

Abstract
Fluorescence correlation microscopy (FCM) was applied to characterize fusion proteins of the green fluorescent protein (GFP) on the cellular as well as molecular level within seconds in an integrated instrument. FCM combines the inherent sensitivity and high spatial resolution of fluorescence correlation spectroscopy with fluorescence imaging and micropositioning, thereby providing a spectrum of molecular information in the cellular context. Signatures of characteristic parameters derived from the autocorrelation functions served to distinguish a GFP fusion protein of the epidermal growth factor receptor from GFP fluorescence in the endoplasmic reticulum and cytoplasm. Diffusion constants measured for free transiently expressed GFP reproduced values reported previously with other techniques. The accessible concentration range extends from millions to only a few thousand molecules per cell, with single molecule detectability in the femtoliter detection volume. The detailed molecular characterization offered by FCM is fully compatible with automation in sample identification and detection, offering new possibilities for highly integrated high-throughput screening.
New techniques are constantly evolving for high-throughput screening (HTS) assays in cellular systems as well as in vitro (1). At present, however, analytical approaches are mutually exclusive, providing information either on the level of the whole cell or on the molecular level with isolated target molecules. In the functional characterization of genomic sequence information and the exploitation of this information for drug screening, experimental strategies integrating both levels of complexity are highly desirable. In vitro screening with isolated potential drug targets carries the risk of introducing artifacts due to exclusion of the physiological cellular context. Furthermore, proteins representing important clinical intervention points in cancer therapy, such as transmembrane receptors, are difficult to generate for use in in vitro model systems. Cellular screens based on reporter readouts alone, however, fail to confirm unequivocally that a specific molecular interaction or mechanism has been targeted. The green fluorescent protein (GFP; ref. 2) of the jellyfish Aequorea victoria can be fused to intracellular proteins. The use of GFP as a fluorescent tag for intracellular protein trafficking and dynamics in vivo has alleviated the requirement for protein purification, external fluorescent labeling, and microinjection, which has hitherto limited the number of accessible experimental systems. However, in vivo HTS using fluorescent techniques will require sensitive and versatile screening technology. Once a potential target is identified and characterized, one would wish to proceed to drug screening on the same instrument.
Fluorescence correlation microscopy (FCM; refs. 3 and 4) has recently been introduced as an integrated experimental approach combining the sensitivity and spatial resolution of confocal fluorescence correlation spectroscopy (FCS; refs. 5 and 6) with high-sensitivity imaging, three-dimensional micropositioning, and micromanipulation in live-cell microscopy. Confocal FCS measures fluctuations in the number of fluorescent molecules diffusing and/or reacting within femtoliter volumes and occurring over a very large temporal range (microseconds to seconds). The method is unique in providing a determination of the absolute number (i.e., concentration) of molecules and their interactions, matching the challenges outlined above. Applications of FCS with immediate relevance to pharmaceutical lead searches include the measurement of diffusion, aggregation, photophysical characteristics, receptor-ligand interactions, DNA hybridization, and enzymatic reactions (7) both at the single-molecule level and in HTS applications (8).
To demonstrate the applicability of FCM in cell biological screening, we analyzed the diffusion of a fusion of GFP with the human epidermal growth factor receptor (EGFR) and defined the nature of GFP fluorescence localized to the ER and cytoplasm in subpopulations of cells, using for comparison transiently expressed free GFP. The EGFR is a prototypic member of the large family of transmembrane growth factor receptors with tyrosine kinase activity (9). Ligand binding leads to activation of the kinase and autophosphorylation of tyrosine residues in the intracellular C-terminal segment. Signal transduction proceeds through binding of cytoplasmic proteins via SH2 and PTB domains (10, 11) to these phosphotyrosines. Screening tasks in such systems demand a rapid quantitation of proteins localized in both cellular compartments, the plasma membrane, and the cytoplasm. By using FCM, protocols can be devised for rapid molecular characterization, which are amenable to automation in such complex screening applications.
MATERIALS AND METHODS
Cell Culture.
Chinese hamster ovary cells (obtained from Y. Yarden, Weizmann Institute, Rehovot, Israel) and transfected cell lines were grown in a 5% CO2 humidified atmosphere at 37°C in DMEM supplemented with 10% FCS, 104 units/liter penicillin G, and 100 mg/liter streptomycin sulfate. Cells were propagated every 3 to 4 days.
Generation of EGFR-GFP Fusion Proteins and Transfected Cell Lines.
The EGFR-GFP fusion protein was generated as described elsewhere (12). The EGFR was derived from an EGFR cDNA (13) in pcDNA3 (Invitrogen; obtained from Y. Yarden) and cloned into the pEGFP-N3 plasmid (CLONTECH). For transient transfection, Chinese hamster ovary cells were seeded in 35-mm Petri dishes onto 12-mm glass coverslips at a confluency of 10-15%. The next day, DMEM was replaced with 0.8-ml Optimem (GIBCO/Life Technologies) supplemented with antibiotics. One microgram of the pEGFP-N3 vector DNA and 3 μl of a noncommercial transfection agent (gift of H. Eibl, Max Planck Institute for Biophysical Chemistry) or 6 μl Lipofectin (GIBCO) were incubated separately with 100 μl of Optimem for 15 min and then for 30 min after mixing the two solutions at room temperature. The cells were transfected in 1 ml total volume for 24 h and used for microscopy 48 h later. Cells stably expressing the fusion construct were generated in the same way with selection in medium supplemented with 0.4 mg/ml G418 (GIBCO) and two rounds of cell sorting (Epics Elite, Coulter) at 2-wk intervals. The structural and functional integrity of the EGFR-GFP fusion protein localized to the plasma membrane was characterized in detail (12).
FCM Measurements.
The FCM apparatus is based on a Zeiss Axiovert 35 microscope with a C-Apochromat 40× 1.2 numerical aperture objective. The hardware specifications are given elsewhere (3). GFP fluorescence was excited at 488 nm and detected with a 500DRLP dichroic mirror (Omega Optical, Brattleboro, VT) and a BP515–545 detection filter (Delta Light & Optics, Lyngby, Denmark). Power densities in the excitation focus (≈16 kW/cm2 for intracellular measurements) were calculated according to Eggeling et al. (14). Autocorrelation functions were generated on-line with an ALV-5000/E dual channel autocorrelator board (ALV-Laservertriebsgesellschaft, Langen, Germany) and fitted off-line with Igor Pro (WaveMetrics, Lake Oswego, OR) to the autocorrelation function given below (Eq. 1). The triplet state lifetime τT, triplet fraction T, the fractional contributions φj, and the characteristic diffusion times τj of components j ≤ 3 were allowed to vary. An offset parameter was included to compensate for artifacts in the autocorrelation related to drift in the fluorescence signal (e.g., photobleaching) with time constants >1 s, a specific problem of the ALV-5000/E. The theory for cellular FCS measurements has been described in detail elsewhere (15). The relevant equation is
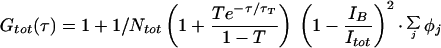 |
1 |
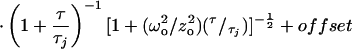 |
where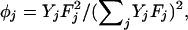 with the fluorescence efficiencies Fj and relative molar fractions Yj of species j. ωo and zo are the axial and radial waists defining the detection volume, Itot the total detected signal, and IB the intensity of uncorrelated background signal (16). The diffusional relaxation constant is related to the diffusion constant D by τ = ωo2/4D, with ωo = 0.3 μm in our experimental configuration. Cells grown on 12-mm coverslips were mounted in an incubation chamber on the microscope stage in HBS (10 mM Na-Hepes, pH 7.4/135 mM NaCl/5 mM KCl/1 mM MgCl2/1.8 mM CaCl2/1% glucose/0.1% BSA) and used at room temperature.
with the fluorescence efficiencies Fj and relative molar fractions Yj of species j. ωo and zo are the axial and radial waists defining the detection volume, Itot the total detected signal, and IB the intensity of uncorrelated background signal (16). The diffusional relaxation constant is related to the diffusion constant D by τ = ωo2/4D, with ωo = 0.3 μm in our experimental configuration. Cells grown on 12-mm coverslips were mounted in an incubation chamber on the microscope stage in HBS (10 mM Na-Hepes, pH 7.4/135 mM NaCl/5 mM KCl/1 mM MgCl2/1.8 mM CaCl2/1% glucose/0.1% BSA) and used at room temperature.
RESULTS
FCM-Based HTS Protocol in the Characterization of GFP Fusion Proteins.
The protocol for a high-throughput characterization of GFP fusion proteins is illustrated in Fig. 1. The experiments presented here proceeded in the same way, except that the spectrum of cells was chosen from one heterogeneous population expressing an EGFR-GFP fusion protein localized at the plasma membrane (Fig. 2A) or GFP fluorescence in the ER (Fig. 2B) or cytoplasm (Fig. 2C) rather than from homogeneous populations of cells expressing different fusion constructs. To enable the fast analysis of rapidly diffusing molecules, the excitation intensity was set relatively high. The signal-to-noise ratio in autocorrelation measurements depends on the number of photons detected per molecule per residence time in the detection volume and is therefore a function of excitation laser power as well as of the diffusion constant (17). For the EGFR-GFP fusion protein, the autocorrelation functions were acquired after an initial phase of laser illumination during which strong photobleaching occurred, indicative of slow-moving or immobile molecules.
Figure 1.

Experimental protocol for FCM-based characterization of GFP fusion proteins. (A) Large numbers of GFP fusion constructs from recloned cDNAs or directly generated as cDNA-GFP fusion expression libraries are transfected into cells seeded in microtiter plates with bottoms of coverslip thickness, compatible for use in high-sensitivity measurements on inverted fluorescence microscopes. (B) The imaging modality of the FCM is used to identify single cells, document the distribution of the molecule, the homogeneity of the cell population, and the cell morphology. In confocal point measurements, the distribution of the molecule is determined along the optical z axis for precise positioning of the confocal measurement volume. Switching between illumination modes requires less than 1 sec and is free of optical readjustments. Subsequently, autocorrelations from the fluctuations of fluorescence in a confocal detection volume are measured, yielding information about the number of molecules and the presence and nature of distinct molecular states. The FCM integrates all these functionalities in an automated and programmable microscope environment. (C) Compilation of the information obtained from one FCM experiment: subcellular distribution of the fluorophore, morphology, and integrity of the cell, number of molecules in the cell, and the presence and nature of distinct molecular states inside the cell. In the case of the EGFR-GFP fusion protein presented here, both slow and fast diffusing components were present. Bar in C = 10 μm.
Figure 2.

FCM-based characterization of GFP fusion proteins localized in (A) the plasma membrane, (B) the ER, and (C) the cytoplasm. (Left) Subcellular distribution in three dimensions. Profiles along the optical axis are superimposed on the fluorescence micrographs; (Center) bright-field images; (Right) autocorrelation functions fitted with three diffusional components, triplet term, and offset. For the later analysis weighted means were calculated for relaxation times differing by less than one order of magnitude (Fig. 3). Bar = 10 μm.
FCM-Based Characterization of Intracellular GFP Fusion Proteins. Fig. 2 represents a compilation of the information obtained for GFP fusion proteins at different locations. The three-dimensional distribution of the fusion proteins in the cells was determined through a combination of intensified whole-field fluorescence imaging and recording of profiles along the optical axis, superior in sensitivity to conventional confocal microscopy for visualizing the low concentrations of fluorophores appropriate for detection with FCS. The morphological integrity of the cells before and after the measurements was documented by bright-field microscopy (Middle), in conjunction with the whole-field epifluorescence images. For each cell type, distinct autocorrelation functions were acquired within measurement times of ≈10 s (Right). The numerical analysis of the autocorrelation functions yields the number of molecules N in the observation volume and delineates the number, nature, and relative fraction of molecular components of heterogeneous systems. In the case of the EGFR-GFP fusion protein, a slow component with a relaxation time of 190 ms contributed strongly to the autocorrelation function. In contrast, for the cytoplasmic protein, a fast component with a relaxation time of 810 μs dominated the signal, consistent with a small freely diffusing protein. For the endoplasmic protein, the fast component dominated; a slow component contributed approximately one-third of the autocorrelation amplitude.
In the presence of mixed populations of fluorescent molecules in different states of aggregation as well as photobleaching, N is at best an estimate of the absolute molecule number. For the rapid and reliable discrimination of the GFP fusion proteins, further analysis concentrated on the autocorrelation times and relative fractions under a given set of experimental conditions. The detailed analysis of all molecular states requires measurements at different laser powers, taking into account those molecules lost during initial photobleaching (4).
Statistical Evaluation of FCM Data. For statistical validation of the results presented in Fig. 2, the relative fractions φj of fast and slow components were plotted vs. the autocorrelation constants τj for the GFP signals in a number of cells of each class (Fig. 3). The autocorrelation functions were fitted to two or three diffusion autocorrelation terms according to Eq. 1. The autocorrelation analysis was model independent in that the number of components was not presumed to correspond to known molecular species. This approach is adequate for screening tasks in which the identity or state of the target molecules is not known a priori, and an automated fingerprinting based on autocorrelation parameters is the primary goal. The effect of photobleaching was compensated by an offset term in most fits. The component with a time constant of 1.2 s in Fig. 2C exemplifies the effect of mild photobleaching on the measured autocorrelation function.
Figure 3.

Statistical analysis of intracellular autocorrelation functions. (A and B) Scatter plots of the relative fraction φj vs. autocorrelation time τj for GFP proteins localized to the plasma membrane, the ER, and the cytoplasm. (C and D) Means and standard deviations calculated for the autocorrelation times and fractional contributions. Error bars exceeding the mean were omitted. (A and C) φj and τj as obtained from the fits; (B and D) φj and τj after binning the autocorrelation times within defined intervals (see text). The bleaching rate constants obtained from monoexponential fits to the count traces (not shown) are included on the abscissa of A highlighted by the arrow. The data represent 18 measurements from 3 cells (plasma membrane), 10 measurements from 2 cells (ER), and 21 measurements from 2 cells (cytoplasm).
The data for all three subcellular locations were clustered in three time domains, ≈10 μs, 0.1 to 5 ms, and 10 ms to a few seconds (Fig. 3). The 10-μs relaxation time is too short to be explained by a diffusional process. An apparent triplet term was included to account for fast nondiffusional fluorescence fluctuations (18). However, additional pH-dependent photophysical processes leading to fluctuations of GFP fluorescence in the 100-μs time range have been recently reported (19). Different but overlapping distributions for the three subcellular locations were obtained, with a 1-ms fraction being dominant for the cytoplasmic GFP and a 100-ms autocorrelation time prevalent for the membrane GFP. The averages and standard deviations of these distributions differed by less than one standard deviation (Fig. 3C). Next, a weighted mean given by 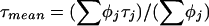 was calculated, and the fractional contributions were added for cases in which two autocorrelation constants for a given autocorrelation function differed by less than one order of magnitude. After this transformation, the fractional contributions of the 1-ms component of plasma membrane GFP and endoplasmic GFP, as well as the 1-ms and the slow component of plasma membrane and cytoplasmic GFP, differed by more than one standard deviation (Fig. 3 B and D). Table 1 lists the diffusion constants that were derived from the diffusional relaxation times as described in Materials and Methods. For all three subcellular locations, the fast diffusional process corresponded well to the diffusion constant of free intracellular GFP (see below). The small diffusion constants were similar to values reported for the diffusion of GFP fusion proteins in the endoplasmic reticulum (20) and for a fusion of GFP with the β2-adrenergic receptor (21).
was calculated, and the fractional contributions were added for cases in which two autocorrelation constants for a given autocorrelation function differed by less than one order of magnitude. After this transformation, the fractional contributions of the 1-ms component of plasma membrane GFP and endoplasmic GFP, as well as the 1-ms and the slow component of plasma membrane and cytoplasmic GFP, differed by more than one standard deviation (Fig. 3 B and D). Table 1 lists the diffusion constants that were derived from the diffusional relaxation times as described in Materials and Methods. For all three subcellular locations, the fast diffusional process corresponded well to the diffusion constant of free intracellular GFP (see below). The small diffusion constants were similar to values reported for the diffusion of GFP fusion proteins in the endoplasmic reticulum (20) and for a fusion of GFP with the β2-adrenergic receptor (21).
Table 1.
Diffusion constants of cellular GFP fusion proteins
| Membrane
|
ER
|
Cytoplasm
|
|
|---|---|---|---|
| Daverage [cm2/s], % | Daverage [cm2/s], % | Daverage [cm2/s], % | |
| Fast | 2.1 ± 0.8 × 10−7 35 ± 7 | 1.2 ± 0.4 × 10−7 62 ± 9 | 2.4 ± 0.8 × 10−7 78 ± 12 |
| Slow | 1.6 ± 0.9 × 10−9 51 ± 14 | 2.9 ± 1.5 × 10−9 29 ± 5 | 5.1 ± 6.6 × 10−9 19 ± 11 |
Given are the average diffusion constants and relative fractions for GFP fusion proteins derived from the autocorrelation times (1-ms and 100-ms processes in Fig. 3D). The fast components correspond well to the diffusion constant of 1.7 ± 0.4 × 10−7 cm2/s for free intracellular GFP. The standard deviations were derived from all the measurements shown in Fig. 3B.
Fast Measurements of Free Cytoplasmic GFP.
The cytoplasmic GFP was identified as a low molecular-weight truncated form of the EGFR-GFP fusion protein on the basis of the nuclear entry and short relaxation time, corresponding to a diffusion constant of 2.4 × 10−7 cm2/s. To further confirm this assumption and demonstrate the reproducibility of FCM in another defined system, cells were transiently transfected with free GFP, which distributes homogeneously throughout the cell. Superposable autocorrelation functions were obtained for repetitive 5-s measurements (Fig. 4A). In two-component fits, either an additional slow or additional fast process was obtained, which affected the fitted time constant for the major component (Fig. 4B). Because all pairs of values fell within one of the categories defined above, weighted averages were calculated, corresponding to a diffusion constant of 1.7 × 10−7 cm2/s. They were nearly identical for all four curves (Fig. 4B), demonstrating again that the model-independent fingerprinting approach was able to confirm unequivocally the identity of the proteins.
Figure 4.

Intracellular autocorrelation functions (A) and fits (B) of repeated 5-s measurements of transiently expressed free GFP in the cytoplasm. The ordinate to the right in B corresponds to the relative fractions φj of the components in the fits. A slow (×) or fast (+) process was obtained in addition to the major component; the arrow points at the weighted means. The average diffusion constant of cytoplasmic GFP was 1.7 ± 0.4 × 10−7 cm2/s, in good agreement with a value reported previously (29).
Concentration Range for the Detection of GFP Fusion Proteins.
The range of working concentrations is a major determinant for the applicability of an assay technology in drug screening and intracellular analyses of molecular dynamics and distribution. In the early phase of a search for compounds binding to a cellular target, micromolar concentrations are desirable, whereas the identification of highly active binders requires nanomolar concentrations. Furthermore, the technique needs to cope with a spectrum of expression levels of intracellular GFP fusion proteins. The sensitivity of FCS reaches down to the single molecule level, corresponding to subnanomolar concentrations. The upper limit is about 1,000 molecules in the detection volume, i.e., concentrations of 0.1 − 1 μM. The autocorrelation amplitude is inversely related to the number of molecules in the detection volume. If the fluctuation amplitudes caused by molecule diffusion are too low, the calculation of the autocorrelation functions is compromised by detection noise. Figs. 2 and 4 include examples of the mean and upper concentration ranges, and in Fig. 5 a measurement at the single-molecule limit is presented. The amplitude of the autocorrelation function >1 as well as the spikes in the count trace most likely corresponded to single EGFR-GFP fusion proteins or aggregates of receptors passing through the detection volume.
Figure 5.

FCM at low levels of intracellular EGFR-GFP fusion proteins. (A) Fluorescence count trace. The peaks in the fluorescence count trace likely represent single EGFR-GFP molecules diffusing through the detection volume. (B) Autocorrelation function and parameters derived from a two-component fit. The number of molecules derived from the autocorrelation amplitude was corrected for the uncorrelated background fluorescence, derived from peak-free count intervals according to Eq. 1. The excitation intensity was 3.8 kW/cm2.
DISCUSSION
Our results demonstrate the realization by FCM of a complex cell biological screening task in the characterization of cellular proteins, benefiting from the imaging capability, the positioning accuracy, and the diminutive confocal measurement volume, as well as the versatility of GFP. GFP fusion proteins localized to different subcellular compartments were discriminated with measurement times of a few seconds on the single-cell level and with single-molecule sensitivity.
The analysis of the autocorrelation curves with a model-independent approach introduced some degree of arbitrariness. We attempted to achieve fits lacking systematic errors by varying the starting conditions. The reproducibility of the fitted values was high. After binning the values within defined intervals, the parameters of plasma membrane EGFR-GFP differed from those for the other two compartments by more than one standard deviation. HTS with FCS based on overlapping distributions of characteristic parameters for in vitro systems has recently been presented (8). A model-independent data analysis of the kind featured here will be even more appropriate if unknown proteins, e.g., from a GFP fusion cDNA expression library, are characterized.
We note that the diffusion constant derived from the long relaxation constant of the EGFR determined by FCM in the present experiments was greater by about one order of magnitude than the values obtained in previous measurements using other techniques (22, 23). This result may be attributable at least in part to the apparent reduction of diffusion constants in the presence of photobleaching (24), especially at the higher laser powers selected for fast measurements of freely diffusing intracellular molecules. However, the experiment in Fig. 5 conducted at one-fourth of the laser power and others carried out at still lower intensities were consistent with the results of Fig. 2. Particular screening tasks or detailed molecular characterizations of slowly diffusing molecules will require adjustment of this critical parameter. Such a fast diffusing component has not been reported so far for an endoplasmic protein or for a transmembrane receptor. Whereas Fluorescence Recovery After Photobleaching (FRAP) detects only recovery of fluorescence by diffusion into the detection volume, FCS is sensitive as well to short-distance movements occurring within the detection volume. Limited proteolytic degradation and release of free GFP into the cytoplasm may also occur. Control experiments with untransfected Chinese hamster ovary cells excluded autofluorescence as a source for the observed autocorrelations (15). Further experiments will be required to define the nature of the fast process in detail.
The work presented here demonstrates the analysis of GFP fusion proteins on the cellular as well as molecular level with FCM. A very similar protocol applies to in vivo drug screens. EGFR and other receptor tyrosine kinases are overexpressed in a number of tumors (25, 26). Because signal transduction proceeds through the interaction of proteins with the phosphotyrosine motifs of these receptors, these receptors and their binding partners have been designated as potential drug targets (26, 27). The discrimination between a free cytoplasmic and a membrane protein presented here will be essential for identifying drugs that inhibit these interactions in vivo. We envisage screens for potential inhibitors of the binding of a cytoplasmic protein to a transmembrane receptor based on changes in the autocorrelation function as well as in the distribution of the proteins in the fluorescence images, thereby integrating the molecular and cellular levels in the lead search. The concentration range of three orders of magnitude will enable the acquisition of dose-response curves of compounds with different biological activities. FCM is compatible with expression of standard cytomegalovirus promotor-driven GFP fusion constructs. However, regulatable expression (28) represents the next step for better control of experimental conditions. FCM will enable not only analyses at low concentrations of GFP fusion proteins, thereby reducing the risk of artifacts because of protein overexpression, but also the quantitation of the number of these probe molecules inside the cell.
With short measurement times, high sensitivity, and automation achieved by a combination of image-based object recognition and motorized high-precision positioning, FCM constitutes a unique tool for meeting the challenge of intracellular HTS.
Acknowledgments
R. B. received a fellowship from the Studienstiftung des deutschen Volkes. The cooperation between T.M.J., G. Vàmosi, and G. Vereb was supported by Deutscher Akademischer Austauschdienst-Hungarian Fellowship Commission joint grant for exchange of researchers no. 91/1997. G. Vereb was supported by Orszgos Tudomnyos Kutatsi Alap (Hungary) grant F025210 and the Alexander von Humboldt Foundation. We thank Didier Marguet, Johns Hopkins University, for plasmids and Rolando Riviera-Pomar for critical reading of the manuscript.
ABBREVIATIONS
- FCM
fluorescence correlation microscopy
- FCS
fluorescence correlation spectroscopy
- GFP
green fluorescent protein
- HTS
high-throughput screening
- EGFR
epidermal growth factor receptor
References
- 1.Silverman L, Campbell R, Broach J R. Curr Opin Chem Biol. 1998;2:397–403. doi: 10.1016/s1367-5931(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 2.Tsien R Y. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 3.Brock R, Jovin T M. Cell Mol Biol. 1998;44:847–856. [PubMed] [Google Scholar]
- 4.Brock, R. & Jovin, T. M. in Fluorescence Correlation Spectroscopy. Theory and Applications, eds. Elson, E. L. & Rigler, R. (Springer, Heidelberg), in press.
- 5.Rigler R, Mets Ü, Widengren J, Kask P. Eur Biophys J. 1993;22:169–175. [Google Scholar]
- 6.Madge D, Elson E, Webb W W. Phys Rev Lett. 1972;29:705–708. [Google Scholar]
- 7.Schwille P, Bieschke J, Oehlenschläger F. Biophys Chem. 1997;66:211–228. doi: 10.1016/s0301-4622(97)00061-6. [DOI] [PubMed] [Google Scholar]
- 8.Koltermann A, Kettling U, Bieschke J, Winkler T, Eigen M. Proc Natl Acad Sci USA. 1998;95:1421–1426. doi: 10.1073/pnas.95.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ullrich A, Schlessinger J. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 10.Mayer B J, Baltimore D. Trends Cell Biol. 1993;3:8–13. doi: 10.1016/0962-8924(93)90194-6. [DOI] [PubMed] [Google Scholar]
- 11.Cowburn D. Curr Opin Struct Biol. 1997;7:835–838. doi: 10.1016/s0959-440x(97)80155-8. [DOI] [PubMed] [Google Scholar]
- 12.Brock R, Hamelers I H L, Jovin T M. Cytometry. 1999;35:353–362. doi: 10.1002/(sici)1097-0320(19990401)35:4<353::aid-cyto8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 13.Ullrich A, Coussens L, Hayflick J S, Dull T J, Gray A, Tam A W, Lee J, Yarden Y, Libermann T A, Schlessinger J, et al. Nature (London) 1984;309:418–425. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- 14.Eggeling C, Widengren J, Rigler R, Seidel C A M. Anal Chem. 1998;70:2651–2659. doi: 10.1021/ac980027p. [DOI] [PubMed] [Google Scholar]
- 15.Brock R, Hink M A, Jovin T M. Biophys J. 1998;75:2547–2557. doi: 10.1016/S0006-3495(98)77699-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mets Ü, Rigler R. J Fluoresc. 1994;4:259–264. doi: 10.1007/BF01878461. [DOI] [PubMed] [Google Scholar]
- 17.Qian H. Biophys Chem. 1990;38:49–57. doi: 10.1016/0301-4622(90)80039-a. [DOI] [PubMed] [Google Scholar]
- 18.Widengren J, Mets Ü, Rigler R. J Phys Chem. 1995;99:13368–13379. [Google Scholar]
- 19.Haupts U, Maiti S, Schwille P, Webb W W. Proc Natl Acad Sci USA. 1998;95:13573–13578. doi: 10.1073/pnas.95.23.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole N B, Smith C L, Sciaky N, Terasaki M, Edidin M, Lippincott-Schwartz J. Science. 1996;273:797–801. doi: 10.1126/science.273.5276.797. [DOI] [PubMed] [Google Scholar]
- 21.Barak L S, Ferguson S S G, Zhang J, Martenson C, Meyer T, Caron M G. Mol Pharmacol. 1997;51:177–184. doi: 10.1124/mol.51.2.177. [DOI] [PubMed] [Google Scholar]
- 22.Livneh E, Benveniste M, Prywes R, Felder S, Kam Z, Schlessinger J. J Cell Biol. 1986;103:327–331. doi: 10.1083/jcb.103.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kusumi A, Sako Y, Yamamoto M. Biophys J. 1993;65:2021–2040. doi: 10.1016/S0006-3495(93)81253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Widengren J, Rigler R. Bioimaging. 1996;4:149–157. [Google Scholar]
- 25.Libermann T A, Nusbaum H R, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield M D, Ullrich A, Schlessinger J. Nature (London) 1985;313:144–147. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 26.Levitzki A. Eur J Biochem. 1994;226:1–13. doi: 10.1111/j.1432-1033.1994.tb20020.x. [DOI] [PubMed] [Google Scholar]
- 27.Al-Obeidi F A, Wu J J, Lam K S. Biopolymers. 1998;47:197–223. doi: 10.1002/(SICI)1097-0282(1998)47:3<197::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 28.Gossen M, Bujard H. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swaminathan R, Hoang C P, Verkman A S. Biophys J. 1997;72:1900–1907. doi: 10.1016/S0006-3495(97)78835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]