

SUMMARY
The Runt domain is the DNA binding domain of the core binding factor (CBF) Runx subunits. The CBFs are transcription factors that play critical roles in hematopoiesis, bone, and neuron development in mammals. A common non-DNA binding CBFβ subunit heterodimerizes with the Runt domain of the Runx proteins and allosterically regulates its affinity for DNA. Previous NMR dynamics studies suggested a model whereby CBFβ allosterically regulates DNA binding by quenching conformational exchange in the Runt domain, particularly in the S-switch region and the βE’-F loop. We sought to test this model, and to this end introduced all possible single amino acid substitutions into the S-switch region and the βE’-F loop, and screened for mutations that enhanced DNA-binding. We demonstrate that one Runt domain mutant, R164N, binds both DNA and CBFβ with higher affinity, but it is less sensitive to allosteric regulation by CBFβ. Analysis of NMR relaxation data shows that the chemical exchange exhibited by the wild-type Runt domain is largely quenched by the R164N substitution. These data support a model in which the dynamic behavior of a network of residues connecting the CBFβ and DNA binding sites on the Runt domain plays a critical role in the mechanism of allosteric regulation. This study provides an important functional link between dynamic behavior and protein allosteric function, consistent with results on other allosterically regulated proteins.
Keywords: RUNX, AML1, core binding factors, Runt domain, CBFβ, allosteric, NMR, X-ray
INTRODUCTION
Allosteric regulation is a critical function of many enzymatic systems, and the role of protein dynamics in both enzyme catalysis and allosteric regulation is now well-documented 1; 2. Notably, changes in dynamic behavior on the microsecond- to-millisecond timescale are frequently associated with function in this regard2; 3; 4; 5; 6; 7; 8; 9; 10; 11. Changes on this timescale are seen for aspartate transcarbamoylase, one of the best-characterized allosteric enzymes 12; 13; 14; 15; 16. Structural studies have defined clear conformational alterations associated with allosteric regulatory changes. Inherent in all of these studies is the idea that proteins are more plastic in the regions that must undergo conformational changes as a result of allosteric regulation—in other words, that there are dynamically connected networks of residues whose dynamic behavior is linked to allosteric regulation.
The DNA-binding Runt domain of the core binding factors (CBFs) is allosterically regulated by a non-DNA binding subunit, CBFβ. CBFs are heterodimeric transcription factors that play critical roles in both mammalian development and human disease 17, and consist of DNA-binding subunits that in mammals are encoded by the Runx1, Runx2, and Runx3 genes, and a common non-DNA-binding CBFβ subunit encoded by Cbfb. The CBFβ subunit is essential for both Runx1 and Runx2 functions in vivo 18; 19; 20; 21; 22. The Runt domain of the Runx subunits is an immunoglobulin s-type fold that binds DNA via loops located at one end of its eight-stranded β-barrel 23; 24; 25; 26. CBFβ heterodimerizes with the Runt domain and increases its affinity for DNA approximately 7–10 fold, and does so without contacting the DNA directly 25; 26; 27; 28; 29; 30. In addition, CBFβ can overcome an auto-inhibited conformation of the full-length Runx1 and Runx2 proteins, stimulating their DNA-binding by at least 40 fold 31; 32. CBFβ also protects the full-length Runx1 protein from ubiquitin-mediated degradation 33.
A comparison of crystal structures of the Runt domain-DNA binary complex, the Runt domain-CBFβ-DNA ternary complex, and the free Runt domain (Figure 1) suggested that CBFβ may allosterically enhance DNA binding by the Runt domain through a specific hydrogen-bonding network that serves to stabilize the flexible regions of the Runt domain important for DNA binding 25; 34; 35. Likewise, comparison of the ternary complex to the Runt domain-CBFβ binary complex showed that DNA appears to order the conformation of the DNA-contacting flexible Runt domain “tail” (aa 170–177) and the βE’-F loop (aa 141–143) 25; 26; 36. NMR studies definitively demonstrated that several regions of the Runt domain are in conformational exchange, and that two regions in particular - the βE’-F loop and βG through the βG-G’ loop (aa 161–166, otherwise known as the S-switch region) (Figures 1 and 2A) undergo conformational exchange in the Runt domain-DNA complex that is quenched in the presence of CBFβ 37. These observations support a model in which CBFβ enhances DNA binding by the Runt domain by locking these dynamic regions into a conformation that most favorably interacts with DNA.
Figure 1. Comparison of structures of the apo Runt domain (1LJM, red), the Runt domain-DNA binary complex (1HJC, green), and the Runt domain-CBFβ-DNA ternary complex (1H9D, blue).
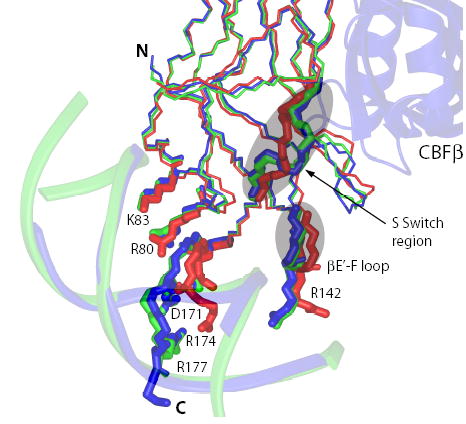
The S-switch region, βE’-F loop, and sidechains of the Runt domain residues involved in interactions with DNA are shown.
Figure 2. Genetic screen for mutations in the Runt domain that enhance DNA binding.
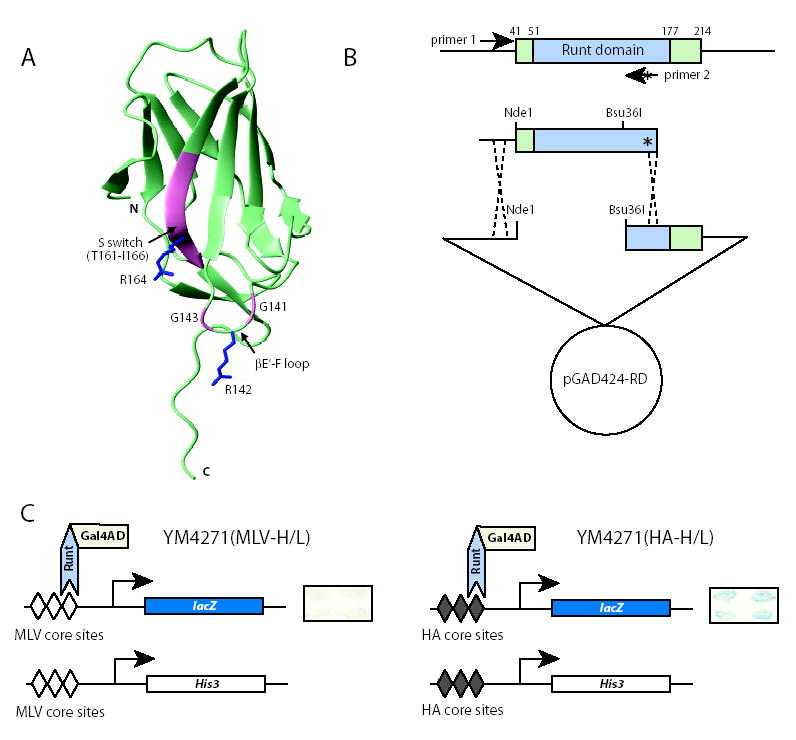
(A) Ribbon diagram of the Runt domain showing the amino acids we subjected to saturation mutagenesis (pink). The sidechains of R142, a DNA contacting residue, and R164 are shown (blue). The βE’-F loop and S-switch region are indicated.
(B) PCR based strategy for introducing all possible codons into the βE’-F loop and S-switch region. The “*” indicates single amino acid substitutions introduced by PCR primer 2. The PCR product was co-transformed into the yeast reporter strain along with the pGAD424-RD plasmid gapped with Ndel and Bsu36I.
(C) Two yeast strains used in yeast one-hybrid assay. Both strains have a His3 selectable marker and a lacZ reporter under the control of three repeats of Runt domain-binding core sites. Expression of a wild-type Runt domain-Gal4 activation domain fusion protein in these two reporter strains results in a robust β-galactosidase (blue) signal in the YM4271(HA-H/L) strain with three high affinity (HA) core sites, and a barely detectable signal in the YM4271(MLV-H/L) strain with three lower affinity core sites from the Moloney murine leukemia virus enhancer (MLV). Examples of β-galactosidase filter assays are shown on the right. We screened for mutant Runt domains that generated a visible β-galactosidase signal in the YM4271(MLV-H/L) strain. We then re-assayed putative positive mutants identified in the YM4271(MLV-H/L) strain in the YM4271(HA-H/L) strain to independently confirm that they conferred higher β-galactosidase, and thus increased DNA binding affinity.
In an effort to provide functional evidence for this model of allosteric regulation, we searched for Runt domain mutants with enhanced DNA binding activity in the absence of CBFβ. We reasoned that if CBFβ increases the affinity of Runt domain for DNA by “locking in” a particular Runt domain conformation, mutations that reduce the conformational exchange exhibited by the Runt domain should result in higher affinity DNA binding. A further prediction is that a more “rigid” Runt domain should be less sensitive to allosteric regulation by CBFβ. Here we show that by introducing single amino acid substitutions into the flexible regions of the Runt domain, the βE’-F loop and the S-switch region, we were able to identify at least one Runt domain mutant (R164N, in the S-switch region) with enhanced DNA-binding activity in the absence of CBFβ that is also less sensitive to allosteric regulation by CBFβ. An X-ray crystal structure of the R164N Runt domain identified no significant structural changes, although additional intramolecular hydrogen bonds are mediated by the N164 sidechain that may contribute to its decreased dynamic behavior. NMR relaxation studies show that the conformational exchange exhibited by the binary Runt domain-DNA complex is quenched in the R164N mutant, further supporting the allosteric regulation model.
RESULTS
Identification of Runt domain mutants with enhanced DNA-binding activity
We used a yeast one-hybrid assay to screen for Runt domain mutants with enhanced DNA-binding affinity. We generated a yeast reporter strain YM4271(MLV-H/L) that contains three low affinity CBF binding sites from the Moloney murine leukemia virus enhancer (MLV) driving the expression of both His3 and lacZ reporter genes (Figure 2C). We also prepared the reporter strain YM4271(HA-H/L) that contains three high affinity core sites (HA) driving His3 and lacZ expression. Expression of a wild-type Runt domain-Gal4 activation domain fusion protein in these two reporter strains resulted in growth in the absence of exogenous histidine and a robust β-galactosidase signal in the YM4271(HA-H/L) strain, and visibly slower growth and a barely detectable signal in the YM4271(MLV-H/L) strain (Figure 2C). The difference in Runt domain binding affinity for the MLV and HA core sites is approximately 10-fold 38, thus the yeast one hybrid assay is sensitive in this 10-fold range of DNA-binding affinity.
We introduced all possible amino acid substitutions into the dynamic regions of the Runt domain, which are the S-switch region (T161, Y162, H163, R164, A165, and I166) and the DNA-contacting βE’-F loop (G141 and G143) (Figure 2A,B) and used the reporter strain with the lower affinity MLV core sites to screen for Runt domain mutants that conferred both enhanced growth in the absence of histidine (His+) and elevated β-galactosidase activity. We did not mutate R142 in the βE’-F loop as it is a DNA contacting side chain.
We could identify no amino acid substitutions at G141, G143, A165, or I166 that conferred increased β-galactosidase activity (Table 1). However, amino acid substitutions in the S-switch region in residues that are less well conserved across species than A165 or I166 did yield yeast colonies with elevated β-galactosidase activities. The mutant we isolated most frequently (four clones in each of two independent screens) was R164N (Table 1).
Table 1.
Summary of mutant screen.
| Number of Independent Isolates |
||||
|---|---|---|---|---|
| Residue | Mutants | Screen 1 | Screen 2 | Total |
| G141 | 0 | 0 | 0 | |
| G143 | 0 | 0 | 0 | |
| T161 | V | 1 | 1 | 2 |
| I | 2 | 0 | 2 | |
| L | 1 | 0 | 1 | |
| F | 2 | 0 | 2 | |
| Y | 0 | 1 | 1 | |
| Y162 | N | 0 | 1 | 1 |
| L | 0 | 3 | 3 | |
| H163 | I | 1 | 0 | 1 |
| E | 2 | 0 | 2 | |
| S | 1 | 1 | 2 | |
| D | 0 | 3 | 3 | |
| R164 | N | 4 | 4 | 8 |
| D | 2 | 1 | 3 | |
| G | 4 | 0 | 4 | |
| H | 1 | 0 | 1 | |
| S | 1 | 0 | 1 | |
| E | 1 | 0 | 1 | |
| C | 0 | 1 | 1 | |
| A165 | 0 | 0 | 0 | |
| I166 | 0 | 0 | 0 | |
To discriminate true positives (enhanced growth and β-galactosidase activity resulting from an increase in DNA binding affinity) from false positives (enhanced growth and β-galactosidase activity due to increased plasmid copy number caused by selection for the His+ phenotype), we retransformed all mutants into the YM4271(MLV-H/L) strain, grew the colonies in the presence of exogenous histidine to eliminate the selection pressure, and performed β-galactosidase filter assays. Only R164N, R164D, and to a lesser extent R164E consistently conferred increased β-galactosidase activity compared to the wild-type Runt domain under these conditions (not shown). We then transformed R164N, R164D, and R164E into the YM4271(HA-H/L) strain that has higher affinity Runt domain binding sites and found that all three mutants yielded stronger β-galactosidase signals than the wild-type Runt domain in this independent reporter strain (Figure 3A). All three mutants also had higher β-galactosidase activities than the wild-type Runt domain in liquid assays (Figure 3B).
Figure 3. Identification and characterization of Runt domain mutants with increased DNA binding affinity.
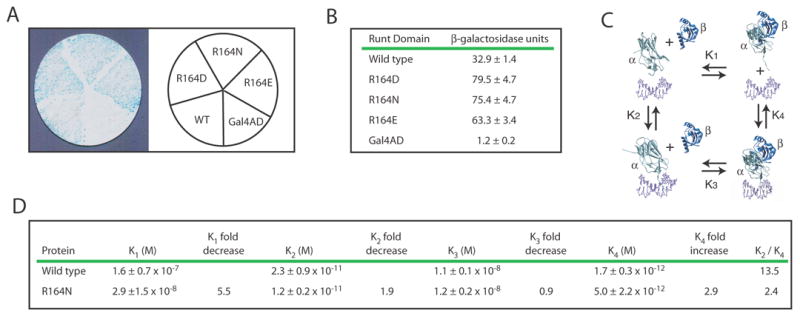
(A) β-galactosidase filter assay for three Runt domain mutants, R164N, R164D, and R164E, in the YM4271(HA-H/L) reporter strain.
(B) Results from the liquid β-galactosidase assay for R164N, R164D, and R164E. β-galactoside units are indicated ± standard deviation. The differences in activity between these mutants and the wild-type Runt domain were significant at P = 0.00001 for R164N, P = 0.00005 for R164D, and P = 0.00001 for R164E.
(C) Thermodynamic box illustrating the dissociation constants (K1, K2, K3, K4) for the interactions between the Runt domain (α), CBFβ (β), and the DNA (unlabeled, purple).
(D) Equilibrium dissociation constants for the wild-type Runt domain and the R164N mutant. The P values for the differences in K2 and K4 were = 0.11 and 0.06, respectively. K2/K4 represents the degree to which CBFβ enhances DNA binding by the Runt domain. Each K value was averaged from at least three binding curves.
Binding of the R164N Runt domain to DNA and CBFβ
Since R164N was the mutant we most frequently isolated, we targeted it for further biochemical characterization. We expressed and purified the R164N mutant protein and measured its equilibrium binding constants (K2, K3, K4) (Figure 3C) by electrophoretic mobility shift assay (EMSA), as described previously 29; 30. K2 is the dissociation constant of the Runt domain for DNA in the absence of CBFβ (Figure 3C). The K2 of R164N (1.2 ± 0.2 x 10−11 M) is 1.9-fold lower than that of the wild-type Runt domain (2.3 ± 0.9 x 10−11 M) (Figure 3D). Although this difference in K2 is not statistically significant, it is consistent with the approximate two fold difference in β-galactosidase activity measured in the yeast liquid assay (Figure 3B), which is significant (P = 0.00001). K3 measures the affinity of CBFβ for the Runt domain-DNA complex. The K3 of R164N (1.2 ± 0.2 x 10−8 M) is the same as that of the wild-type Runt domain (1.1 ± 0.1 x 10−8 M). K4 is the dissociation constant of the Runt domain-CBFβ heterodimer for DNA, and appears to be 3-fold higher for the R164N mutant than for the wild-type Runt domain (P = 0.06). K1, the dissociation constant of the Runt domain for CBFβ in the absence of DNA can be calculated from the formula K2K3=K1K4 and is about 5–6 fold lower than that of the wild-type Runt domain. The K2/K4 ratio shows that CBFβ can enhance the affinity of the wild-type Runt domain for DNA by about 13.5 fold, whereas CBFβ can only enhance the affinity of the R164N mutant for DNA by approximately 2.4-fold. In summary, the R164N mutation increases the affinity of the Runt domain alone for DNA, decreases the affinity of the Runt domain-CBFβ heterodimer for DNA, increases the affinity of the Runt domain for CBFβ in solution, and renders the Runt domain less sensitive to allosteric regulation by CBFβ.
The R164N Runt domain is in the closed S-switch conformation
R164 is located in the S-switch region, which in crystal structures of the free Runt domain was found in the so-called “closed” conformation 34; 35. In crystal structures of the Runt domain-DNA and Runt domain-CBFβ binary complexes, and in the ternary complex, the S-switch region is in the “open” conformation 25; 26; 36 (Figure 4B). The open conformation of the S-switch region is believed to confer enhanced DNA binding by causing a realignment of the DNA-binding βE’-F loop (Figures 1 and 4B), and this realignment is thought to be mediated, in part, by the formation of several additional hydrogen bonds between the βE’-F loop and the S-switch region 34; 35. The realignment of the βE’-F loop allows the interaction of two DNA contacting residues within that loop, R142 and G143, with two bases in the minor groove and with a phosphate group on the DNA backbone, respectively 25; 26; 34; 35. The other loop that undergoes a major conformational change in the binary and ternary complexes is the βC-D loop, which shifts towards the βE’-F loop (Figures 1 and 4B). CBFβ contacts Y113 and S114 in the βC-D loop, causing a change in its conformation that could be propagated to loop βE’-F.
Figure 4. X-ray crystal structure of the R164N Runt domain.
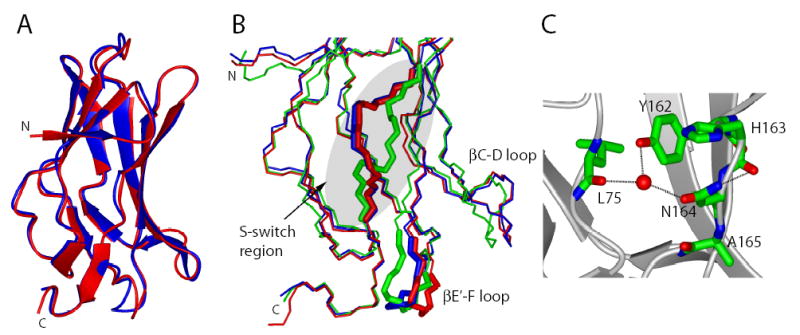
(A) Ribbon representation of the wild-type Runt domain (red, 1LJM) superimposed on the R164N Runt domain (blue).
(B) Close up of the S-switch and βE’-F loop region (designated as thick sticks) in the wild-type (red) and R164N Runt domain (blue). The Runt domain structure originating from the complex with DNA (1HJC), and representing the open conformation of the S-switch region is shown for comparison (green). The βC-D loop is also indicated.
C) The intramolecular interactions of the N164 side chain of R164N Runt domain. Atoms are colored according to atom type (C-green, N-blue, O-red); the water molecule is represented by a red sphere; hydrogen bonds are shown as dashed lines.
We crystallized the R164N mutant Runt domain in the absence of DNA to determine whether the S-switch region was in the closed or open conformation. The R164N Runt domain crystallized in space group C2 and the structure was refined at 2.60 Å resolution (Table 2). The asymmetric unit contained two molecules (monomers A and B) and residues 50–173 were clearly visible in the σA-weighted 2mFobs − DFcalc electron density map contoured at 1σ. No interpretable electron density was observed for five N-terminal amino acids and for C-terminal residues beyond residue 173. The overall structure of the R164N Runt domain (Figure 4A) is very similar to that of the double cysteine → serine mutant of the free Runt domain (C72S;C81S) (encoded as 1EAO in PDB, the rms for the backbone atoms is 0.23 Å) 35 as well as to the free wild-type Runt domain structure (encoded as 1LJM in PDB; rms 0.43Å) 34 (Figure 4A). Only very small conformational differences can be observed in some loop regions.
Table 2.
Crystallographic data for the R164N mutant of the Runt domain.
| Experimental Data | Refinement Details | ||
|---|---|---|---|
| Wavelength (λ) | 0.97940 | Resolution (Å) | 50.00-2.60 |
| Space group | C2 | Reflections (working) | 7186 |
| Unit cell parameters (Å) | Reflections (test) | 776 | |
| a | 91.95 | Rwork (%)c | 20.9 |
| b | 46.50 | Rfree (%)c | 26.8 |
| c | 63.55 | Number of waters | 48 |
| β | 90.66 | Number of ions | 4 Cl− |
| Resolution (Å) | 50.00-2.60 (2.64-2.60)a | Rms deviation from ideal geometry | |
| Unique reflections | 7962 | Bonds (Å) | 0.022 |
| Completeness (%) | 94.6 | Angles (°) | 2.238 |
| Rsym (%)b | 12.4 (33.9) | Average B factor (Å2) | 49.2 |
| Average I/σ (I) | 19.6 (2.3) | ||
The numbers in parentheses describe the relevant value for the highest resolution shell.
Rsym = ∑|Ii − <I>|/∑I, where Ii is the intensity of the i-th observation and <I> is the mean intensity of the reflections.
R = ∑||Fobs| − |Fcalc||/∑|Fobs|, crystallographic R factor, and Rfree = ∑||Fobs| − |Fcalc||/∑|Fobs|, where all reflections belong to the test set of randomly selected data.
We performed a detailed comparison of the S-switch region (aa 161–166) and the βE’-F loop (aa 141–143) in the R164N versus wild-type Runt domains. The S-switch region in the R164N Runt domain is in the “closed” conformation (Figure 4B). The S-switch is not involved in any crystal contacts, so the observed conformation likely reflects that found in solution. Thus the two-fold increase in DNA binding affinity apparently does not result from “locking” the S-switch region into what is thought to be its higher-affinity DNA-binding open conformation 34; 35. The N164 amide participates in intramolecular hydrogen bonding interactions with the backbone carbonyl of H163 (Figure 4C) (in monomer B of the R164N Runt domain dimer, N164 forms a hydrogen bond with the backbone of A165), and the N164 side chain is involved in a hydrogen bond with the backbone of L75 and the side chain hydroxyl of Y162 through a water molecule (Figure 4C). These interactions do not exist in the wild-type Runt domain structure. The additional intramolecular hydrogen bonds involving N164 would be predicted, if anything, to further stabilize the closed conformation of the S-switch region in the R164N mutant Runt domain.
The conformation of the βE’-F loop (residues 141–143, Figure 4B), which is involved in interactions with DNA, is affected by the crystal packing in our R164N Runt domain structure. This makes any interpretation of the observed conformation in this region tenuous. In addition, the βE’-F loop makes minimal contacts with other portions of the protein and has been shown to adopt different conformations in the various structures of the isolated Runt domain, suggesting it is quite flexible in the absence of DNA. Indeed, this is one of the regions of the protein that is not observable in NMR spectra of the free Runt domain due to exchange-broadening. The structure of the βC-D loop most closely resembles the closed conformation seen in the free Runt domain structures 34; 35 (Figure 4B).
The R164N mutation quenches exchange in the dynamic regions of the Runt domain
We determined whether the change in the R164N Runt domain’s allosteric properties correlated with changes in its dynamic behavior. Previous studies of the wild-type Runt domain bound to DNA clearly showed the presence of exchange for a number of critical regions, including the S-switch region and the βE’-F loop 37. In order to characterize the dynamic behavior of the R164N Runt domain bound to DNA, we measured 15N T1 and T2 relaxation rates and fit these to derive R1 (1/T1) and R2 (1/T2) rate constants. Bracken and co-workers demonstrated that the product R1* R2 can be used as a very sensitive and reliable indicator of chemical exchange 39, and we used this successfully in our previous studies to provide an independent identification of residues undergoing exchange 37. We collected relaxation data on two different samples of the R164N mutant Runt domain bound to DNA and calculated the R1* R2 data. In previous studies, resonance overlap and severe exchange broadening precluded analysis of a significant portion of the S-switch region (βG-G’ loop, aa 161–166) 37. Because the R164N mutation is in the βG-G’ loop, we felt it was important to obtain data for at least one residue in that loop and prepared samples of specifically 15N-Tyr labeled wild-type and R164N Runt domain complexed to DNA for this purpose. There are only two tyrosines in the Runt domain and one, Y162, is a good reporter for the dynamics in this region. Y162 was not fully analyzed in our previous study of the wild-type Runt domain because of resonance overlap 37. R1* R2 values for Y162 in the wild-type and R164N Runt domains (complexed to DNA) are shown in Figure 5A. Residues with no exchange will yield an R1* R2 value of ~25 at the magnetic field strength employed for the relaxation measurements (500 MHz) and the overall correlation time of the complex. Whereas the wild-type Runt domain had an R1* R2 value of 34.1, the R1* R2 value for R164N was 24.0, indicating there is no exchange in the mutant.
Figure 5. NMR relaxation results for the R164N Runt domain.
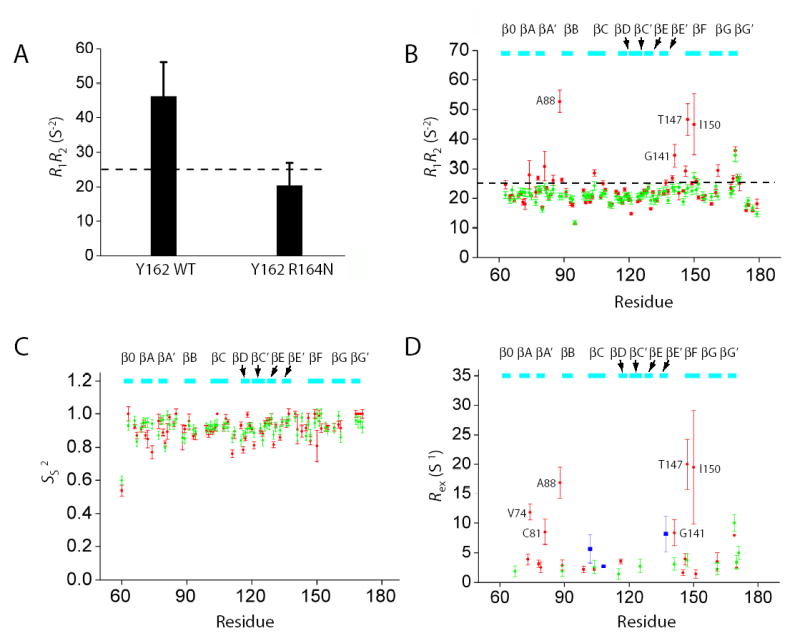
(A) 15N R1* R2 values for Y162 in the wild-type Runt domain-DNA and R164N Runt domain-DNA complexes measured in 15N-Tyr specifically labeled samples at 500 MHz. Error bars represent the fitted error for the R1 and R2 measurements propagated to the R1* R2 values. The dashed line indicates the theoretical value of R1*R2 for τc=15 ns, 11 T, and S2=1.0. The difference between the wild-type and R164N Runt domain was significant at P < 0.0001 by Student’s t-test.
(B–D) Relaxation data (15N R1* R2, Ss2, and Rex) for the wild-type and R164N Runt domain (complexed to DNA) measured from [U-2H,15N]-labeled samples at 500 MHz. The dashed line in Panel B indicates the theoretical value of R1*R2 for τc=15 ns, 11 T, and S2=1.0. Data for the wild-type protein are from Yan et al. 37. For Panels B–D, wild-type protein data are in red and R164N data are in green. Data are only shown for residues that could be analyzed in both proteins. Five residues could not be adequately fit using ModelFree and are not shown in panels C and D. Residues showing Rex in the CBFβ-Runt domain-DNA complex are shown in blue in panel D.
In order to assess whether this change in the S-switch dynamics is transmitted to other portions of the protein, we collected T1, T2, and heteronuclear NOE data on a [U-2H,15N]-labeled (>98% 2H) sample of the R164N Runt domain (complexed to DNA) and calculated the R1* R2 data. As seen in Figure 5B, there is a clear reduction in the number of residues displaying exchange, including residues in the DNA-contacting βA’-B (A88) and βE’-F (G41) loops, and in the βF strand (T147, I150). We have also fit the relaxation data using ModelFree to derive model-free parameters to describe the dynamic behavior of the backbone. We observe little change in the nanosecond-picosecond dynamics (as indicated by Ss2) between the R164N Runt domain-DNA and wild-type Runt domain-DNA complexes (Figure 5C). In contrast, there is a substantial reduction in the number of residues displaying significant Rex, a measure of the contribution of chemical exchange to the transverse relaxation rate, as well as in the magnitude of the remaining Rex terms (Figure 5D). These data are consistent with large-scale quenching of the dynamic behavior that is observed in the wild-type Runt domain. It should be noted that the observed changes in Rex could arise from changes in the exchange rate constants, relative populations, or chemical shifts. The close agreement between HSQC spectra of the wildtype and R164N Runt domain-DNA complexes (not shown) argues against chemical shift changes, as does the virtually identical structures of the two proteins (Figure 4A), strongly suggesting changes in rate constant or populations as the basis for the observed differences.
DISCUSSION
We recently showed that CBFβ binding reduces the backbone dynamics exhibited by several regions of the Runt domain, including the S-switch region 37. The S-switch region exists in two different conformations in the crystal structures of the Runt domain: in the closed conformation in the free, unliganded Runt domain, and in the open conformation in the Runt domain-DNA, Runt domain-CBFβ, and Runt domain-CBFβ-DNA complexes 25; 26; 34; 35; 36. Our previous NMR studies showed that the S-switch region in the Runt domain-DNA complex displays exchange behavior, presumably as a result of conformational exchange between the closed and open state, and we hypothesized that CBFβ enhances DNA binding by the Runt domain by stabilizing the open conformation 37. To test the hypothesis that quenching conformational exchange is integral to the process of allosteric regulation, we decided to search for Runt domain mutants with enhanced DNA binding activity in the absence of CBFβ. In theory, the most informative mutant would be one that binds DNA equally well with or without CBFβ (i.e., K2 = K4). Although we were unable to identify a mutant with this property, we did identify one mutant, R164N approximating this behavior that displayed reduced sensitivity to allosteric regulation by CBFβ.
Using NMR relaxation measurements, we demonstrated that the R164N mutation largely quenches the exchange observed in the S-switch region, the βA’-B and βE’-F loops, and in the βF strand of the protein. The increased affinity for DNA associated with this change in dynamics is consistent with our model that these regions of the protein are undergoing conformational exchange, and that quenching this exchange increases DNA binding affinity37. We previously showed by calorimetric measurements that CBFβ’s enhancement of DNA binding by the Runt domain is primarily enthalpically driven37. We posited that stabilizing the regions of the Runt domain that contact DNA results in more energetically favorable interactions between the two molecules. We see quenching of several of the same regions of the Runt domain in the R164N Runt domain-DNA complex, notably in the S-switch region as well as the βA’-B and βE’-F loops. However, as seen in the Rex data in Figure 5D, the degree of quenching observed for the R164N mutant is clearly not as extensive as that seen upon the addition of CBFβ. This suggests that the R164N mutation moves the conformational ensemble to something that more closely resembles that found in the CBFβ-Runt domain-DNA complex, but it does not move as far along this path. These data could also be explained by a model in which the open and closed conformations each have ensembles of substates and the effect of R164N is to reduce this ensemble for the closed state, resulting in the observed change in dynamic behavior and a more stable energetically favorable interaction with the DNA.
Since the structure of the isolated R164N Runt domain is virtually identical to that of the wild-type Runt domain, we expect that the R164N Runt domain binds DNA the same as the wild-type protein. In support of this, the HSQC spectra of the Runt domain-DNA and R164N Runt domain-DNA complexes overlay extremely well (not shown). On this basis, we hypothesize that, similar to CBFβ, the R164N mutation has stabilized regions of the protein contacting the DNA, resulting in an enhancement of the enthalpic contribution to binding. However, contrary to our expectations, the S-switch region of the R164N mutant was stabilized in the closed conformation in our X-ray crystal structure rather than in the open conformation seen in structures of the Runt domain bound to DNA or CBFβ. As this region of the protein shows significant flexibility, it is possible the crystal structure does not reflect the solution behavior of the protein. However, the observation that this region of the protein was not involved in crystal contacts argues against this. Alternatively, we can speculate that while the decrease in dynamics has led to enhanced interactions of the protein with DNA, this has occurred in a conformation that is not completely optimal for DNA binding. Thus, we have not recovered all of the DNA binding affinity normally conferred by CBFβ with this single mutation. This is consistent with the observation that the changes in dynamics observed for R164N are not as extensive as that seen for CBFβ addition.
It is worth noting that K3, the dissociation constant for CBFβ to the Runt domain-DNA complex, is virtually identical for the wild type and R164N Runt domain, whereas K1, K2, and K4 differ substantially. As mentioned above, it has been shown that both DNA and CBFβ induce similar changes in the conformation of the Runt domain. This implies that either a wild type or R164N Runt domain that has been pre-organized structurally on the DNA is in an optimal conformation for CBFβ binding. If the R164N mutation, perhaps through the additional sidechain interactions seen in the crystal structure, shifts the conformational ensemble towards the DNA-bound ensemble, it should increase the affinity of the Runt domain for both CBFβ and DNA, which is what we observed. In a two (or more)-state model of this, the reduced chemical exchange observed is consistent with a shift in the conformational ensemble from one with significant populations of both free and DNA-bound conformational species to one that is more highly populated by DNA-bound conformations. The K4 measurements show weaker binding for the CBFβ-R164N Runt domain complex. We can only speculate on the cause of this, however it is important to note that while the R164N mutation enhances binding to DNA, it does not do so to the same extent that CBFβ enhances DNA binding by wild-type Runt domain. This suggests that the R164N Runt domain could be shifted to a conformational substate that is more like the DNA-bound conformation, but from which the optimal conformation for DNA binding can only be achieved with greater energetic cost, perhaps as a result of the sidechain interactions of N164 observed in the crystal structure. This could result from the sidechain interactions of N164 imparting a higher energetic cost for converting the S-switch to the open conformation necessary for high affinity DNA binding.
Although residue 164 in the Runt domain is an R or a K in most species, in the nematodes C. elegans and Meloidogyne hapla it is an N and D, respectively, and in the spider Cupenniussalei salei it is an E 40. We isolated all three of these residues (N, D, E) in our screen, showing that they all have higher binding affinity. Modeling D and E substitutions at position 164 to check their potential interactions in the S-switch region suggested that these residues could also make stabilizing interactions with other portions of the protein. Therefore, both D164 and E164 are potentially able to stabilize the closed conformation of the Runt domain, which could result in a reduction of the conformational exchange of the S-switch region for these mutants as well.
Recent NMR studies of allosteric systems have clearly demonstrated the critical role of protein dynamics in allosteric regulation 2; 3; 4; 5; 6; 7; 8; 9; 10; 11; 37. From these studies, there is an emerging view that allosteric communication between sites in a protein is not caused by the propagation of binary conformational switches through a protein, but rather through perturbation of a conformational ensemble 41. Indeed, a modeling study of dihydrofolate reductase using such an ensemble view successfully predicted changes in portions of the protein associated with allosteric communication between sites 42. This behavior results not only from enthalpic contributions, which are easily identified through inspection of the high-resolution structure (i.e., changes in the numbers and types of interactions), but also from significant entropic (possibly dynamic) contributions, which are difficult, if not impossible, to deduce from structure alone but can be characterized using NMR relaxation methods. Our results with the R164N Runt domain provide experimental validation for this ensemble view of allosteric regulation. The observed changes in dynamic behavior we see for the R164N Runt domain corresponds to a significant reduction in the conformational ensemble of the Runt domain that results in a loss of its ability to be allosterically regulated.
MATERIALS AND METHODS
PCR-based mutagenesis of the Runt domain and genetic screen for DNA binding activity
We introduced amino acid substitutions into regions of the Runt domain (aa 41–214) that undergo conformational exchange in the Runt domain-DNA complex 37. The amino acids we targeted for mutagenesis were G141 and G143 in the βE’-F loop, and T161, Y162, H163, R164, A165 and I166 in the S switch region (Figure 2A). PCR primers covering each of these codons that contained a mixture of nucleotides at each position of the codon (primer sequences available upon request) were used in conjunction with a primer hybridizing to sequences in the pGAD424-RD plasmid upstream of the Runt domain coding region to generate the 64 possible codons for all 20 amino acids. The PCR product was co-transformed with a pGAD424-RD plasmid that was gapped by NdeI+Bsu36I digestion (Figure 2B) into a yeast dual-reporter strain YM4271(MLV-H/L). YM4271(MLV-H/L) was constructed from YM4271 (Clontech), by introducing two independent reporter genes, lacZ and His3, under the control of three repeats of the CBF binding sites from the Moloney murine leukemia virus enhancer (MLV) [5′-(TCTGTGGTAAG)3-3′] (Figure 2C) 43. The transformants were directly plated on SC His− Leu− plates with 30 or 45mM 3-amino-1, 2, 4-triazole and incubated at 30°C. After 6 days, the largest colonies on the plates (His+) were picked and inoculated onto SC Leu− plates. After incubating at 30°C for 2 more days, filter assays for β-galactosidase activity were carried out following the manufacturer’s protocol (Clontech, Mountain View CA). Colonies with strong β-galactosidase activities (His+ lacZ+) were then single-colony purified by streaking onto new SC Leu− plates, and the plasmids isolated and sequenced. Plasmids with Runt domain mutations were then transformed into either YM4271(MLV-H/L) or another reporter yeast strain, YM4271(HA-H/L), to confirm that β-galactosidase activities were indeed elevated. The YM4271(HA-H/L) strain contains three high affinity core sites (HA) (5′-(TTTGCGGTTAG)3-3′) driving His3 and lacZ (Figure 2C).
Purification of Runt domain mutants
The NdeI-BamHI mutant Runt domain fragment (residues 41–214) from the pGAD424 vector was subcloned into the bacterial pET-3c (Novagen, Darmstadt Germany) expression vector. 15N Tyr- and [U-2H,15N]-labeled (>98% 2H) mutant proteins were expressed and purified and the Runt domain-DNA complex prepared as described previously 24; 30.
EMSA Dissociation Constant Measurements
Equilibrium dissociation constants were measured by electrophoretic mobility shift assay (EMSA) as previously described 29; 30, using an 18bp DNA sequence containing the core site from the SL3-3 murine leukemia virus enhancer (5′- GGATATCTGTGGTTAAGCA –3′).
X-ray crystallography
We introduced the R164N mutation into the Runt domain 46–185 (C72S;C81S) construct used previously for crystallization of the isolated Runt domain 35. The purified protein was dialyzed against 25mM HEPES buffer, 150 mM NaCl, pH = 7.5. An initial search for crystals of the R164N Runt domain was carried out with the Hampton I and Hampton II Crystal screens (Hampton Research, Aliso Viejo CA). The best crystals were obtained with 12 mg/ml protein with Hampton I, condition 15: 22% PEG 8000, 0.1M Sodium Cacodylate pH=6.5, 0.2M Ammonium Sulfate. Drops were formed with 1 μl of protein solution and 1 μl of reservoir solution and were overlaid with 15 μl of a 1:1 mixture of silicon and mineral oil. The best crystals were obtained by microseeding using the sitting drop vapor-diffusion technique at room temperature.
Crystals used for data collection were briefly soaked in the crystallization buffer containing 12.5% ethylene glycol and frozen by immersion in liquid nitrogen. Data for the R164N Runt domain crystals were collected at beamline 19ID at APS, and processed and scaled using HKL2000 44. Crystallographic details, including unit cells and data statistics, are shown in Table 2. The structure was solved by molecular replacement method using AMORE 45, with the Runt domain structure (PDB code 1EAO) as a search model. The atomic models were refined with REFMAC5 46 from the CCP4 suite 47 to 2.6 Å resolution. Manual model building was performed in O 48. Details of the refinement are provided in Table 2.
NMR Spectroscopy
All measurements were performed on a Varian Inova 500 MHz NMR spectrometer equipped with an actively shielded triple resonance probe. Samples of 0.5 mM R164N Runt domain-DNA complex were prepared and spectra recorded at 40°C as described previously 24; 30; 37. 15N T1, 15N T2, and {1H}15N NOE measurements were carried out using the sequences of Farrow et al. 49 on samples of 15N-Tyr-labeled wild-type and R164N Runt domain as well as on a [U-2H,15N]-labeled (>98% 2H) sample of R164N Runt domain.
Analysis of NMR relaxation data
T1 and T2 values were calculated by least-square fitting peak heights to a single exponential decay 49. Errors in T1 and T2 were estimated from 500 Monte Carlo simulations with error ranges set to 5% of measured peak intensities 50; 51. NOE values were calculated as the ratio of saturated peak heights over unsaturated peak heights with errors set to 5% of peak heights 49; 50. The average errors of T1, T2 and NOE, respectively, are 3.7%, 4.5%, and 7.1%. The rotational diffusion tensor was determined following a previously described procedure 37. The crystal structure of the R164N Runt domain (PDB code to be assigned) was used for the modeling of axially symmetrical and fully anisotropic global tumblings. An axially symmetrical model was adopted based on an F-test on the χ2 values. The best-fit τm is 15 ns and D||/D⊥ 1.34. The internal dynamics and Rex were obtained by fitting the relaxation data and the diffusion tensor to an extended Lipari-Szabo model 52; 53; 54 using ModelFree 4.0 following a described model selection procedure 55.
Acknowledgments
This work was supported by RO1 CA75611 to NAS and R01 CA108056 to JHB.
References
- 1.Hammes GG. Multiple conformational changes in enzyme catalysis. Biochemistry. 2002;41:8221–8. doi: 10.1021/bi0260839. [DOI] [PubMed] [Google Scholar]
- 2.Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13:748–57. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–8. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C, Karpowich N, Hunt JF, Rance M, Palmer AG. Dynamics of ATP-binding cassette contribute to allosteric control, nucleotide binding and energy transduction in ABC transporters. J Mol Biol. 2004;342:525–37. doi: 10.1016/j.jmb.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–33. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 6.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1520–3. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 7.Hawkins RJ, McLeish TC. Coupling of global and local vibrational modes in dynamic allostery of proteins. Biophys J. 2006;91:2055–62. doi: 10.1529/biophysj.106.082180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarkson MW, Gilmore SA, Edgell MH, Lee AL. Dynamic coupling and allosteric behavior in a nonallosteric protein. Biochemistry. 2006;45:7693–9. doi: 10.1021/bi060652l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu J, Cistola DP, Li E. Analysis of ligand binding and protein dynamics of human retinoid X receptor alpha ligand-binding domain by nuclear magnetic resonance. Biochemistry. 2006;45:1629–39. doi: 10.1021/bi051474j. [DOI] [PubMed] [Google Scholar]
- 10.Igumenova TI, Lee AL, Wand AJ. Backbone and side chain dynamics of mutant calmodulin-peptide complexes. Biochemistry. 2005;44:12627–39. doi: 10.1021/bi050832f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McElroy C, Manfredo A, Wendt A, Gollnick P, Foster M. TROSY-NMR studies of the 91kDa TRAP protein reveal allosteric control of a gene regulatory protein by ligand-altered flexibility. J Mol Biol. 2002;323:463–73. doi: 10.1016/s0022-2836(02)00940-3. [DOI] [PubMed] [Google Scholar]
- 12.Howlett GJ, Blackburn MN, Compton JG, Schachman HK. Allosteric regulation of aspartate transcarbamoylase. Analysis of the structural and functional behavior in terms of a two-state model. Biochemistry. 1977;16:5091–100. doi: 10.1021/bi00642a023. [DOI] [PubMed] [Google Scholar]
- 13.Hammes GG, Wu CW. Kinetics of allosteric enzymes. Annu Rev Biophys Bioeng. 1974;3:1–33. doi: 10.1146/annurev.bb.03.060174.000245. [DOI] [PubMed] [Google Scholar]
- 14.Sakash JB, Kantrowitz ER. The contribution of individual interchain interactions to the stabilization of the T and R states of Escherichia coli aspartate transcarbamoylase. J Biol Chem. 2000;275:28701–7. doi: 10.1074/jbc.M005079200. [DOI] [PubMed] [Google Scholar]
- 15.Chan RS, Sakash JB, Macol CP, West JM, Tsuruta H, Kantrowitz ER. The role of intersubunit interactions for the stabilization of the T state of Escherichia coli aspartate transcarbamoylase. J Biol Chem. 2002;277:49755–60. doi: 10.1074/jbc.M208919200. [DOI] [PubMed] [Google Scholar]
- 16.Lipscomb WN. Aspartate transcarbamylase from Echerichia coli: activity and regulation. Adv Enzymol Relat Areas Mol Biol. 1994;68:67–151. doi: 10.1002/9780470123140.ch3. [DOI] [PubMed] [Google Scholar]
- 17.Ito Y. Oncogenic potential of the RUNX gene family: ‘overview’. Oncogene. 2004;23:4198–208. doi: 10.1038/sj.onc.1207755. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki K, Yagi H, Bronson RT, Tominaga K, Matsunashi T, Deguchi K, Tani Y, Kishimoto T, Komori T. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc Natl Acad Sci USA. 1996;93:12359–12363. doi: 10.1073/pnas.93.22.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Stacy T, Miller JD, Lewis AF, Huang X, Bories JC, Bushweller JH, Alt FW, Binder M, Marín-Padilla M, Sharpe A, Speck NA. The CBFβ subunit is essential for CBFα2 (AML1) function in vivo. Cell. 1996;87:697–708. doi: 10.1016/s0092-8674(00)81389-6. [DOI] [PubMed] [Google Scholar]
- 20.Miller J, Horner A, Stacy T, Lowrey C, Lian JB, Stein G, Nuckolls GH, Speck NA. The core-binding factor ß subunit is required for bone formation and hematopoietic maturation. Nat Genet. 2002;32:645–649. doi: 10.1038/ng1049. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida CA, Furuichi T, Fujita T, Fukuyama R, Kanatani N, Kobayashi S, Satake M, Takada K, Komori T. Core-binding factor ß interacts with Runx2 and is required for skeletal development. Nat Genet. 2002;32:633–638. doi: 10.1038/ng1015. [DOI] [PubMed] [Google Scholar]
- 22.Kundu M, Javed A, Jeon JP, Horner A, Shum L, Eckhaus M, Muenke M, Lian JB, Yang Y, Nuckolls GH, Stein GS, Liu PP. The hematopoietic transcription factor, Cbfß, interacts with Cbfa1 and plays a critical role in bone developent. Nat Genet. 2002;32:639–644. doi: 10.1038/ng1050. [DOI] [PubMed] [Google Scholar]
- 23.Nagata T, Gupta V, Sorce D, Kim WY, Sali A, Chait BT, Shigesada K, Ito Y, Werner MH. Immunoglobulin motif DNA-binding and heterodimerization for the PEBP2/CBF Runt-domain. Nat Struct Biol. 1999;6:615–619. doi: 10.1038/10658. [DOI] [PubMed] [Google Scholar]
- 24.Berardi M, Sun C, Zehr M, Abildgaard F, Peng J, Speck NA, Bushweller JH. The Ig fold of the core binding factor α Runt domain is a member of a family of structurally and functionally related Ig fold DNA binding domains. Structure Fold Des. 1999;7:1247–1256. doi: 10.1016/s0969-2126(00)80058-1. [DOI] [PubMed] [Google Scholar]
- 25.Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, Shiina M, Sato K, Kumasaka T, Yamamoto M, Ishii S, Ogata K. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFβ. Cell. 2001;104:755–767. doi: 10.1016/s0092-8674(01)00271-9. [DOI] [PubMed] [Google Scholar]
- 26.Bravo J, Li Z, Speck NA, Warren AJ. The leukaemia-associated AML1 (Runx1)-CBFβ complex functions as a DNA-induced molecular clamp. Nat Struct Biol. 2001;8:371–377. doi: 10.1038/86264. [DOI] [PubMed] [Google Scholar]
- 27.Kamachi Y, Ogawa E, Asano M, Ishida S, Murakami Y, Satake M, Ito Y, Shigesada K. Purification of a mouse nuclear factor that binds to both the A and B cores of the polyomavirus enhancer. J Virol. 1990;64:4808–4819. doi: 10.1128/jvi.64.10.4808-4819.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang S, Wang Q, Crute BE, Melnikova IN, Keller SR, Speck NA. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Mol Cell Biol. 1993;13:3324–3339. doi: 10.1128/mcb.13.6.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang YY, Shi J, Zhang L, Davis A, Bravo J, Warren AJ, Speck NA, Bushweller JH. Energetic and functional contribution of residues in the core binding factor β (CBFβ) subunit to heterodimerization with CBFα. J Biol Chem. 2000;275:39579–39588. doi: 10.1074/jbc.M007350200. [DOI] [PubMed] [Google Scholar]
- 30.Tang YY, Crute BE, Kelley JJ, III, Huang X, Yan J, Shi J, Hartman KL, Laue TM, Speck NA, Bushweller JH. Biophysical characterization of interactions between the core-binding factor α and β subunits and DNA. FEBS L. 2000;470:167–172. doi: 10.1016/s0014-5793(00)01312-0. [DOI] [PubMed] [Google Scholar]
- 31.Gu TL, Goetz TL, Graves BJ, Speck NA. Autoinhibition and partner proteins, CBFβ and Ets-1, modulate DNA binding by CBFα2(AML1) Mol Cell Biol. 2000;20 :91–103. doi: 10.1128/mcb.20.1.91-103.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanno T, Kanno Y, Chen LF, Ogawa E, Kim WY, Ito Y. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor α subunit revealed in the presence of the β subunit. Mol Cell Biol. 1998;18:2444–2454. doi: 10.1128/mcb.18.5.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y. Dimerization with PEBP2β protects RUNX1/AML1 from ubiquitin-proteosome-mediated degradation. EMBO J. 2001;15:723–733. doi: 10.1093/emboj/20.4.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartfeld D, Shimon L, Couture GC, Rabinovich D, Frolow F, Levanon D, Groner Y, Shakked Z. DNA recognition by the RUNX1 transcription factor is mediated by an allosteric transition in the RUNT domain and by DNA bending. Structure. 2002;10:1395–1407. doi: 10.1016/s0969-2126(02)00853-5. [DOI] [PubMed] [Google Scholar]
- 35.Bäckström S, Wolf-Watz M, Grundström C, Härd T, Grundström T, Sauer UH. The RUNX1 Runt domain at 1.25 Å resolution: a structural switch and specifically bound chloride ions modulate DNA binding. J Mol Biol. 2002;322:259–272. doi: 10.1016/s0022-2836(02)00702-7. [DOI] [PubMed] [Google Scholar]
- 36.Warren AJ, Bravo J, Williams RL, Rabbitts TH. Structural basis for the heterodimeric interaction between the acute leukaemia-associated transcription factors AML1 and CBFβ. EMBO J. 2000;19:3004–3115. doi: 10.1093/emboj/19.12.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan J, Liu Y, Lukasik SM, Speck NA, Bushweller JH. CBFbeta allosterically regulates the Runx1 Runt domain via a dynamic conformational equilibrium. Nat Struct Mol Biol. 2004;11:901–6. doi: 10.1038/nsmb819. [DOI] [PubMed] [Google Scholar]
- 38.Lewis AF, Stacy T, Green W, Taddesse-Heath L, Hartley JW, Speck NA. Core-binding factor influences the disease specificity of Moloney murine leukemia virus. J Virol. 1999;73:5535–5547. doi: 10.1128/jvi.73.7.5535-5547.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kneller JM, Lu M, Bracken C. An effective method for the discrimination of motional anisotropy and chemical exchange. J Am Chem Soc. 2002;124:1852–1853. doi: 10.1021/ja017461k. [DOI] [PubMed] [Google Scholar]
- 40.Rennert J, Coffman JA, Mushegian AR, Robertson AJ. The evolution of Runx genes I. A comparative study of sequences from phylogenetically diverse model organisms. BMC Evol Biol. 2003;3:4. doi: 10.1186/1471-2148-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Freire E. Can allosteric regulation be predicted from structure? Proc Natl Acad Sci U S A. 2000;97:11680–2. doi: 10.1073/pnas.97.22.11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pan H, Lee JC, Hilser VJ. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc Natl Acad Sci U S A. 2000;97:12020–5. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z, Yan J, Matheny CJ, Corpora T, Bravo J, Warren AJ, Bushweller JH, Speck NA. Energetic contribution of residues in the Runx1 Runt domain to DNA binding. J Biol Chem. 2003;278:33088–33096. doi: 10.1074/jbc.M303973200. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.Navaza J. Amore - an automated package for molecular replacement. Acta Cryst. 1994;A50:157–163. [Google Scholar]
- 46.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 47.Bailey S. The Cdp4 suite – programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 48.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 49.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 50.Luginbuhl P, Pervushin KV, Iwai H, Wuthrich K. Anisotropic molecular rotational diffusion in 15N spin relaxation studies of protein mobility. Biochemistry. 1997;36:7305–12. doi: 10.1021/bi963161h. [DOI] [PubMed] [Google Scholar]
- 51.Kamath U, Shriver JW. Characterization of thermotropic state changes in myosin subfragment-1 and heavy meromyosin by UV difference spectroscopy. J Biol Chem. 1989;264:5586–92. [PubMed] [Google Scholar]
- 52.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 1 Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 53.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 2 Analysis of experimental results. J Am Chem Soc. 1982;104:4559–4570. [Google Scholar]
- 54.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-5 nuclear magnetic relaxation of proteins. J Am Chem Soc. 1990;112:144–163. [Google Scholar]
- 55.Mandel AM, Akke M, Palmer AG., 3rd Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–63. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]