

Abstract
Lysosome-associated membrane proteins 1 and 2 (LAMP-1 and LAMP-2) are delivered to phagosomes during the maturation process. We used cells from LAMP-deficient mice to analyze the role of these proteins in phagosome maturation. Macrophages from LAMP-1- or LAMP-2-deficient mice displayed normal fusion of lysosomes with phagosomes. Because ablation of both the lamp-1 and lamp-2 genes yields an embryonic-lethal phenotype, we were unable to study macrophages from double knockouts. Instead, we reconstituted phagocytosis in murine embryonic fibroblasts (MEFs) by transfection of FcγIIA receptors. Phagosomes formed by FcγIIA-transfected MEFs obtained from LAMP-1- or LAMP-2- deficient mice acquired lysosomal markers. Remarkably, although FcγIIA-transfected MEFs from double-deficient mice ingested particles normally, phagosomal maturation was arrested. LAMP-1 and LAMP-2 double-deficient phagosomes acquired Rab5 and accumulated phosphatidylinositol 3-phosphate, but failed to recruit Rab7 and did not fuse with lysosomes. We attribute the deficiency to impaired organellar motility along microtubules. Time-lapse cinematography revealed that late endosomes/lysosomes as well as phagosomes lacking LAMP-1 and LAMP-2 had reduced ability to move toward the microtubule-organizing center, likely precluding their interaction with each other.
Keywords: acidification, FC receptor, macrophage, maturation
Introduction
Phagocytosis is an essential component of the innate immune response. Phagocytic vacuoles acquire degradative and microbicidal properties through a complex series of interactions with endomembranes. This process, collectively termed phagosome maturation, culminates in the fusion of phagosomes with lysosomes, yielding a hybrid organelle enriched in hydrolytic enzymes, reactive oxygen species and antimicrobial peptides that promote the killing and degradation of internalized microorganisms. Fusion with lysosomes also delivers to the phagosomal membrane integral proteins that are critical for function, such as the V-ATPase that renders the lumen acidic (Lukacs et al, 1990). Two of the most abundant lysosomal components are the lysosome-associated membrane proteins 1 and 2 (LAMP-1 and LAMP-2, respectively) (Eskelinen et al, 2003). LAMP-1 and LAMP-2 exhibit considerable sequence homology and have similar domain structure and biochemical properties. LAMPs are transmembrane proteins with a large, heavily glycosylated luminal domain and a short cytosolic tail (Eskelinen et al, 2003). LAMPs form a continuous carbohydrate lining on the inner leaflet, generating a glycocalyx. Therefore, LAMPs were believed to function in the maintenance of the structural integrity of the lysosomal membrane by protecting it from the hostile luminal environment. However, recent observations are inconsistent with this notion. Depletion of N-glycans with endoglycosidase H caused rapid degradation of LAMP-1 and LAMP-2, yet no changes in lysosomal integrity were noted (Kundra and Kornfeld, 1999). This suggested that LAMPs must have alternative functions. Accordingly, LAMP-2 was proposed to serve as a receptor for chaperone-mediated autophagy of cytosolic proteins (Cuervo and Dice, 1996) and was implicated in MHC class II presentation of cytoplasmic antigens (Zhou et al, 2005).
Generation of lamp-1 and lamp-2 single- and double-knockout (DKO) mice revealed additional details of LAMP function. Lamp-1-deficient mice have a near-normal phenotype, with no significant alterations in their lysosomal properties (Andrejewski et al, 1999). However, they display elevated levels of LAMP-2, which is thought to compensate for the loss of LAMP-1 (Andrejewski et al, 1999). By contrast, lamp-2-deficient mice exhibit elevated postnatal mortality, and the surviving mice are small (Tanaka et al, 2000). Several tissues show accumulation of autophagic vacuoles (Tanaka et al, 2000; Eskelinen et al, 2002). Moreover, hepatocytes from lamp-2-deficient mice display elevated secretion of lysosomal enzymes, improper cathepsin D processing and abnormal retention of mannose-6-phosphate receptors in autophagic vacuoles (Eskelinen et al, 2002). Thus, LAMP-2 may be involved in autophagy and lysosome biogenesis. Consistent with the notion that LAMP-1 and LAMP-2 may have redundant function, deletion of both lamp-1 and lamp-2 causes embryonic lethality (Eskelinen et al, 2004). As in lamp-2-deficient mice, fibroblasts isolated from DKO embryos accumulate abnormally high amounts of autophagic vacuoles and were found to accumulate non-esterified cholesterol in endosomes/lysosomes.
Although phagolysosomes are highly enriched in both LAMP-1 and LAMP-2, it remains unclear whether these glycoproteins play any role in phagosome maturation and/or in microbial killing. We took advantage of the availability of cells lacking either one or both LAMPs to assess their function in phagolysosome biogenesis.
Results
Effect of lamp-1 and lamp-2 deficiency on phagocytosis and phagosome maturation in macrophages
Primary macrophages were elicited from wild-type (WT) mice and from lamp-1 or lamp-2 single-knockout mice, and their phenotype was verified by immunostaining for LAMP-1 and LAMP-2 (Figure 1A, C and E). The phagocytic ability of the macrophages was tested by exposing them to IgG-opsonized latex beads. LAMP-1- and LAMP-2-deficient macrophages ingested beads at least as avidly as did WT cells (Figure 1A, C and E). The phagocytic index (beads/cell), determined by counting at least 250 cells per genotype, was 0.88 for WT, 0.95 for lamp-1−/− and 1.33 for lamp-2−/− macrophages.
Figure 1.
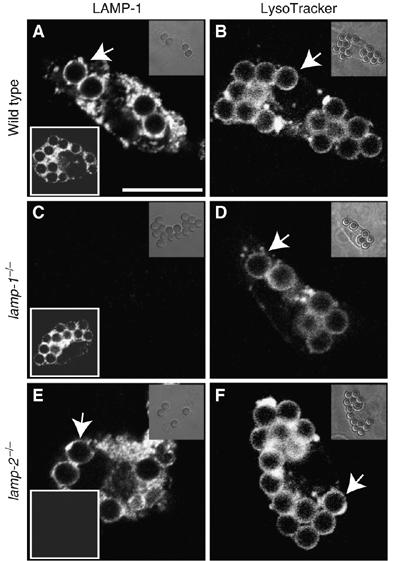
Phagocytosis and phagosome maturation by primary macrophages. Macrophages isolated from the peritoneum of WT (A, B), lamp-1−/− (C, D) and lamp-2−/− mice (E, F) were exposed to IgG-opsonized beads. Maturation was allowed to proceed for 60 min and macrophages were immunostained for LAMP-1 (main panels in A, C, E) and LAMP-2 (lower left insets in A, C, E) or labeled with LysoTracker (B, D, F). Arrows indicate LysoTracker-enriched phagosomes. Images are representative of at least three separate experiments of each type. Scale bar=10 μm.
We next analyzed whether LAMP-1 or LAMP-2 are required for phagolysosome biogenesis. Labeling with the acidotropic dye LysoTracker was used to quantify the efficiency of lysosome fusion with phagosomes 60 min after ingestion of beads. As illustrated in Figure 1B, most of the phagosomes formed by macrophages isolated from WT mice merged with lysosomes. Virtually identical results were obtained in macrophages from LAMP-1- (Figure 1D) or LAMP-2-deficient mice (Figure 1F). Upwards of 90% of the phagosomes accumulated LysoTracker in all cases. Therefore, deletion of either LAMP-1 or LAMP-2 alone had no detectable effect on the ability of macrophages to ingest particles, nor did it impair the formation of phagolysosomes. The latter conclusion is supported by the presence of LAMP-2 in phagosomes of LAMP-1-deficient cells (Figure 1C, inset), and vice versa (Figure 1E).
Effect of simultaneous deletion of lamp-1 and lamp-2
Because of their structural and biochemical similarities, LAMP-1 and LAMP-2 may have overlapping functions. This functional redundancy could account for our failure to detect significant effects on phagosome formation and maturation in single-knockout cells. In principle, this limitation could be overcome by studying phagocytosis in macrophages from DKO mice. However, simultaneous deletion of the lamp-1 and lamp-2 genes produces an embryonic lethal phenotype (Eskelinen et al, 2004), precluding isolation of professional phagocytes (i.e. macrophages or neutrophils). As an alternative, we generated ‘engineered phagocytes' by transfection of phagocytic receptors into immortalized embryonic fibroblasts (MEFs) obtained from WT or LAMP-deficient mice. Heterologous transfection of the FcγIIA receptors (FcR) was shown earlier to confer phagocytic capability to fibroblastic cells (Indik et al, 1995), and the resulting phagosomes undergo a maturation sequence similar to that of professional phagocytes (Downey et al, 1999). We found transfection efficiency to be similar in WT and mutant fibroblasts (not shown); the phenotypic properties of the transfectants are illustrated in Figure 2. As expected, WT fibroblasts express both LAMP-1 and LAMP-2 (Figure 2A and B) and, following transfection with FcR acquire the ability to engulf IgG-opsonized beads (Figure 2C). Fibroblasts from LAMP-1 or LAMP-2 single-knockout mice (Figure 2D, E, G and H) and DKO mice (Figure 2J and K) lack the appropriate gene products. FcR-transfected fibroblasts lacking either LAMP-1 or LAMP-2 were competent to perform phagocytosis (Figure 2F and I), in good agreement with the results using primary macrophages. Importantly, MEFs from DKO mice were also capable of internalizing beads (Figure 2L). There were no gross differences in phagocytic ability between WT, single knockout or DKO MEFs. Hence, we concluded that neither LAMP-1 nor LAMP-2 plays a role in FcR-mediated phagosome formation. The persistence of phagocytosis enabled us to analyze the process of maturation in DKO cells.
Figure 2.
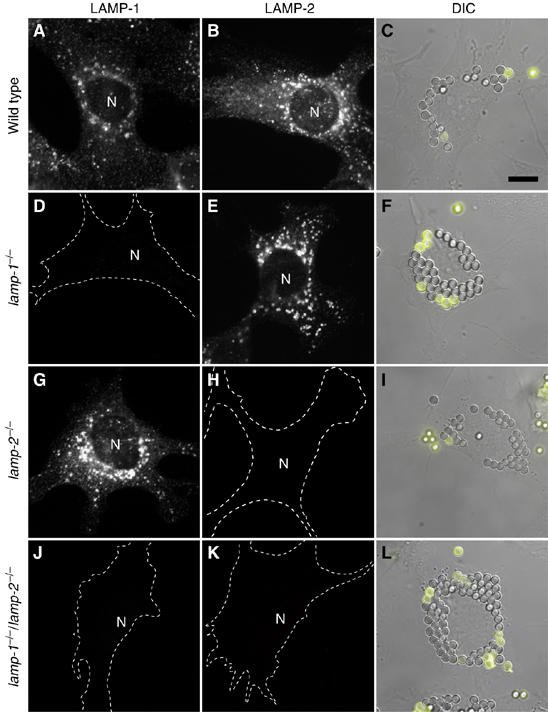
LAMP immunostaining and phagocytosis by Fc receptor-transfected MEFs. MEFs from WT (A–C), lamp-1−/− (D–F), lamp-2−/− (G–I) and lamp-1−/−/lamp-2−/− (J–L) mice were stained for LAMP-1 (A, D, G, J) or LAMP-2 (B, E, H, K). Cell outlines were traced when immunostaining was negative. N=nucleus. WT (C), lamp-1−/− (F), lamp-2−/− (I) and lamp-1−/−/lamp-2−/− fibroblasts (L) were transfected with FcR. After 24 h, transfectants were exposed to beads and phagocytosis allowed to proceed for 15 min. External beads were identified by the addition of anti-IgG antibodies (yellow). Images are representative of at least 3 experiments of each type. Scale bar=10 μm.
We proceeded to measure phagosome–lysosome fusion in transfected MEFs using LysoTracker. Accumulation of LysoTracker in these cells was precluded by treatment with NH4Cl and concanamycin A (Figure 3A), indicating that it functions as an acidotropic probe, suitable for assessment of phagolysosome formation. As found earlier for COS cells, phagosomes formed by FcR-transfected WT fibroblasts accumulated LysoTracker (Figure 3B). Similar results were obtained with two separate clones of WT fibroblasts: 90±2% of the phagosomes in clone no. 3 and 79±2% in clone no. 5 had fused with lysosomes after 60 min of maturation (Figure 3H; means±s.e., n=4 for each clone). The occurrence of phago-lysosomal fusion was validated by EM, which demonstrated delivery of electron-dense material characteristic of lysosomes to the lumen of phagosomes (Figure 3F, enlarged in F′). These data imply that engineered phagocytic fibroblasts are capable of supporting phagosome maturation in a manner analogous to primary macrophages and are useful for analyzing the role of LAMPs in phagolysosome biogenesis.
Figure 3.
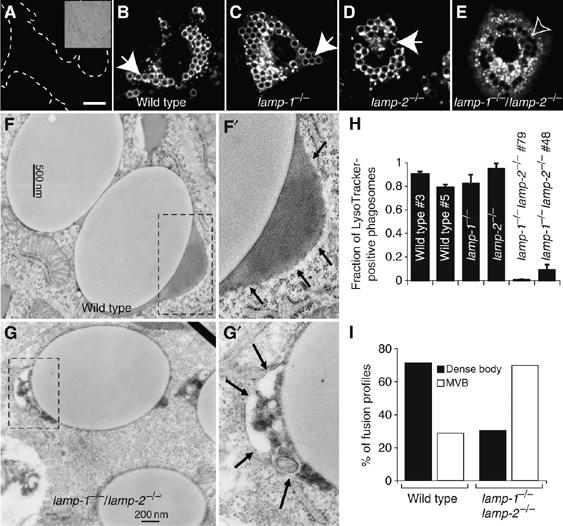
Phagosome maturation in MEFs expressing FcγIIA receptors. (A) WT fibroblasts were treated with 10 mM NH4Cl and 250 nM concanamycin A and labeled with LysoTracker. Dashed lines show cell outline. Inset: corresponding DIC. WT (B, F), lamp-1−/− (C), lamp-2−/− (D) and lamp-1−/−/lamp-2−/− MEFs (E, G) were transfected with FcR. After 16–24 h, transfectants were exposed to beads, and after 15 min of engulfment, phagosomes were allowed to undergo maturation for 60 min. In (A–E), LysoTracker was added and cells were visualized by confocal microscopy. Closed arrows: LysoTracker-positive phagosome; open arrows: LysoTracker-negative phagosomes. Scale bar=10 μm. WT (F, F′) and lamp-1−/−/lamp-2−/− (G, G′) phagosomes were analyzed by EM. Dashed boxes were enlarged and are shown in panels F′ and G′. Arrows point to the region of interest. (H) Quantitation of the fraction of LysoTracker-positive phagosomes in WT and lamp1−/−/lamp2−/− (two clones each) and lamp1−/− and lamp2−/− MEFs. Accumulation or absence of LysoTracker around internalized latex beads was determined by confocal microscopy. Data are means±s.e. of four experiments (at least 100 phagosomes from ⩾20 cells per experiment. (I) Quantitation of phagosomes undergoing fusion with dense bodies (lysosomes, black bars) or multivesicular bodies (MVB, open bars) in WT and lamp-1−/−/lamp-2−/− MEFs. Dense body indicates fusion partners that contained homogeneous electron-dense material. MVB indicates fusion partners that contained internal membranes, mostly small vesicles. 230 and 221 phagosome fusion profiles were scored for WT and lamp-1−/−/lamp-2−/− fibroblasts, respectively.
Phagosomes formed by transfected MEFs isolated from single-knockout mice also fused effectively with lysosomes (Figure 3C and D). The estimates of LysoTracker-positive phagosomes, 82±7% (n=4) and 95±4% (n=4) for LAMP-1- and LAMP-2-deficient fibroblasts, respectively, are indistinguishable from those in WT cells (Figure 3H). Thus, the data from transfected fibroblasts recapitulated the findings obtained with primary macrophages.
Strikingly different results were obtained in MEFs derived from DKO mice. As shown in Figure 3E, phagosomes formed by cells lacking both LAMP-1 and LAMP-2 failed to stain with LysoTracker, despite normal accumulation of the probe by lysosomes. Similar results were obtained using two different DKO clones, one transformed with SV40 large T antigen and another that became spontaneously immortalized, suggesting that clonal variation is not responsible for the maturation defect. Only 0.6±0.3% (n=4) and 9±4% (n=4) of phagosomes matured in clones no. 79 and no. 48, respectively (Figure 3H). The failure of such phagosomes to fuse with lysosomes was confirmed by EM (Figure 3G). Electron-dense homogeneous contents, characteristic of lysosomes, were found in the lumen of 71.3% of WT phagosome fusion profiles (Figure 3F and I), but only rarely in fusion profiles of LAMP-1- and LAMP-2-deficient phagosomes (Figure 3G and I). Instead, multivesicular structures, typical of endosomes, were observed in 69.7% of the fusion profiles of DKO phagosomes (Figure 3G, enlarged in G′). Therefore, either LAMP-1 or LAMP-2 alone can support phagosome maturation, whereas loss of both LAMPs markedly inhibits phagosome–lysosome fusion.
LAMPs were proposed to maintain lysosomal membrane integrity. Therefore, the number of functional lysosomes may have been compromised in DKO fibroblasts, possibly accounting for the defective phago-lysosome formation. To rule out this possibility, we determined the relative proportion of lysosomes by integrating the total fluorescence from LysoTracker in WT and DKO fibroblasts, using identical conditions for image acquisition. The accumulation of LysoTracker was in fact greater in the LAMP-deficient cells than in their WT counterparts (e.g. Figure 6), implying that the defect in phagosomal acidification was not due to a deficit of lysosomes or their inability to acidify. To ensure that the phagosome membrane was not compromised in the absence of LAMP, we expressed a marker that, in macrophages, is recruited at an early stage and persists on the phagosomal membrane even after phago-lysosome fusion. TC10, a GTPase that is constitutively associated with membranes by virtue of its acylation, was found to be recruited to phagosomes shortly after sealing and remained associated with both WT and DKO phagosomes even after 60 min (Supplementary Figure 1). Additionally, close examination by EM revealed that the phagosome membrane was continuous and seemingly intact in both genotypes (Figure 3F and G). Thus, maturation arrest cannot be attributed to loss of phagosomal membrane integrity.
Figure 6.
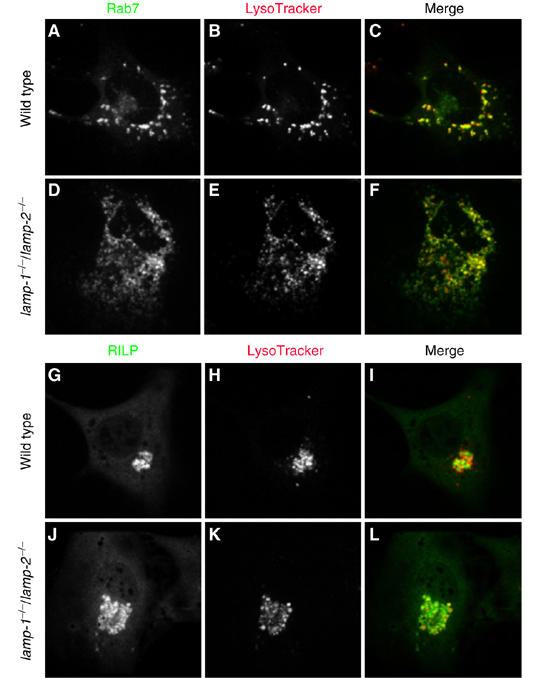
Colocalization of Rab7 and RILP with LysoTracker-labeled compartments. WT (A–C, G–I) and lamp-1−/−/lamp-2−/− fibroblasts (D–F, J–L) were transfected with cDNA encoding GFP-Rab7 (A–F) or GFP-RILP (G–L). After 24 h, acidic compartments were labeled with LysoTracker and the cells visualized by confocal microscopy. Rab7 distribution is shown in (A) and (D); RILP distribution is shown in (G) and (J); LysoTracker is shown in (B), (E), (H) and (K); merged images are shown in (C), (F), (I) and (L). Images are representative of ⩾3 experiments.
Determination of the stage of maturation arrest
Before fusing with lysosomes, phagocytic vacuoles interact with early and late endosomes. We used organelle-specific markers to delineate the stage where maturation arrested when LAMP-1 and LAMP-2 were deleted. Two early endosome markers were used: Rab5 (Chavrier et al, 1990) and the PX domain of p40phox, which binds to phosphatidylinositol 3-phosphate (PI(3)P; Kanai et al, 2001). The number of phagosomes that recruited Rab5 (Figure 4A, B and K; Supplementary Figure 2A and C) 5 min after particle ingestion was similar in WT and DKO fibroblasts (71±3 and 75±2%, respectively; n=3). Similarly, accumulation of PI(3)P (Figure 4C, D and K; Supplementary Figure 2B and C) was comparable in WT and LAMP-deficient phagosomes (88±3 and 83±1%; n=3). Rab5 and PI(3)P were subsequently lost at comparable rates from both WT and LAMP-deficient cells (Supplementary Figure 2G-I). These data imply that lamp-1/lamp-2 deficiency does not alter the transient interaction between phagosomes and early endosomes.
Figure 4.
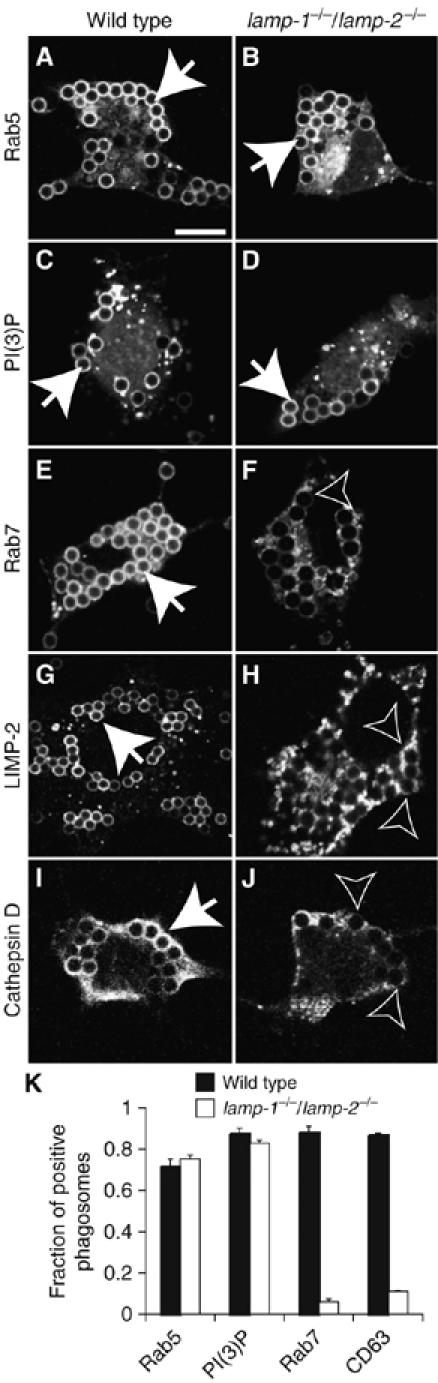
Acquisition of endocytic markers. WT (A, C, E, G, I) and lamp-1−/−/lamp-2−/− MEFs (B, D, F, H, J) were transfected with FcR and after 24 h were exposed to IgG-opsonized beads. Phagosome maturation was allowed to proceed for 5 min in WT and lamp1−/−/lamp2−/− fibroblasts expressing GFP-Rab5 (A, B, respectively) or GFP-PX (C, D, respectively). Alternatively, maturation was allowed to proceed for 60 min in WT and lamp1−/−/lamp2−/− MEFs expressing GFP-Rab7 (E, F). WT and lamp-1−/−/lamp-2−/− MEFs were stained for LIMP-2 (G and H, respectively) and cathepsin D (I, J). (K) Quantitation of the fraction of phagosomes positive for endocytic markers in WT (black bars) and lamp1−/−/lamp2−/− fibroblasts (open bars). Data are means±s.e. of four experiments with ⩾100 phagosomes from ⩾20 different cells. Closed arrows: phagosomes positive for the indicated marker; open arrows: phagosomes negative for the markers. Scale bar=10 μm.
To assess the fusion of phagosomes with late endosomes, we used a similar strategy, expressing markers of late endosomes: Rab7 (Chavrier et al, 1990), Rab-interacting lysosomal protein (RILP) (Cantalupo et al, 2001; Harrison et al, 2003) and CD63 (Fukuda, 1991; Escola et al, 1998; Kobayashi et al, 1999). As expected, most WT phagosomes recruited Rab7 (88±3%; n=3, Figure 4E and K), CD63 (87±0.5%; n=3, Figure 4K), and also co-stained for both CD63 and RILP (Supplementary Figure 3A-C). In stark contrast, lamp-1/lamp-2 deficiency severely inhibited acquisition of late endosomal markers. Only 6±1% (n=3) of LAMP-deficient phagosomes recruited Rab7 (Figure 4F and K), whereas 11±3% (n=3) acquired CD63 (Figure 4K; Supplementary Figure 3D-F). Because Rab7, RILP and CD63 are normally present on late endosomes, these findings suggest that the phagosomes from LAMP-deficient cells fail to merge with these organelles.
To confirm that lamp-1/lamp-2-deficient phagosomes were unable to interact with lysosomes, we immunostained two endogenous markers of lysosomes: LIMP-2 (LGP85), a lysosomal membrane protein (Kuronita et al, 2002), and cathepsin D, a luminal protease (Godbold et al, 1998). As expected, WT phagosomes acquired both LIMP-2 and cathepsin D within 60 min of formation (Figure 4G and I). By contrast, lamp-1/lamp-2-deficient phagosomes were devoid of either LIMP-2 or cathepsin D (Figure 4H and J). To test if lamp-1/lamp-2-null phagosomes were diverted to an autophagic pathway, we expressed a marker of autophagic organelles, LC3 (Kabeya et al, 2000). Neither WT nor lamp-1/lamp-2-deficient phagosomes acquired LC3 (Supplementary Figure 4). Collectively, these data demonstrate that LAMP deficiency impairs phagolysosome biogenesis, without diverting the phagosomes to the autophagic pathway.
Reconstitution of LAMP-1 and LAMP-2
To confirm that the maturation defect is strictly attributable to the loss of LAMP expression, we tried to rescue maturation by exogenously expressing LAMP-1 or LAMP-2. FcR and either LAMP-1 or LAMP-2 were cotransfected into DKO fibroblasts, and expression was confirmed by immunoblotting (Supplementary Figure 5A). Because not all the cells in the population acquire the transgene in transient transfections, the LAMP levels attained were lower than those found in WT populations. Nevertheless, we observed that heterologous expression of LAMP-2 partially restored Rab7 recruitment to 53.5±7.3% of the phagosomes in the transfectants (n=3; Supplementary Figure 5C and D), whereas LAMP-1 was able to rescue only 22.3±6.2% of phagosomes (Supplementary Figure 5B and C). Heterologous transfection of LAMP-1 may not have reached a sufficiently high level of protein expression to rescue the defect in phagolysosome biogenesis. Regardless, these data are consistent with previously published data indicating that LAMP-2 may be functionally more effective than LAMP-1 (Tanaka et al, 2000; Eskelinen et al, 2002) and confirm that inhibition of phagosome maturation can be strictly attributed to the loss of LAMP expression.
Effect of knockdown of lamp-1 and lamp-2 in macrophages
To ensure that our conclusions regarding the role of LAMP isoforms in phagosome maturation apply not only to engineered, fibroblast-derived phagocytes but also to professional phagocytes, we used siRNA to knock down the expression of LAMP-1 and LAMP-2 in RAW264.7 macrophages. Immunoblotting of samples treated with a combination of siRNAs targeting both LAMP isoforms or with scrambled siRNA indicated that, despite the use of electroporation, silencing of the lamp-1 and lamp-2 genes was incomplete (40–50% reduction; Figure 5I). Microscopic examination of immunostained samples revealed that, rather than a homogeneous partial decrease across the entire population, the siRNA treatment caused a marked reduction of the expression of both LAMP-1 (Figure 5F) and LAMP-2 (Figure 5G) in a subpopulation of the cells, whereas the rest remained largely unaffected, which was not observed in cells transfected with scrambled siRNA (Figure 5B and C). We believe that this heterogeneity is due to inhomogeneous delivery of the siRNA. We proceeded to assay phagosome maturation in macrophages treated with siRNA, scoring only those cells with little or no residual LAMP immunostaining. The cells were simultaneously stained for LIMP-2, which was unaffected by the siRNA mixture (Figure 5E), to score phagolysosomal fusion. As shown in Figure 5J, 61±6% (n=3) of the phagosomes from macrophages transfected with scrambled siRNA acquired LIMP-2 after 60 min. In sharp contrast, phagosomes from macrophages with reduced LAMP showed a paucity (12±2%; n=3) of LIMP-2 (Figure 5J). These data recapitulate the observations made in fibroblasts, validating the conclusions in a native phagocytic system.
Figure 5.
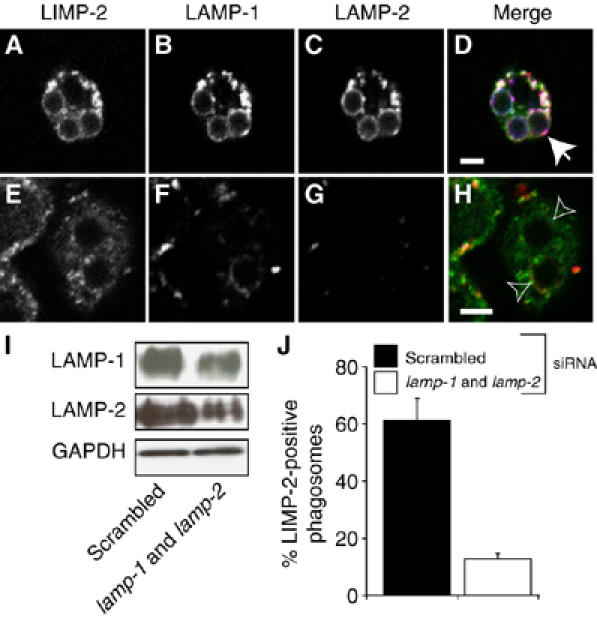
Phagosome maturation in macrophages treated with siRNA against lamp-1 and lamp-2. RAW 264.7 macrophages were transfected with either scrambled siRNA (A–D, left lane in I) or siRNA against lamp-1 and lamp-2 (E–H, right lane in I). (A–H) Macrophages treated with scrambled (A–D) or lamp-1 and lamp-2 siRNA (E, F) were exposed to beads and maturation allowed to proceed for 60 min. Cells were stained for LIMP-2 (A, E; green in D) to assess phagolysosome fusion and for LAMP-1 (B, F; red in D) and LAMP-2 (C, G; blue in D) to determine knockdown effectiveness. Merged images are shown in (D) and (H). Closed arrows: phagosomes enriched with LIMP-2; open arrows: phagosomes negative for LIMP-2. (I) Whole-cell lysates were probed by immunoblotting for LAMP-1 and LAMP-2 72 h later. Blots were stripped and reprobed for GAPDH as loading control. (J) Quantitation of the effect of LAMP-1 and LAMP-2 knockdown on phagolysosome biogenesis. Phagolysosomes were identified by LIMP-2 staining. Data represent means±s.e. from three experiments with ⩾30 phagosomes from ⩾10 cells. Scale bar=3 μm.
Targeting and activation of Rab7
Rab7 recruitment and activation were shown to be necessary for completion of phagosome maturation (Harrison et al, 2003; Jager et al, 2004). The defective formation of phago-lysosomes could be caused by an inability of Rab7 to associate and/or become activated on late endosomes/lysosomes of LAMP-deficient cells. We therefore compared the distribution and state of activation of Rab7 in WT and DKO cells. As shown in Figure 6A and C, a large fraction of Rab7 is associated with vesicular structures that colocalize extensively with LysoTracker in WT cells, as expected from the acidic nature of late endosomes/lysosomes. Similarly, most of the Rab7 was membrane-associated in LAMP-deficient cells (Figure 6D), although the Rab7-bearing vesicles were more widely distributed throughout the cells. Nevertheless, as in the control cells, the Rab7 vesicles showed substantive colocalization with LysoTracker in DKO cells (Figure 6F). Rab7 was also found to colocalize with cathepsin D in both cell types (not shown). These observations indicate that Rab7 is present in late endosomes/lysosomes of LAMP-deficient cells.
Association with late endosomes/lysosomes suggests that Rab7 is active in the LAMP-deficient cells, as the GTP-bound form is preferentially membrane-bound. To verify this, we analyzed the subcellular distribution of RILP, an effector of Rab7 that binds exclusively to its active conformation (Cantalupo et al, 2001). GFP-RILP was expressed in WT and DKO fibroblasts and its localization compared to that of LysoTracker. As shown in Figure 6G and I, RILP not only colocalized extensively with acidic organelles, but also induced their clustering near the nucleus. Clustering results from the linkage of late endosomes/lysosomes bearing active Rab7 to dynein, a centripetal microtubule-associated motor (Cantalupo et al, 2001). Of note, the LysoTracker-positive organelles of DKO fibroblasts similarly associated with RILP and clustered beside the nucleus (Figure 6J and L), implying that Rab7 was present and active on late endosomes/lysosomes of LAMP-deficient cells. Thus, the defect in phagosome maturation observed in these cells cannot be attributed to a general Rab7 mistargeting or dysfunction. Accordingly, expression of Rab7(Q67L), a constitutively active mutant that causes enlargement of late endosomes (Bucci et al, 2000), was unable to restore acidification of phagosomes in DKO cells (Supplementary Figure 6).
Distribution and motility of lysosomes and phagosomes
We noted earlier that the distribution of Rab7-postive organelles appeared different in WT and LAMP-deficient cells (Figure 6). The statistical and functional significance of this difference is explored in more detail in Figure 7. Acidic organelles, stained using LysoTracker, were generally clustered in the perinuclear region of WT cells (Figure 7A), whereas they were consistently dispersed more widely throughout the LAMP-deficient cells (Figure 7B). A quantitative analysis of the distribution of lysosomes with respect to the nucleus was made by high-throughput imaging using the Cellomics system. Lysosomes were labeled with rhodamine-dextran using a prolonged pulse-chase protocol, whereas the nucleus was identified using Höechst 33342. The distance between all identifiable lysosomes and the nearest edge of the nucleus was quantified in 509 individual WT cells and in 876 DKO cells by a specially designed algorithm. The data were binned in 1.58 μm units and expressed as a fraction of the total lysosomal content. As summarized in Figure 7C, the largest fraction of WT lysosomes resided within 1.58 μm of the nucleus and their density decayed sharply as the distance from the nucleus increased. By contrast, the highest density of LAMP-deficient lysosomes was found >3 μm away from the nucleus. Identical results were obtained for lysosomes labeled by LysoTracker. These data confirmed that lysosomes are more diffusely distributed in the cytosol of cells lacking LAMP-1 and LAMP-2.
Figure 7.
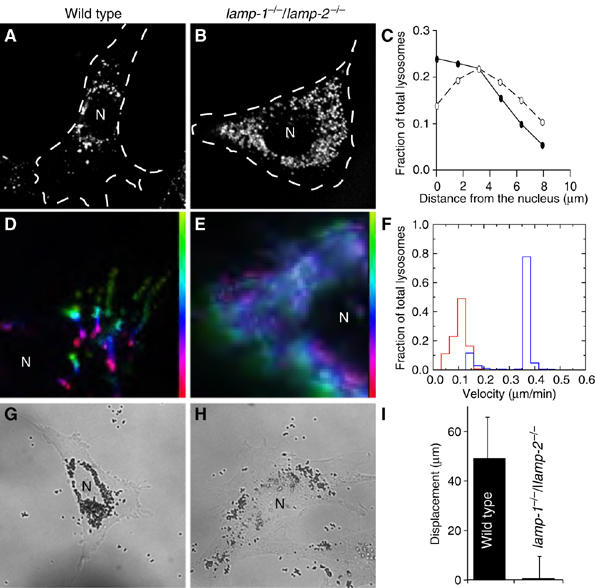
Determination of lysosome and phagosome motility. Acidic compartments of WT (A, D) and lamp-1−/−/lamp-2−/− MEFs (B, E) were loaded with LysoTracker. Their distribution at steady state was recorded by epifluorescence microscopy (A, B) and the cell outline, determined by DIC, is shown by dotted line. In (C), the distribution of individual lysosomes at rest was determined relative to the nucleus using the Cellomics system. The distribution of lysosomes, expressed as a fraction of the total, is plotted as a function of distance (μm) from the nucleus in WT (solid symbols) and lamp-1−/−/lamp-2−/− cells (open symbols). Data are means of three experiments, scoring ⩾500 cells per experiment. The error bars (±s.e.) were smaller than the symbol and are not visible. In (D) and (E), the response of lysosomes to cytosolic alkalinization was documented using time-lapse microscopy. The path of a given lysosome is shown by a pseudocolor scale (vertical color bar) where yellow-green indicates the starting position and red the final position attained 10 min after alkalinization. White indicates lysosomes that did not move. (F) Summary of automated determinations of lysosomal velocities. Late endosome/lysosomes of WT (blue) and lamp-1−/−/lamp-2−/− (red) were labeled with LysoTracker. Retrograde transport upon alkalinization of the cytosol was determined for a period of 10 min as described in Materials and Methods. WT (G) and lamp-1−/−/lamp-2−/− cells (H) were exposed to 0.8 μm beads and after 3 h, their distribution was imaged by DIC. (I) Quantitation of displacement of internalized beads from the plasma membrane over 30 min. Data are means+s.e. of three experiments, scoring ⩾18 phagosomes per cell. N=nucleus.
In normal cells, lysosomes are maintained in the perinuclear region by microtubule-associated motors. Dynein, which mediates retrograde movement of lysosomes along microtubules (Jordens et al, 2001), is thought to predominate over kinesin(s), which are responsible for anterograde transport (Hollenbeck and Swanson, 1990). The more peripheral distribution of LAMP-deficient lysosomes could be caused by defective dynein function and/or to excessive kinesin activity in the DKO cells. The contribution of anterograde and retrograde motors can be analyzed separately by varying the intracellular pH. Heuser (1989) found that kinesin activity predominates when the cytosol is acidic, resulting in lysosomal displacement toward the cell edge. Conversely, under alkalinizing conditions, dynein activity becomes dominant and lysosomes are mobilized toward the nucleus. We compared the mobility of labeled lysosomes by time-lapse cinematography. Acidification caused marginalization of lysosomes in both cell types, although the effect was less marked in LAMP-deficient cells, because their lysosomes were initially more dispersed. Subsequent alkalinization reversed the direction of lysosomal movement. The lysosomes of both WT and DKO cells moved centripetally, whereas the overall displacement of the LAMP-deficient lysosomes was reduced (Figure 7D and E). Quantitation of lysosomal speed using particle-tracking software revealed that WT lysosomes displayed a bimodal distribution (Figure 7F); the majority of the lysosomes moved rapidly (mean velocity 0.37±0.03 μm/min), with a smaller subpopulation that moved more slowly (0.14±0.03 μm/min). By contrast, only a minute fraction of the LAMP-deficient lysosomes moved rapidly, whereas the vast majority were considerably slower (0.10±0.03 μm/min). These data indicate that the dynein-mediated transport of lysosomes is depressed in LAMP-knockout cells, likely accounting for the altered lysosomal distribution.
Following their formation at the cell periphery, phagosomes travel toward the centriole in a microtubule- and dynein-dependent manner, and this displacement is required for phago-lysosome fusion (Harrison et al, 2003). We considered the possibility that phagosome motility might also be impaired in lamp-null cells. Fibroblasts expressing FcR were allowed to ingest beads, which were tracked in real time by a combination of DIC and fluorescence microscopy. Small (0.8 μm) beads were used to facilitate displacement and prevent crowding. In WT cells, phagosomes traveled 49±17 μm over 30 min, whereas lamp-deficient phagosomes traveled only 0.6±9 μm (Figure 7I). The result of this differential speed was most apparent after longer periods, when steady state had been reached. After 3 h, particles internalized by WT fibroblasts had gathered around the nucleus (Figure 7G), whereas LAMP-deficient phagosomes remained scattered about the cytoplasm (Figure 7H). Thus, phagosome motility is also impaired in lamp-null cells and, in combination with the lysosomal mobility defect, likely contributes to the maturation arrest.
Discussion
Phagosome–lysosome fusion bestows on the phagocytic vacuole the lytic properties essential for efficient removal of internalized pathogens. Fusion with lysosomes results in delivery to phagosomes of an assortment of luminal and membrane proteins. Despite the fact that LAMP-1 and LAMP-2 are possibly the most abundant integral membrane proteins of phago-lysosomes, their role in phagosome function has remained elusive. Our data indicate that LAMPs are essential for successful completion of phagosome maturation, specifically, for the transition from early to late phagosomes. In addition, we noted that the absence of both LAMP-1 and LAMP-2 delayed transport of fluid-phase markers from early endosomes to lysosomes. These observations, together with the reported requirement of LAMPs for proper formation of autolysosomes (Eskelinen et al, 2004; Jager et al, 2004), highlight an important role for LAMPs in the dynamics of the endocytic pathway. However, the absence of LAMPs does not produce a global, nonselective impairment of endocytic traffic. Unlike fluid-phase uptake, internalization and sorting of receptors, such as the mannose-6-phosphate receptor and its ligand, arylsulfatase A, were found to be normal in DKO cells (Eskelinen et al, 2004). Differences in the regulation of fluidphase and receptor-mediated endocytosis have been reported: transport of the EGF receptors from early to late endosomes/lysosomes requires PI(3)P, whereas fluid phase does not (Petiot et al, 2003). Thus, LAMPs may be required for a PI(3)P-independent pathway. Accordingly, inhibition of the class III phosphatidylinositol 3-kinase produces only a partial inhibition of LAMP acquisition by phagosomes (Vieira et al, 2001).
We investigated the mechanism whereby LAMPs contribute to the maturation process. It was reported that lysosomes from DKO mice retain abnormally high levels of cholesterol (Eskelinen et al, 2004), raising the possibility that the defects in maturation may be an indirect result of abnormal lipid metabolism. This hypothesis was further buttressed by the observation that acute loading with cholesterol using U18666A inhibited lysosome motility (Lebrand et al, 2002). However, several lines of evidence indicate that cholesterol accumulation alone is unlikely to account for the impairment of phagosome maturation. Firstly, lamp-1/lamp-2-deficient lysosomes are scattered throughout the cytosol (Figure 7B), which contrasts sharply with the observations of Lebrand et al (2002), who found that in cells treated with U18666A lysosomes became tightly concentrated in the juxtanuclear region. This difference in localization implies that ablation of LAMP-1 and LAMP-2 and cholesterol accumulation have diametrically opposed effects on lysosome motility. Secondly, reduction of cholesterol in DKO fibroblasts to WT levels did not relocate lysosomes to the perinuclear region. Thirdly and most importantly, cholesterol depletion did not restore normal traffic along the endocytic pathway (Supplementary Figure 7). Jointly, these observations support the notion that the defects in phagosome maturation do not result from cholesterol accumulation.
Rab5 is thought to associate with phagosomes shortly after they seal and is thought to be essential for subsequent acquisition of Rab7, which in turn is necessary for phago-lysosome fusion (Vieira et al, 2003). Fusion with late endosomes/lysosomes is thought to mediate delivery of LAMPs to the maturing phagosome. Unexpectedly, we observed that ablation of LAMPs precluded delivery of Rab7 (Figure 4F and K), indicating that Rab7 association with phagosomes is itself LAMP-dependent. This implies that LAMPs and Rab7 work in a concerted fashion to direct maturation. To clarify the relationship between these factors, we expressed a dominant-negative allele of Rab7 and found that acquisition of LAMP-1 and LAMP-2 is inhibited only partially (not shown), suggesting that not all LAMP molecules rely on Rab7 for recruitment to phagosomes and that LAMPs may act upstream and independently of Rab7 (Figure 8, step 2). This interpretation is supported by earlier evidence that LAMPs appear on phagosomes containing Leishmania (Scianimanico et al, 1999) despite their lack of Rab7.
Figure 8.
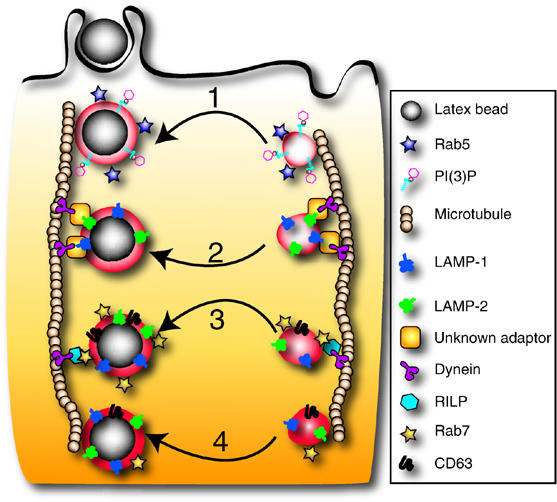
Schematic representation of the proposed role of LAMPs in maturation. Following sealing, the phagosome (left vertical path) is propelled into the cell where a series of successive fusion events with endocytic vesicles (right vertical sequence) occurs. Phagosome maturation begins with fusion to early endosomes (1) that coincides with acquisition of PI(3)P and Rab5. These are needed for subsequent interactions with late endosomes. We propose that LAMP-1 and LAMP-2 can be acquired independently and before Rab7 (2). LAMP isoforms promote docking of phagosomes onto microtubules and centripetal movement (downward) by association with dynein/dynactin directly or via an unknown adaptor. LAMPs then mediate fusion with traditional late endosomes carrying Rab7 (3). Maturation culminates with phagosome-lysosome fusion (4), forming a microbicidal organelle.
The best-characterized effector of Rab7 is RILP. RILP plays a critical role in phagosome maturation by recruiting dynein–dynactin, a motor complex that displaces phagosomes along microtubules toward the juxtanuclear region (Harrison et al, 2003), where fusion with lysosomes takes place. As LAMPs regulate accretion of Rab7 on phagosomes, recruitment of RILP is impaired and the inward mobility of phagosomes is subsequently inhibited (Figure 7G and I). In addition, however, lysosomal distribution and motility were found to be abnormal despite the fact that lysosomes recruit Rab7 and associate with RILP, implying that the GTPase is in the active configuration. We conclude that, in addition to the RILP–dynein complex, lysosomes require additional factors to attain normal distribution and motility and that LAMPs themselves, or molecules associated with them, are involved in the process. It is conceivable that a tripartite complex involving Rab7, RILP and LAMP is required for optimal association of late endosomes/lysosomes with dynein/dynactin. Alternatively, phagosomes and late endosomes/lysosomes may interact with dynein/dynactin by more than one mechanism: the conventional Rab7/RILP bridge, plus a second complex involving LAMPs and either RILP or an unknown adaptor (Figure 8). The joint presence of both systems would be required for optimal motility. These considerations are summarized in Figure 8.
It is unclear how LAMPs are physically linked to the microtubular motor complex. LAMPs expose only a small C-terminal tail of 11 amino acids to the cytoplasm. Although short, the cytoplasmic tail of LAMP-2 can interact with cytosolic proteins, such as RNAse A and GAPDH (Dice and Terlecky, 1990; Cuervo and Dice, 1996), and may directly interact with adaptors or GTPases. Alternatively, the interaction may involve the transmembrane domain of LAMP. Regardless of the detailed mode of association with dynein/dynactin, it is clear that organellar movement toward the microtubule-organizing center is essential for successful phago-lysosome formation. Elimination of lysosome transport by disruption of microtubules (Blocker et al, 1998) or by interference with RILP or dynein function (Harrison et al, 2003) impairs contact and fusion between lysosomes and phagosomes. Thus, our findings can be most simply explained by a model whereby LAMP-1 and LAMP-2 are required for normal organellar motility, which in turn is essential for phago-lysosome fusion.
Materials and methods
Cell culture, plasmids and transfection
Generation of lamp-1−/−, lamp-2−/− and lamp-1−/−/lamp-2−/− mice was described (Andrejewski et al, 1999; Tanaka et al, 2000; Eskelinen et al, 2004). Primary peritoneal macrophages were isolated using thioglycolate. Isolation and immortalization of MEFs were reported elsewhere (Eskelinen et al, 2004). RAW264.7 murine macrophages were obtained from the American Type Culture Collection and cultured in DMEM containing 5% FBS. Fibroblasts plated on glass coverslips were transfected with FUGENE-6 according to the manufacturer's instructions (Roche). cDNA encoding myc-FcγIIA receptor was a gift of Dr A Schreiber (University of Pennsylvania School of Medicine, Philadelphia, PA). The vector encoding EGFP-Rab5 was reported earlier (Scott et al, 2002). The p40phox-PX domain vector (Kanai et al, 2001) was provided by Dr M Yaffe (MIT, Cambridge, MA). CD63 cDNA was the kind gift of Dr GM Griffiths (William Dunn School of Pathology, Oxford, UK). EGFP-tagged WT and mutant Rab7 were published elsewhere (Bucci et al, 2000). LAMP-1 and LAMP-2 vectors were as in Eskelinen et al (2004).
Phagocytosis assay and pulse-chase protocols
Latex beads (Bangs Laboratories Inc.) were opsonized with 1 mg/ml human IgG (Sigma). Cells were washed and overlaid with serum-free DMEM and opsonized beads. Phagocytosis was synchronized by spinning at 300 g for 1 min. To induce internalization, cells were incubated at 37°C. External beads were labeled with fluorophore-conjugated donkey anti-human IgG (1:500) on ice in PBS for 1 min. Cells were placed again in serum-free DMEM at 37°C and further incubated to monitor maturation. To assess the transition from early endosomes to lysosomes, cells were pulsed overnight with 250 μg/ml tetramethylrhodamine-dextran (Molecular Probes), followed by a 1 h chase in dye-free medium to ensure delivery of dextran to lysosomes. Labeling with LysoTracker was according to Molecular Probes.
Gene silencing with siRNA
siRNA to murine LAMP-1 (L-048173-00-0005) and LAMP-2 (L-059036-00-0005) and scrambled siRNA (D-0011210-01) were from Dharmacon (Lafayette, CO). siRNA was transfected using the Amaxa electroporation system (Cologne, Germany), following the manufacturer's guidelines. Two million cells and 1.5 μg of siRNA were used per electroporation. Knockdown of LAMP-1 and LAMP-2 was confirmed by immunostaining with both LAMP-1 and LAMP-2 antibodies. Cells were first exposed to rat antibodies to mouse LAMP-1 (ID4B) at 1:20 followed by Cy2-conjugated secondary antibodies. Rat antibodies to LAMP-2 were preincubated with substoichiometric amounts of Cy5-conjugated donkey anti-rat F(ab) fragments (Jackson Laboratories) at 37°C for 1 h, then exposed to cells already stained for LAMP-1 for 1 h.
Immunostaining, epifluorescence and confocal microscopy
Cells were fixed with 4% paraformaldehyde in PBS for 30 min, washed with 100 mM glycine, permeabilized and blocked for 1 h in 0.1% Triton X-100 (v/v)/5% milk powder (w/v) in PBS. All steps were at room temperature. Permeabilized cells were incubated with primary antibodies for 1 h followed by three washes with PBS and incubation with secondary antibodies for 1 h and three more washes. Antibodies to mouse LAMP-1 (ID4B) and LAMP-2 (ABL-93) from the Developmental Hybridoma Bank were used at 1:20. Before immunostaining with LIMP-2 or cathepsin D, cells were permeabilized with 0.03% saponin in PBS for 1 h, then blocked with 3% BSA (w/v) in PBS for 1 h. Antibodies against LIMP-2 were provided by Dr Y Tanaka (Kyushu University, Fukuoka, Japan) and used at 1:50. Cathepsin D antibodies (Kasper et al, 1996) were used at 1:50. All fluorophore-conjugated donkey anti-rat secondary antibodies and fragments thereof were used at 1:1000 and purchased from Jackson Laboratories. Images were acquired using a Leica DM IRE2 microscope with a × 100 oil immersion objective and standard filter sets. Images were captured using a Hamamatsu Orca-ER camera and processed using Open Lab V3.4 (Improvision). Confocal microscopy was performed using an LSM 510 microscope (Zeiss).
Electron microscopy
Fibroblasts were fixed in 2% glutaraldehyde in 0.2 M Hepes, pH 7.4, for 2 h. After postfixation in 1% osmium tetroxide, the cells were stained with 2% uranyl acetate, dehydrated in ethanol and flat-embedded in Epon. Thin sections were stained with uranyl acetate and lead citrate and examined with a Jeol JEM 1200EX electron microscope.
Immunoblotting
Lysates were analyzed by SDS–PAGE, transferred to nitrocellulose, blocked for 1 h at room temperature in 5% (w/vol) milk or 5% (w/vol) BSA in PBS with 0.01% (vol/vol) Tween 20, then probed with primary antibody for 1 h. Antibodies for LAMP-1 and LAMP-2 were used at 1:100 and antibodies for GAPDH (Chemicon) at 1:10 000. After several washes in 0.01% Tween 20 in PBS, blots were probed with HRP-conjugated secondary antibodies (1:5000–1:10 000) for 1 h, washed and visualized by enhanced chemiluminescence (Amersham).
Quantification of lysosome and phagosome distribution and motility
Lysosomes were labeled with LysoTracker or rhodamine-dextran as described above. To identify cells, their nuclei were stained with 12.5 μg/ml of Höechst 33342 (Fluka) for 15 min at 37°C. The distribution of lysosomes as a function of distance from the nucleus was determined with a KineticScan HCS Reader (Cellomics). The fluorescence intensity of lysosomes was integrated within concentric rings positioned at 1.58 μm intervals from the edge of the nucleus using a specially designed algorithm. Lysosome motility was assayed after altering the cytoplasmic pH as in Heuser (1989). Time-lapse images were acquired by fluorescence microscopy. Lysosome mobility was computed analyzing time-stacks, using the fluorescence image correlation microscopy approach of Gelles et al (1988) and Petersen et al (1993). Images were segmented into domains moving with approximately the same velocity, but containing enough features to allow unambiguous matching of the domain with a displaced segment in the time sequence. The magnitude of displacement was calculated for each segment and weighted with the intensity of its signal. Histograms of displacements were constructed and average velocities were computed by summing weighted displacements.
Phagosome motility was measured using 0.8 μm latex beads. Cells were transfected with a fluorescent construct modeled after the tail of H-Ras (Michaelson et al, 2002) to label the membrane in order to identify internalized beads, which appeared as bright rings of fluorescence.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Figure 9
Supplementary Figure 10
Supplementary Figure 11
Acknowledgments
We thank Arja Strandell (University of Helsinki), Marlies Rusch (University of Kiel) and David K Evans for their assistance in the completion of this project. This work was supported by grants from the Canadian Cystic Fibrosis Foundation (CCFF), the Canadian Institutes for Health Research, the Deutsche Forschungsgemeinschaft SA683/6-1 and the Fonds der Chemischen Industrie. KKH is the recipient of a CCFF Graduate Studentship. SG is the current holder of the Pitblado Chair in Cell Biology.
References
- Andrejewski N, Punnonen EL, Guhde G, Tanaka Y, Lullmann-Rauch R, Hartmann D, von Figura K, Saftig P (1999) Normal lysosomal morphology and function in LAMP-1-deficient mice. J Biol Chem 274: 12692–12701 [DOI] [PubMed] [Google Scholar]
- Blocker A, Griffiths G, Olivo JC, Hyman AA, Severin FF (1998) A role for microtubule dynamics in phagosome movement. J Cell Sci 111 (Part 3): 303–312 [DOI] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B (2000) Rab7: a key to lysosome biogenesis. Mol Biol Cell 11: 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C (2001) Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J 20: 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M (1990) Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell 62: 317–329 [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273: 501–503 [DOI] [PubMed] [Google Scholar]
- Dice JF, Terlecky SR (1990) Targeting of cytosolic proteins to lysosomes for degradation. Crit Rev Ther Drug Carrier Syst 7: 211–233 [PubMed] [Google Scholar]
- Downey GP, Botelho RJ, Butler JR, Moltyaner Y, Chien P, Schreiber AD, Grinstein S (1999) Phagosomal maturation, acidification, and inhibition of bacterial growth in nonphagocytic cells transfected with FcgammaRIIA receptors. J Biol Chem 274: 28436–28444 [DOI] [PubMed] [Google Scholar]
- Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ (1998) Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem 273: 20121–20127 [DOI] [PubMed] [Google Scholar]
- Eskelinen EL, Illert AL, Tanaka Y, Schwarzmann G, Blanz J, Von Figura K, Saftig P (2002) Role of LAMP-2 in lysosome biogenesis and autophagy. Mol Biol Cell 13: 3355–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, Tanaka Y, Lullmann-Rauch R, Hartmann D, Heeren J, von Figura K, Knecht E, Saftig P (2004) Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell 15: 3132–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL, Tanaka Y, Saftig P (2003) At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol 13: 137–145 [DOI] [PubMed] [Google Scholar]
- Fukuda M (1991) Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. J Biol Chem 266: 21327–21330 [PubMed] [Google Scholar]
- Gelles J, Schnapp BJ, Sheetz MP (1988) Tracking kinesin-driven movements with nanometre-scale precision. Nature 331: 450–453 [DOI] [PubMed] [Google Scholar]
- Godbold GD, Ahn K, Yeyeodu S, Lee LF, Ting JP, Erickson AH (1998) Biosynthesis and intracellular targeting of the lysosomal aspartic proteinase cathepsin D. Adv Exp Med Biol 436: 153–162 [DOI] [PubMed] [Google Scholar]
- Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S (2003) Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol 23: 6494–6506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J (1989) Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol 108: 855–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Swanson JA (1990) Radial extension of macrophage tubular lysosomes supported by kinesin. Nature 346: 864–866 [DOI] [PubMed] [Google Scholar]
- Indik ZK, Park JG, Hunter S, Mantaring M, Schreiber AD (1995) Molecular dissection of Fc gamma receptor-mediated phagocytosis. Immunol Lett 44: 133–138 [DOI] [PubMed] [Google Scholar]
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL (2004) Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 117: 4837–4848 [DOI] [PubMed] [Google Scholar]
- Jordens I, Fernandez-Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, Janssen H, Wubbolts R, Neefjes J (2001) The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein–dynactin motors. Curr Biol 11: 1680–1685 [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB (2001) The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol 3: 675–678 [DOI] [PubMed] [Google Scholar]
- Kasper D, Dittmer F, von Figura K, Pohlmann R (1996) Neither type of mannose 6-phosphate receptor is sufficient for targeting of lysosomal enzymes along intracellular routes. J Cell Biol 134: 615–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Beuchat MH, Lindsay M, Frias S, Palmiter RD, Sakuraba H, Parton RG, Gruenberg J (1999) Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat Cell Biol 1: 113–118 [DOI] [PubMed] [Google Scholar]
- Kundra R, Kornfeld S (1999) Asparagine-linked oligosaccharides protect Lamp-1 and Lamp-2 from intracellular proteolysis. J Biol Chem 274: 31039–31046 [DOI] [PubMed] [Google Scholar]
- Kuronita T, Eskelinen EL, Fujita H, Saftig P, Himeno M, Tanaka Y (2002) A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J Cell Sci 115: 4117–4131 [DOI] [PubMed] [Google Scholar]
- Lebrand C, Corti M, Goodson H, Cosson P, Cavalli V, Mayran N, Faure J, Gruenberg J (2002) Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J 21: 1289–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Rotstein OD, Grinstein S (1990) Phagosomal acidification is mediated by a vacuolar-type H(+)-ATPase in murine macrophages. J Biol Chem 265: 21099–21107 [PubMed] [Google Scholar]
- Michaelson D, Ahearn I, Bergo M, Young S, Philips M (2002) Membrane trafficking of heterotrimeric G proteins via the endoplasmic reticulum and Golgi. Mol Biol Cell 13: 3294–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen NO, Hoddelius PL, Wiseman PW, Seger O, Magnusson KE (1993) Quantitation of membrane receptor distributions by image correlation spectroscopy: concept and application. Biophys J 65: 1135–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiot A, Faure J, Stenmark H, Gruenberg J (2003) PI3P signaling regulates receptor sorting but not transport in the endosomal pathway. J Cell Biol 162: 971–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scianimanico S, Desrosiers M, Dermine JF, Meresse S, Descoteaux A, Desjardins M (1999) Impaired recruitment of the small GTPase rab7 correlates with the inhibition of phagosome maturation by Leishmania donovani promastigotes. Cell Microbiol 1: 19–32 [DOI] [PubMed] [Google Scholar]
- Scott CC, Cuellar-Mata P, Matsuo T, Davidson HW, Grinstein S (2002) Role of 3-phosphoinositides in the maturation of Salmonella-containing vacuoles within host cells. J Biol Chem 277: 12770–12776 [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P (2000) Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406: 902–906 [DOI] [PubMed] [Google Scholar]
- Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S (2001) Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol 155: 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Bucci C, Harrison RE, Trimble WS, Lanzetti L, Gruenberg J, Schreiber AD, Stahl PD, Grinstein S (2003) Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol Cell Biol 23: 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH, Brutkiewicz RR, Blum JS (2005) Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity 22: 571–581 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Figure 9
Supplementary Figure 10
Supplementary Figure 11