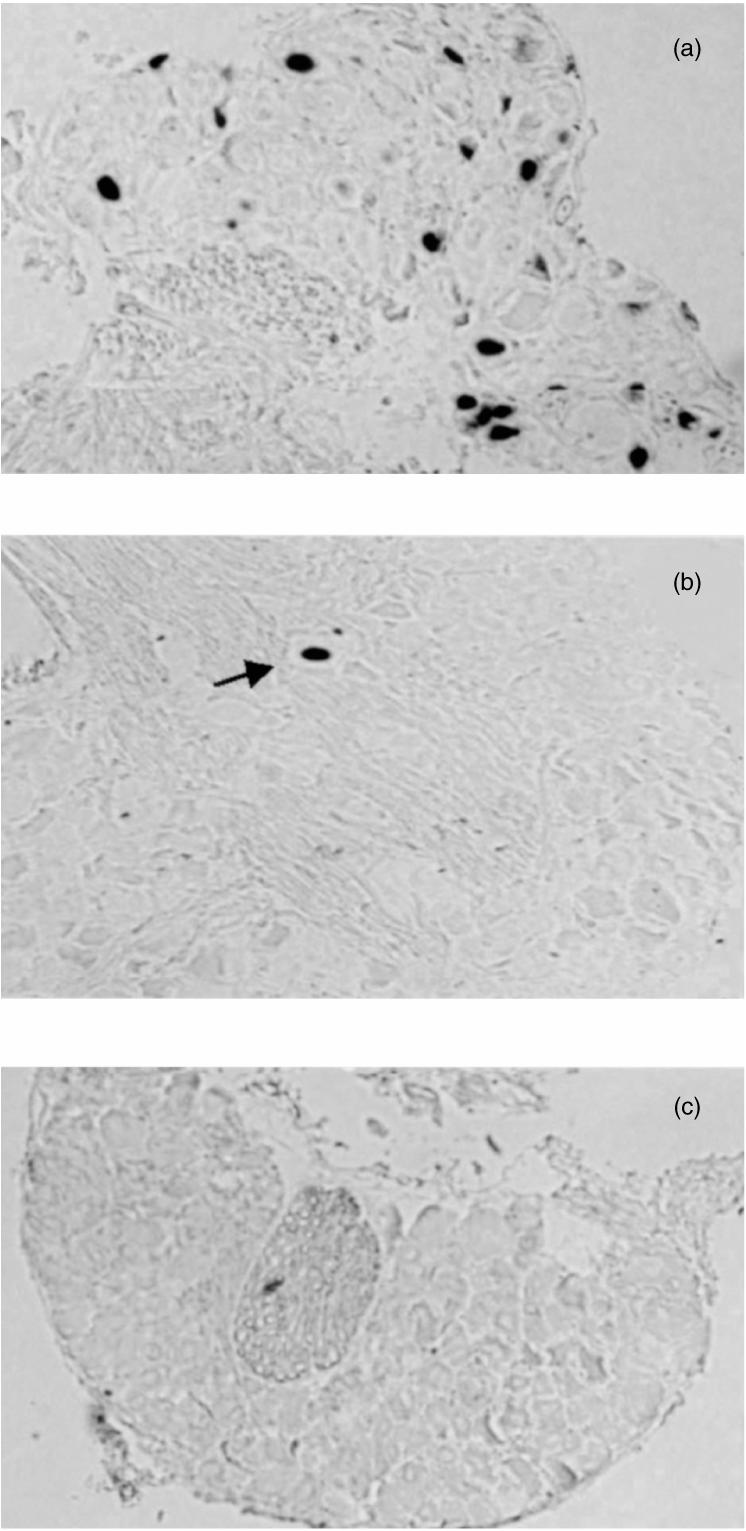
In situ hybridization to detect latently infected neurons. Mice were inoculated with 106 PFU HSV-1 (SC16) by the ear pinna route after pretreatment daily for 7 days with either 3·5 µg human recombinant Flt-3L or MSA. One month later ganglia were fixed and wax embedded. Then, 5-µm sections were analysed by means of a DIG-labelled major LAT riboprobe as described in the Materials and Methods. Representative sections are shown for each treatment group. (a) MSA-treated CIII showing characteristic neuronal staining. The brown/black signal is confined to the nucleus. (b) Flt-3L-treated CIII showing one rare isolated positive neuron (arrowed). (c) Mock-infected CIII showing background staining. Magnification × 100. Similar sections were used to enumerate LAT-positive cells for the data shown in Table 1.
Table 1.
Effect of Flt-3L on HSV-1 latency
| Tissue | ||||
|---|---|---|---|---|
| Treatment | LTG | RTG | LCIII | RCIII |
| Flt-3L | 0 | 0 | 0 | 0 |
| (n = 5) | ||||
| MSA | 13·6 ± 4·8 | 2·8 ± 1·2 | 10·1 ± 2·1 | 2·7 ± 1·0 |
| (n = 4) | ||||
| Mock-infected | 0 | 0 | 0 | 0 |
| (n = 5) | ||||
Latent virus (1 month post-inoculation) was detected by in situ hybridization using the major LAT probe (pSLAT-2). Alternate sections were taken through the whole tissue and the results are shown as the mean number of LAT-positive neurons per section per group ± standard deviation. LTG, left trigeminal ganglia; RTG, right trigeminal ganglia; LCIII, left third cervical dorsal root ganglia; RCIII, right third cervical dorsal root ganglia.
Table 2.
The effect of Flt-3L on HSV-1 reactivation following conventional co-cultivation
| Tissue | ||||
|---|---|---|---|---|
| Treatment | LTG | RTG | LCIII | RCIII |
| Flt-3L | < 0·5 | < 0·5 | < 0·5 | < 0·5 |
| (n = 5) | ||||
| MSA | 3·9 ± 0·5 | 3·1 ± 0·6 | 3·6 ± 0·4 | 2·7 ± 0·6 |
| (n = 4) | ||||
| Mock-infected | < 0·5 | < 0·5 | < 0·5 | < 0·5 |
| (n = 5) | ||||
Latent virus (1 month post-inoculation) was detected by conventional co-cultivation. The results are shown as log10 PFU per tissue per group ± standard deviation. Limit of detection for this test was 0·5 log10 PFU. All samples that scored < 0·5 log10 PFU are below the level of detection. LTG, left trigeminal ganglia; RTG, right trigeminal ganglia; LCIII, left third cervical dorsal root ganglia; RCIII, right third cervical dorsal root ganglia.
Discussion
Flt-3L is a haemopoietic cytokine, which acts upon both myeloid and lymphoid progenitor cells. An important function of Flt-3L is its capacity to induce the expansion of DC and NK cells in lymphoid and non-lymphoid tissues.27 In this study we have investigated if Flt-3L can provide protective immunity to HSV-1 in BALB/c mice which are normally highly susceptible to infection with this virus. Our present data confirm that Flt-3L treatment of mice resulted in a significant increase in the percentage and number of DC in the spleen and liver. The percentage of CD11c+ DC rose 45% and 15% in the spleen and liver, respectively, after treatment with Flt-3L. The absolute number of DC was increased up to approximately 20-fold in both tissues compared to control levels. Myeloid DC were the predominant subset of these APC in both the spleen and liver before and after Flt-3L treatment. However, following Flt-3L treatment the spleen showed a greater expansion of the lymphoid subset whilst the liver showed a greater expansion of the myeloid subset. A modest expansion of NK cells (DX5+) was seen in the spleen but not the liver of Flt-3L-treated mice. Spleen NK cells were increased up to five-fold compared to control levels. Interestingly, CD4+ NK cells were absent from the liver of Flt-3L-treated mice. Studies by others have shown that DC and NK cells are functionally competent following their in vivo induction by Flt-3L. Maraskovsky et al.17 have shown that DC with immature and mature APC function can be isolated from the spleens of Flt-3L-treated mice. Shaw et al.18 have shown that NK cells from Flt-3L-treated mice are mature, resting, cytolytic and reactive to IL-2 by generating lymphokine-activated killer activity. Collectively, these reports and our data presented here support the notion that DC and NK cells share a common progenitor cell and that Flt-3L treatment enhances the number of functionally relevant forms of these cells.
We have tested the effect in vivo of Flt-3L in two murine models of HSV-1 infection, namely ear pinna inoculation and neck scarification. The neck scarification model of HSV-1 infection is more severe than the ear inoculation model, as shown by the respective mortality curves. This difference in severity may be because more sensory neurons are exposed to the virus using the scarification technique compared to the single discrete inoculation site in the pinna model. As a consequence, the quantity of retroaxonally transported virus able to reach the central nervous system and cause fatal encephalitis is expected to be greater in the scarification model. Despite this difference, our data show clearly that Flt-3L is able to induce sufficient protective immunity to reduce mortality in both HSV-1 murine models. Prophylactic treatment with Flt-3L also resulted in a significant decrease in detectable latent virus, in the cervical dorsal root ganglia and trigeminal ganglia as measured by co-cultivation and in situ hybridization. It remains to be established if this is due to a decrease in the level of acute virus available to establish latency or a direct immune effect on HSV-1-infected neurons. The effect on pinna thickness and weight loss was not alleviated by the Flt-3L treatment in the ear model, however, the increase in pinna thickness was reduced by Flt-3L in the neck scarification model. A possible explanation is that the pinna is a site of primary viral replication in the ear model, but a site of secondary viral replication in the neck scarification model. In the neck model, virus travels in a retrograde direction from sensory neurons innervating the skin to the cervical dorsal root ganglia and subsequently to the brain stem and trigeminal ganglia before reaching the ear pinna. This is reflected in the difference in timing of ear pinna inflammation between the two models which occurs later in the neck model.
The protective effect of Flt-3L against HSV-1 infection seen here may be mediated by enhancement of the innate and/or adaptive immune system. Whilst both the innate and adaptive immune systems contribute to the generation of effective immunity against HSV-1, it is T-cell-mediated immunity which ultimately controls anti-viral immunity and which is responsible for the generation of memory cells.29 We are currently investigating the contribution of both the innate and adaptive immune system in protection against HSV infection following Flt-3L treatment. The role of NK cells in resistance to HSV-1 infections has previously been unclear. In vivo depletion studies suggested that NK cells were not important30 whilst adoptive transfer studies indicated that these cells contributed towards resistance.31 Recent experiments using gene-knockout and congenic mouse strains have now clarified the role of NK cells in resistance to HSV-1 infection. Firstly, mice lacking NK and T cells are more susceptible to HSV-1 infection compared to mice which lack T cells alone.32 Secondly, BALB/c mice congenic for the C57BL/6 NK complex (NKC) gene Rhs1r were found to be 10-fold more resistant to HSV-1 infection in comparison to wild-type BALB/c controls. Treatment of congenic BALB/c.B6. Rhs1r mice with anti-NK1.1 antibody abrogated this resistance.33 DC are now seen as accessory cells responsible for NK-cell activation. Recent data have shown that precursor DC, human pDC2 and the related plasmacytoid cells, produce large amounts of IFN-α after exposure to HSV-1,33 and in response to influenza virus infection and CD40 ligation,12 respectively. IFN-α, along with IFN-β and cytokines such as IFN-γ, IL-15 and IL-18, are among the main effectors of the innate immune system and initiate a cascade of biological effects including NK-cell activation.34 These observations strongly suggest a role for NK cells in immunity to HSV-1 which, like antigen-specific T-cell immunity, is initiated and regulated by DC.
A feature of our data is that increased numbers of DC produced by Flt-3L may initiate the enhanced immunity to HSV. It remains to be established how DC acquire and handle cytopathic viruses such as HSV-1 during their initiation of the adaptive and innate immune response. Recent observations have suggested that DC phagocytose apoptotic or necrotic epithelial cells as part of a constitutive process which includes migration from the periphery to draining lymph nodes.35,36 At a time of infection at these sites, there is likely to be extensive death of virus-infected cells. Uptake of these cells by DC would serve two purposes. Firstly, phagocytosis of virus-laden cells would be an efficient mechanism by which DC accumulated virus. Secondly, the inflammatory signals released by virus-infected or dying cells would serve as DC maturation/migratory stimuli. DC would still need to be protected from cytopathic damage or viral attempts to subvert antigen presentation. Human DC directly infected with influenza virus restrict viral replication and its cytopathic effect through the action of the protein MxA.37 DC may express other proteins with a similar protective mechanism during their carriage of other cytopathic viruses. Cells, including DC, infected with HSV-1 do show reduced expression of cell surface MHC class I expression.38,39 The mechanism of MHC class I down-regulation is mediated by the HSV protein (ICP)47, which binds to the transporter associated with antigen processing (TAP), and blocks the transport of antigenic peptides into the endoplasmic reticulum.40,41 The resultant inhibition of MHC class I–antigen complex formation on the surface of HSV-1-infected cells will result in impaired recognition by CD8+ cytotoxic T lymphocytes. However, the absence of MHC class I expression on cells will increase their susceptibility to NK-mediated cytolysis.42 Virus-carrying DC may therefore be killed by NK cells which may represent the normal course of activation of these effector cells during the early events of the innate immune response.43
Acknowledgments
This work was supported by a studentship (J.R.S.) from the BBSRC and by funds from the Wellcome Trust. We would like to thank Immunex Corporation for the supply of human recombinant Flt-3L and useful discussions with Dr Stuart Lyman.
References
- 1.Roizman B. The Herpesviridae. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. 3. Philadelphia: Lippincott-Raven; 1996. pp. 352–358. [Google Scholar]
- 2.Fraser N, Valyi-Nagy T. Viral, neuronal, and immune factors which may influence herpes simplex virus (HSV) latency and reactivation. Microb Pathol. 1993;15:83–91. doi: 10.1006/mpat.1993.1059. [DOI] [PubMed] [Google Scholar]
- 3.Maggioncalda J, Mehta A, Su YH, Fraser NW, Block TM. Correlation between herpes simplex virus type 1 rate of reactivation from latent infection and the number of infected neurons in trigeminal ganglia. Virology. 1996;225:72–81. doi: 10.1006/viro.1996.0576. 10.1006/viro.1996.0576. [DOI] [PubMed] [Google Scholar]
- 4.Hoffmann J, Kaftos FC, Janeway CA, Ezekowitz RA. Phylogenetic perspectives in innate immunity. Science. 1999;284:1313–18. doi: 10.1126/science.284.5418.1313. 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- 5.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–96. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 6.Grouard G, Rissoan M-C, Filgueria L, Durand I, Banchereau J, Liu Y-J. The engimatic plasmacytoid T cells develop into dendritic cells with interleukin (IL) -3 and CD40-ligand. J Exp Med. 1997;185:1101–11. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olweus J, BitMansour A, Warnke R, Thompson PA, Carballido J, Picker IJ, Lund-Johansen F. Dendritic cell ontogeny: a human dendritic cell lineage of myeloid origin. Proc Natl Acad Sci USA. 1997;94:12551–6. doi: 10.1073/pnas.94.23.12551. 10.1073/pnas.94.23.12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rissoan MC, Soumelis V, Kadowaki N, Grouard G, Briere F, de Waal Malefyt R, Lui YJ. Reciprocal control of T helper and dendritic cell differentiation. Science. 1999;283:1183–6. doi: 10.1126/science.283.5405.1183. 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- 9.Palucka K, Banchereau J. Linking innate and adaptive immunity. Nature Med. 1999;5:868–70. doi: 10.1038/11303. [DOI] [PubMed] [Google Scholar]
- 10.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 11.Siegal FP, Kadowaki N, Shodell M, et al. The nature of the principal type I interferon-producing cells in human blood. Science. 1999;284:1835–7. doi: 10.1126/science.284.5421.1835. 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 12.Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nature Med. 1999;5:919–23. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 13.Geldhof AB, Moser M, Lespagnard L, Thielemans K, Baetselier D. Interleukin-12-activated natural killer cells recognize B7 costimulatory molecules on tumor cells and autologous dendritic cells. Blood. 1998;91:196–206. [PubMed] [Google Scholar]
- 14.Lanier L. NK cell receptors. Annu Rev Immunol. 1998;16:359–93. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Fontecha A, Assarsson E, Carbone E, Karre K, Ljunggren H-G. Triggering of murine NK cells by CD40 and CD86 (B7-2) J Immunol. 1999;162:5910–16. [PubMed] [Google Scholar]
- 16.Kitamura H, Iwakabe K, Yahata T, et al. The natural killer T (NKT) cell ligand α-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL) -12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med. 1999;189:1121–8. doi: 10.1084/jem.189.7.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maraskovsky E, Brasel K, Teepe M, Roux ER, Lyman SD, Shortman K, McKenna HJ. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. 1996;184:1953–62. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw SG, Maung AA, Steptoe RJ, Thomson AW, Vujanovic NL. Expansion of functional NK cells in multiple tissue compartments of mice treated with Flt3-ligand: implications for anti-cancer and anti-viral therapy. J Immunol. 1998;161:2817–24. [PubMed] [Google Scholar]
- 19.Boyum A. Separation of white blood cells. Nature (London) 1964;204:793. doi: 10.1038/204793a0. [DOI] [PubMed] [Google Scholar]
- 20.Hill TJ, Field HJ, Blyth WA. Acute and recurrent infection with herpes simplex virus in the mouse: a model for studying latency and recurrent disease. J Gen Virol. 1975;28:341–53. doi: 10.1099/0022-1317-28-3-341. [DOI] [PubMed] [Google Scholar]
- 21.Field HJ, Tewari D, Sutton D, Thackray AM. Comparison of efficacies of famciclovir and valaciclovir against herpes simplex virus type 1 in a murine immunosuppression model. Antimicrob Agents Chemother. 1995;39:1114–19. doi: 10.1128/aac.39.5.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blyth WA, Harbour DA, Hill TJ. Pathogenesis of zosteriform spread of herpes simplex virus in the mouse. J Gen Virol. 1984;65:1477–86. doi: 10.1099/0022-1317-65-9-1477. [DOI] [PubMed] [Google Scholar]
- 23.Field HJ, Bell SE, Elion GB, Nash AA, Wildy P. Effect of acycloguanosine treatment on acute and latent herpes simplex infections in mice. Antimicrob Agents Chemother. 1979;15:554–61. doi: 10.1128/aac.15.4.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thackray AM, Field HJ. Differential effects of famciclovir and valaciclovir on the pathogenesis of herpes simplex virus in a murine infection model including reactivation from latency. J Infect Dis. 1996;173:291–9. doi: 10.1093/infdis/173.2.291. [DOI] [PubMed] [Google Scholar]
- 25.Thackray AM, Field HJ. Famciclovir and valaciclovir differ in the prevention of herpes simplex virus type 1 latency in mice: a quantitative study. Antimicrob Agents Chemother. 1998;42:1555–62. doi: 10.1128/aac.42.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arthur J, Efstathiou S, Simmons A. Intranuclear foci containing low abundance herpes simplex virus latency-associated transcripts visualized by non-isotopic in situ hybridization. J Gen Virol. 1993;74:1363–70. doi: 10.1099/0022-1317-74-7-1363. [DOI] [PubMed] [Google Scholar]
- 27.Antonysamy MA, Thomson AW. Flt3 ligand (FL) and its influence on immune reactivity. Cytokine. 2000;12:87–100. doi: 10.1006/cyto.1999.0540. 10.1006/cyto.1999.0540. [DOI] [PubMed] [Google Scholar]
- 28.Brasel K, McKenna HJ, Morrissey PJ, Charrier K, Morris AE, Lee CC, Williams DE, Lyman SD. Hematologic effects of flt3 ligand in vivo in mice. Blood. 1996;88:2004–12. [PubMed] [Google Scholar]
- 29.Simmons A, Tscharke DC. Anti-CD8 impairs clearance of Herpes Simplex Virus from the nervous system; implications for the fate of virally infected neurons. J Exp Med. 1992;175:1337–44. doi: 10.1084/jem.175.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spits H, Lanier LL, Phillips JH. Development of human T and natural killer cells. Blood. 1995;85:2654–70. [PubMed] [Google Scholar]
- 31.Bendelac A, Rivera MN, Park SH, Roark JH. Mouse CD1-specific Nk1 T cells; development specificity and function. Annu Rev Immunol. 1997;15:535–62. doi: 10.1146/annurev.immunol.15.1.535. [DOI] [PubMed] [Google Scholar]
- 32.Bukowski JF, Welsh RM. The role of natural killer cells and interferon in resistance to acute infection of mice with herpes simplex virus type 1. J Immunol. 1986;136:3481–5. [PubMed] [Google Scholar]
- 33.Simmons A, Scalzo A, Pereira R. Rhs1: a genetic locus which controls the severity of herpes simplex virus infection. 1999. . 24th International Herpesvirus Workshop, Boston. Abstract 9.003.
- 34.Biron C, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in anti-viral defense; function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 35.Huang FP, Platt N, Wykes M, Major JR, Powell TJ, Jenkins CD, MacPherson GG. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J Exp Med. 2000;191:435–44. doi: 10.1084/jem.191.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–34. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cella M, Salio M, Sakakibara Y, Langen H, Julkunen I, Lanzavecchia A. Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J Exp Med. 1999;189:821–9. doi: 10.1084/jem.189.5.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson DC, Hill AB. Herpesviruses and immune evasion. Curr Top Microbiol Immunol. 1998;232:149–77. doi: 10.1007/978-3-642-72045-1_8. [DOI] [PubMed] [Google Scholar]
- 39.Salio M, Cella M, Sauter M, Lanzavecchia A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur J Immunol. 1999;29:3245–53. doi: 10.1002/(SICI)1521-4141(199910)29:10<3245::AID-IMMU3245>3.0.CO;2-X. 10.1002/(sici)1521-4141(199910)29:10<3245::aid-immu3245>3.3.co;2-o. [DOI] [PubMed] [Google Scholar]
- 40.Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. Herpes simplex virus turns off TAP to evade host immunity. Nature. 1995;375:411–15. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 41.Galocha B, Hill A, Barnett BC, Dolan A, Raimondi A, Cook RF, Brunner J, McGeoch DJ, Ploegh Hl. The active site of ICP47, a herpes simplex virus-encoded inhibitor of the major histocompatibility complex (MHC) -encoded peptide transporter associated with antigen processing (TAP), maps to the NH2-terminal 35 residues. J Exp Med. 1997;185:1565–72. doi: 10.1084/jem.185.9.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimizu Y, DeMars R. Demonstration by class I gene transfer that reduced susceptibility of human cells to natural killer cell-mediated lysis is inversely correlated with HLA class I antigen expression. Eur J Immunol. 1989;19:447–51. doi: 10.1002/eji.1830190306. [DOI] [PubMed] [Google Scholar]
- 43.Shah PD, Gilbertson SM, Rowley DA. Dendritic cells that have interacted with antigen are targets for natural killer cells. J Exp Med. 1985;162:625–36. doi: 10.1084/jem.162.2.625. [DOI] [PMC free article] [PubMed] [Google Scholar]