

Abstract
The bacterial pathogen, Listeria monocytogenes, grows in the cytoplasm of host cells and spreads intercellularly using a form of actin-based motility mediated by the bacterial protein ActA. Tightly adherent monolayers of MDCK cells that constitutively express GFP-actin were infected with L. monocytogenes, and intercellular spread of bacteria was observed by video microscopy. The probability of formation of membrane-bound protrusions containing bacteria decreased with host cell monolayer age and the establishment of extensive cell-cell contacts. After their extension into a recipient cell, intercellular membrane-bound protrusions underwent a period of bacterium-dependent fitful movement, followed by their collapse into a vacuole and rapid vacuolar lysis. Actin filaments in protrusions exhibited decreased turnover rates compared with bacterially associated cytoplasmic actin comet tails. Recovery of motility in the recipient cell required 1–2 bacterial generations. This delay may be explained by acid-dependent cleavage of ActA by the bacterial metalloprotease, Mpl. Importantly, we have observed that low levels of endocytosis of neighboring MDCK cell surface fragments occurs in the absence of bacteria, implying that intercellular spread of bacteria may exploit an endogenous process of paracytophagy.
Keywords: Listeria monocytogenes, ActA, actin, epithelial cells, bacterial pathogenesis
Listeria monocytogenes is a Gram-positive bacterial pathogen that causes food poisoning, meningitis, and spontaneous abortions in its human host. It invades the host through the epithelial lining of the gut (Stamm et al. 1982). After adhering to a host cell, the bacterium induces a local cytoskeletal rearrangement that results in uptake of the bacterium into a primary vacuole (Tilney and Portnoy 1989; Gaillard et al. 1987). Using listeriolysin O (LLO)1, a pore-forming hemolytic protein, and other factors (Bielecki et al. 1990; Gaillard et al. 1987), the bacterium escapes from the vacuole into the host cytoplasm, where it divides and, using proteins of the host cytoskeleton, acquires intracellular actin-based motility (Dabiri et al. 1990; Sanger et al. 1992; Theriot et al. 1992). This form of motility is required for efficient intercellular spread from the cytoplasm of one infected host cell into the cytoplasm of another (Tilney and Portnoy 1989; Mounier et al. 1990). This mechanism of intercellular spread in endothelial tissue has been posited to explain how L. monocytogenes crosses the transplacental and blood-brain barriers (Greiffenberg et al. 1998; Parida et al. 1998).
L. monocytogenes accomplishes intercellular spread by abducting the host cell's actin-based motility components to propel itself into the host cell's plasma membrane and infect adjacent cells. The membrane-associated bacterial protein ActA is necessary and sufficient for such motility (Kocks et al. 1995; Smith et al. 1995b; Cameron et al. 1999). Distributed in a gradient along the bacterial surface (Kocks et al. 1993), ActA nucleates actin filaments and catalyzes their elongation (Smith et al. 1996). The ensuing polymerization and cross-linking of these filaments cooperate to exert force on the bacterium much as they exert force on a protruding lamellipodium (Sanger et al. 1992; Theriot et al. 1992). The organism is pushed forward, leaving behind a comet-like tail consisting of short (∼0.2 μm), highly cross-linked actin filaments (Tilney and Portnoy 1989; Tilney et al. 1990; Zhukarev et al. 1995) whose depolymerization is regulated by the surrounding environment (Theriot et al. 1992; Carlier et al. 1997; Rosenblatt et al. 1997).
When these actin comet tails propel the bacterium into its host's plasma membrane, the membrane is distended, forming long protrusions into the extracellular space. In tissue culture cells, these structures are nonproductive and simply extend many microns into the media; however, in vivo, infected tissues are less likely to border on a free space, and thus, these bacterium-containing membrane protrusions have a high probability of entering a neighboring cell. When a protrusion from one cell is taken up by another, intercellular spread of bacteria occurs.
Intercellular spread has not previously been documented in real time, despite extensive observation of intracellular movement of bacteria by video microscopy. Thus, very little is known about the mechanism or kinetics of this critical process of L. monocytogenes infection. Transmission electron micrographs have fortuitously captured a few intermediate steps that show membrane-bound protrusions of bacteria inside neighboring cells. The subsequent uptake of these protrusions, leaving the bacterium enclosed in a secondary vacuole, has been inferred from these images (Tilney and Portnoy 1989; Mounier et al. 1990).
This secondary vacuole is distinct from the primary vacuole in that it is surrounded by a double membrane, one from the donor cell and one from the recipient (Tilney and Portnoy 1989). The bacterial protein LLO is required for bacterial escape from the secondary vacuole (Gedde, M.M., D.E. Higgins, L. Tilney, and D.A. Portnoy, manuscript submitted for publication), and disruption of either or both bacterially secreted phospholipase Cs (PC-PLC and PI-PLC) significantly and synergistically curtails the efficiency of bacterial escape from the secondary vacuole (Vazquez-Boland et al. 1992; Smith et al. 1995a). At least one regulator of this process has been identified, a metalloprotease (Mpl), which proteolytically activates PC-PLC upon acidification of the secondary vacuole (Marquis et al. 1997). After escape from this vacuole, the bacterium is believed to reacquire motility and restart its intracellular life cycle.
To increase our chances of observing intercellular spread by video microscopy, we chose to infect a columnar epithelial cell line. This guaranteed that most of the cellular surface area would be in contact with neighboring cells, thereby promoting the chance of intercellular uptake of bacterium-induced membrane protrusions. MDCK cells form a polarized monolayer of epithelial cells in tissue culture (for review, see Marrs and Nelson 1998). We used clones of MDCK cells that express a GFP-tagged form of actin to follow the movement of bacteria in living cells, to document interactions of bacteria with the plasma membrane, and to record intercellular uptake of membrane protrusions containing bacteria by a recipient cell. Using this system, we were frequently able to observe intercellular spread of bacteria. Our observations of the physical and temporal kinetics of this process suggest a high degree of host cell cooperativity at each step.
Materials and Methods
Chemicals were purchased from Sigma-Aldrich unless otherwise indicated. All photographic images were compiled using Adobe Photoshop, Adobe Illustrator, and/or Canvas (Deneba Systems) software. The printed images are representative of the original data.
Cell Lines and CDNA Constructs
Human nonmuscle β-actin (these data are available from GenBank/EMBL/DDBJ under accession number M10277) was cloned by PCR from a human cDNA library and was fused in-frame with the enhanced GFP gene into the BamHI-XhoI sites of the vector pEGFP-C1 (Clontech Inc.) to generate a fusion between the COOH terminus of EGFP and the NH2 terminus of beta-actin in the expressed protein. PCR-derived sequences were confirmed by dideoxy sequencing. MDCK clone II/G cells were obtained from the laboratory of Gerrit van Meer (Academic Medical Center, University of Amsterdam, Department of Cell Biology and Histology, The Netherlands) and have been described previously (Grindstaff et al. 1998). MDCK cells were transfected with pEGFP-actin using the lipofectamine protocol from GIBCO BRL. Clones were selected in 400 μg/ml G418 (Geneticin; GIBCO BRL). Low passage aliquots of drug-resistant MDCK clones were frozen and stored in liquid nitrogen: clones were passaged in DME containing 10% FCS (Gemini) and 400 μg/ml G418 for a maximum of 4–6 wk.
For experiments, all cells were grown in DME containing 10% FBS and antibiotic-antimycotic (GIBCO BRL) at 37°C in a humidified incubator containing 5% CO2. Antibiotic-antimycotic was removed from cultures used for bacterial infections.
Immunoblotting
For the preparation of lysates, 5 × 105 MDCK cells were plated on 35-mm tissue-culture dishes and extracted after 24 h in culture. For the preparation of SDS lysates, MDCK cells were extracted in hot SDS buffer containing 1% SDS, 10 mM Tris-HCl, pH 7.5, and 2 mM EDTA, and then scraped from the petri dish with a rubber policeman. Samples were boiled for 15 min and insoluble material was removed by centrifugation at 12,000 g for 15 min. For preparation of Triton X-100 lysates, MDCK cells were extracted for 15 min at 4°C with CSK buffer (0.5% Triton X-100, 10 mM Pipes, pH 6.8, 50 mM NaCl, 300 mM sucrose, and 3 mM MgCl2) containing a protease inhibitor mix (1 mM Pefabloc, 1 mM benzamidine, and 10 μg/ml each of aprotinin, pestatin, and leupeptin). Pefabloc, pepstatin, and leupeptin were purchased from Boehringer Mannheim Biochemicals. After extraction, cells were scraped from the petri dish with a rubber policeman and insoluble material was pelleted by centrifugation at 12,000 g for 30 min. Pellets were washed twice with CSK buffer, resuspended in SDS buffer, boiled 10 min, and insoluble material was removed by centrifugation at 12,000 g for 15 min. Protein concentrations were determined using the BCA protein assay reagent kit (Pierce Chemical Co.). Portions of the lysates were boiled for 5 min after adding one-third volume of 4× SDS reducing sample buffer and separated by SDS-PAGE in 7.5% polyacrylamide gels (Laemmli 1970). Lysates were immunoblotted with anti-actin monoclonal antibody (clone C4; Boehringer Mannheim; 1:500) and HRP secondary antibody as described previously (Barth et al. 1997).
Phalloidin Staining
1 × 105 MDCK cells were plated on collagen-coated coverslips in 35-mm tissue-culture dishes and fixed after 24 h in culture. Cells were washed once in Dulbecco's PBS and fixed for 20 min at RT with 2% formaldehyde in Dulbecco's PBS, or cells were washed once, extracted 5 min at 4°C with CSK buffer, washed again, and fixed with 2% formaldehyde. Cells were washed with blocking buffer, incubated 10 min with rhodamine-phalloidin (Molecular Probes; 1 unit per coverslip) in blocking buffer, washed, and processed for microscopy as described previously (Barth et al. 1997). Photomicrographs were taken with an Axiophot inverted fluorescence microscope (Carl Zeiss Inc.).
Bacterial Culture and Infection Procedures
L. monocytogenes strain 10403S (Bishop and Hinrichs 1987) was grown 12–15 h in 2 ml BHI (Difco) at room temperature without agitation. Cells were pelleted and resuspended in PBS (0.9 mM CaCl2, 2.7 mM KCl, 1.5 mM KH2PO4, 0.5 mM MgCl2, 137 mM NaCl, and 8.1 mM Na2HPO4) or DMEM containing 10% FBS, and then 50–100 μl was added to cells growing on glass coverslips in 2 ml DMEM with 10% FBS. After 60–90 min, cells were rinsed three times with PBS and fresh DMEM/FBS added. 30 min later, in most cases, gentamicin sulfate was added to a concentration of 30–50 μg/ml.
Fluorescence Video Microscopy and Analysis
Infected cells grown on glass coverslips were observed 5–20 h after infection after mounting on a stage whose temperature was maintained at 37°C. Cells were overlaid with phenol red-free DMEM with 10% FBS and buffered with 20 mM Hepes (pH 7.3), and covered with a thin layer of silicone DC-200 fluid (Serva) to prevent evaporation. Observations were performed on a Nikon Diaphot-300 inverted microscope equipped with phase contrast and epifluorescence optics. Time–lapse video microscopy was achieved with an intensified charge-coupled device (CCD) camera (Dage-MTI; GenIISys/CCD-c72) or a cooled CCD camera (NDE/CCD; Princeton Instruments) and Metamorph (Universal Imaging) software. Phase contrast/fluorescence image pairs were recorded every 10–30 s, with eight video frames averaged per image (intensified CCD) or 50-ms exposures (cooled CCD).
Immunofluorescence Deconvolution Microscopy
Cells grown on glass coverslips were fixed in 3% formaldehyde for 30 min at room temperature in PBS after 7–10 h of infection. Cells were then washed twice in PBS at room temperature and extracted with CSK buffer at room temperature for 10 min. Cells were washed twice in PBS at room temperature for 10 min. Cells were blocked in PBS containing 1% BSA, 1% calf serum, and 50 mM NH4Cl for 1 h at room temperature, then in primary antibody solution containing mouse anti-E-cadherin antibody (Transduction Laboratories) for 2 h at room temperature or overnight at 4°C. Cells were then washed three times in PBS with 0.2% BSA and incubated in Texas red–conjugated mouse secondary antibody solution for 1 h at room temperature. Cells were washed three times in PBS and 0.2% BSA and mounted in 90% glycerol in 20 mM TRIS.
Three-dimensional images were recorded with a cooled CCD camera (Photometrics Ltd.). Optical sections (1,023 × 1,023 pixels) of thickness 0.10–0.25 μm were recorded with a 100× oil immersion objective (Olympus Corp.). All aspects of image collection were controlled by a Silicon Graphics workstation (Silicon Graphics Corp.), on a deconvolution microscopy system (Applied Precision).
Metabolic Inhibition
Infected cells were observed on the microscopy workstation described for video microscopy. DMEM + 10% FBS with 0.5% (wt/vol) methyl cellulose was added to the heated chamber and protrusions imaged. Cellular ATP synthesis was inhibited by addition of 50 μM 2,4-dinitrophenol and 10 mM 2-deoxy-d-glucose. Inhibitors were removed by rinsing in fresh DMEM + 10% FBS with 0.5% methyl cellulose.
Fluorescence Labeling of the Plasma Membrane
TRITC.
Cells were incubated 1–3 h in 1–10 μM TRITC in DMEM containing 10% FBS and antibiotic-antimycotic (GIBCO BRL). Cells were rinsed 1–2 times over the next 1–5 h with fresh media, then trypsinized and replated with unlabeled cells, and grown at least 12 h before infection.
Lipophilic Dye.
Cells were suspended in 2 ml LISS buffer (239 mM sucrose and 5 mM Hepes, pH 7.3), to which was added 4–10 μl of a 0.25% (wt/vol) stock solution of 3, 3′-dioctadecyloxacarbocyanine perchlorate (DiOC18; Molecular Probes) in dimethylformamide (DMF). Stock solution was prepared by mixing DiO and stearylamine octadecylamine in a 5:1 (wt/wt) ratio and dissolved in chloroform, heated to 50°C, precipitated on ice with two volumes of methanol, and pelleted by centrifugation. The pellet was dried then dissolved in 1,000 μl DMF, heated to 50°C, and centrifuged. Cells were incubated for 20 min with gentle agitation, rinsed twice with LISS, and plated in DMEM + 10% FBS.
Inhibition of Vacuolar Acidification
TRITC-labeled and unlabeled cells were plated together and incubated 12–40 h. After infection, DMEM + 10% FBS containing 1 μM bafilomycin A in DMSO was added. This concentration inhibits acidification of endocytic vacuoles in mammalian cells (Yoshimori et al. 1991). Cells were incubated at least 20 min before observation of protrusion dynamics by video microscopy.
Metabolic Labeling
Infection of J774 and metabolic labeling were performed as previously described with some modifications (Marquis et al. 1997). J774 cells were infected with the wild-type strain 10403S, isogenic mutants DP-L2296 (Δmpl) (Marquis et al. 1995), or DP-L1942 (ΔactA) (Brundage et al. 1993). The cells were washed with PBS at 30 min post-infection (p.i.), and gentamicin (10 μg/ml) was added at 1 h p.i. Cells were starved for methionine at 3.5 h p.i., and host protein synthesis was blocked with anisomycin (30 μg/ml) and cycloheximide (22.5 μg/ml) 15 min before labeling. At 4 h p.i., cells were pulse-labeled with 35S-methionine (90 μCi/35-mm dish) for 10 min, and chased with cold methionine (2 mM) and chloramphenicol (20 μg/ml) for 15 min. At that point, the medium was replaced with a potassium buffer, pH 6.5 (133 mM KCl, 1 mM MgCl, 15 mM Hepes, 15 mM MOPS), cold methionine, and chloramphenicol, with or without nigericin (10 μM) (Alpuche Aranda et al. 1992). Treatment with nigericin allows the pH to equilibrate across all membranes. After 15 min, samples were washed with cold PBS, lysed in 2× sample buffer, boiled, and separated by 10% SDS-PAGE.
Fluorescence Recovery after Photobleaching
Photobleaching of cells was performed and analyzed on the Nikon microscopy workstation described above. Infected cells were prepared as described in that section. Bleaching was performed by narrowing the field diaphragm on a 100-W mercury arc lamp, exposing the region to be bleached for 3–5 min through a Chroma FITC filter. For protrusions, bleached regions were chosen so as to exclude the entire cell body except for the protrusion. Clouds were chosen for bleaching based on proximity to cell edges, to minimize photodamage to the cell body and the GFP it contained. Immediately after bleaching, lamp intensity was attenuated to 10% by a neutral density filter and 8-frame averaged images were collected every 7 s to follow recovery.
For analysis, the integrated pixel intensity of each cloud or protrusion was measured using the Measure Brightness function of Metamorph. Additionally, the intensity of a nearby unbleached region of cytoplasm was measured for each frame and used to normalize intensities of regions of analysis, to compensate for fluctuations in lamp intensity and photobleaching due to image capture. The standard intensity never fell below 90% of its original value.
To characterize the exponential recovery of fluorescence, normalized intensities were fit to the following perturbation-relaxation equation:
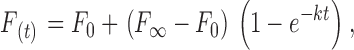 |
1 |
after Salmon et al. 1984. F(t) is the normalized fluorescence ratio at time t after photobleaching, and k is the first-order rate constant, which describes the rate of recovery. F0 is the fluorescence intensity measured immediately after bleaching, and F∞ is the asymptotic value to which fluorescence intensity recovers after bleaching. Data was fitted to and the half-time (t1/2) to recovery from F0 to F∞ was calculated using the equation:
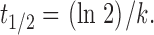 |
2 |
Analysis of Infectious Foci
Infected cells grown on glass coverslips were fixed in 3% formaldehyde for 30 min at room temperature and then incubated in FITC-phalloidin in PBS for 30 min at room temperature. They were then rinsed with PBS for 5 min and mounted on slides in 90% glycerol in 20 mM Tris-HCl. Cells were viewed with the Nikon microscopy workstation described above, using a 100× oil immersion objective. Primarily infected cells were defined as (in descending order of importance): (a) cells with the most bacteria, (b) cells in the center of an infectious focus, and (c) cells with the greatest fraction of bacteria possessing tails. Secondarily infected cells were cells with fewer bacteria surrounding the perimeter of the primarily infected cell, and tertiarily infected cells were at least one cell away from the primarily infected cell and contained fewer bacteria than other cells in the focus. After 8 h, primary, secondary, and tertiary infections could no longer be unambiguously defined.
Online Supplemental Material
Video 1. Supplement to Fig. 1 c. MDCK canine kidney cells, which constitutively express green fluorescent protein fused with actin, are infected with Listeria monocytogenes. Available at http://www.jcb.org/cgi/content/full/146/6/1333/F1/DC1
Figure 1.
MDCK clones expressing GFP-actin. (a) Western blot stained for actin showing ratio of GFP-actin to actin in the GFP-actin MDCK clone. The upper blot was exposed for 10 s; the lower overexposed for 20 min to enable detection of GFP-actin. The ratio of actin to GFP-actin is ∼50:1. Note that GFP-actin is present in both the Triton X-100–soluble and –insoluble fractions though slightly more is soluble compared with wild-type actin. (b) GFP-actin is present in both actin filaments and the soluble actin monomer pool. (Top) GFP-actin colocalizes with filamentous actin as stained by phalloidin; in addition, monomeric GFP-actin is apparent as a diffuse signal. (Bottom) Extraction leaves behind only filamentous actin. (c) Sample time–lapse video microscopy frames, showing paired phase and fluorescent images of GFP-actin MDCK cells infected with L. monocytogenes. The upper cell is not expressing GFP-actin at detectable levels, expression levels vary significantly from cell to cell. (First panel) Black arrowhead, comet tail associated with a moving cytoplasmic bacterium; white arrowheads, extracellular protrusions, which move erratically in and out of focus. Bar, 10 μm. Video available at http://www.jcb.org/cgi/content/full/146/6/1333/F1/DC1 (d) Distribution of average bacterial velocities in GFP-actin MDCK cells (n = 48; 2,855 points tracked).




Video 2. Supplement to Fig. 3. Ricochets and protrusions can be seen in this monolayer, which was confluent for ∼72 h preinfection. Ricochets are marked by yellow R's and protrusions by red P's. Available at http://www.jcb.org/cgi/content/full/146/6/1333/F3/DC1
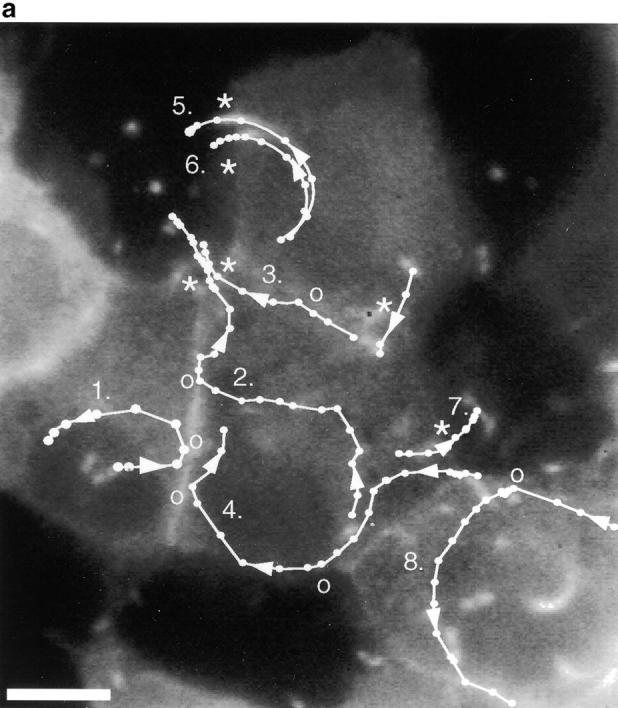
Figure 3.
Protrusion likelihood is dependent on host cell. (a) Bacterial paths in a confluent monolayer aged ∼72 h and infected ∼10 h. GFP-actin is expressed at undetectable levels in some cells. Only a fraction of bacterial paths are shown. Arrows denote direction, o's indicate ricochet sites, and asterisks (*) mark protrusions. Dots indicate position with respect to time (20 s intervals). Note that bacteria no. 2 and no. 3 perform both a ricochet and, minutes later, a protrusion, demonstrating that absence of protrusion is not due to a bacterial deficiency. (b) Dependence of ricochet likelihood on monolayer age (≥22 cells, each containing ≥2 bacteria, for each condition). Error bars are SD. Video available at http://www.jcb.org/cgi/content/full/146/6/1333/F3/DC1 Bar, 10 μm.

Video 3. Supplement to Fig. 4. The majority of fitful movement occurs along the axis of the protrusion. Speeded up 120×. Available at http://www.jcb.org/cgi/content/full/146/6/1333/F4/DC1
Figure 4.
Protrusions undergo bacterially directed fitful movement. (a) White dots indicate bacterial path with respect to time (each dot, 25 s). In this frame, the bacterium is located by the third dot from the top of the image. It slows down as it crosses the plasma membrane at the fourth dot, forming a protrusion. Bar, 2 μm. (b) Initially, the protrusion extends relatively rapidly, then hesitates for ∼2 min and recovers fitful movement for about 4 min before ceasing motility entirely. (c) For each step the bacterium takes, most of the motility is directed along the initial axis of protrusion. Video available at http://www.jcb.org/cgi/content/full/146/6/1333/F4/DC1


Video 4. Supplement to text. In the presence of methyl cellulose, addition of ATP synthesis inhibitors quickly stops all protrusion movement. One protrusion still drifts; however, the chamber was not free of flow, and on average protrusions moved less than 1 μm in 10 min. Washing with inhibitor-free media restores fitful movement. Available at http://www.jcb.org/cgi/content/full/146/6/1333/DC2
Video 5. Supplement to Fig. 6. Collapse of the protrusion from an ovoid to spherical geometry. A long protrusion moving into a non-GFP-expressing cell collapses. Available at http://www.jcb.org/cgi/content/full/146/6/1333/F6/DC1
Figure 6.

Protrusion geometry change. A protrusion extending from an GFP-actin expressing cell into a cell expressing undetectable levels of GFP-actin. (a) The protrusion is originally oblong and clearly connected to the donor cell by a slender, actin-positive stalk, in which it remains for an average time of 35 min. (b) It collapses >150 s to a roughly spherical vacuole surrounding the bacterium. Bar, 5 μm. Video available at http://www.jcb.org/cgi/content/full/146/6/1333/F6/DC1
Video 6. Supplement to Fig. 8 (a–c). Vacuole lysis. Three protrusions/vacuoles can be seen spreading from one cell to another. Shortly after collapse of the protrusion (especially apparent in the lowest protrusion), the membrane signal disappears but the actin signal remains. Phase contrast: lower right corner. Available at http://www.jcb.org/cgi/content/full/146/6/1333/F8/DC1
Figure 8.

Direct observation of vacuolar lysis. (a) A mixed population of cells, some of which are membrane labeled and/or expressing GFP-actin, were observed by simultaneous video microscopy in two fluorescent channels and by phase contrast. A bacterium moved from the lower right cell (with both actin and membrane labeled) into a protrusion into the center (unlabeled) cell. The white box shows the portion of the field in b and c. Bacterial position is indicated by arrows. (b) The double membrane vacuole is apparent in all three channels. (c) 30 s later, the membrane label is gone (third panel), and phase contrast reveals a morphological change from the spherical vacuole to the free, rod-shaped bacterium. A decrease in GFP-actin fluorescence is observed, but the bacterium remains in focus. Bars, 4 μm. Video available at http://www.jcb.org/cgi/content/full/146/6/1333/F8/DC1 (d) In this example (as in 37% of cases examined), membrane dissolution is immediately preceded by a flash of GFP-actin intensity ∼2.5 min after the protrusion collapses to a roughly spherical geometry (at t = 0:00 min). Videos available at http://www.jcb.org/cgi/content/full/146/6/1333/F8/DC1
Video 7. Supplement to Fig. 8 d. Actin flash. Here, a protrusion is formed, undergoes fitful movement, stillness and collapse, followed shortly by a bright flash of actin intensity temporally coincident with average times for vacuole lysis. The membrane was not labeled in this case. Available at http://www.jcb.org/cgi/content/full/146/6/1333/F8/DC1
Results
MDCK Cells Expressing GFP-Actin Provide an Experimentally Tractable System for Study of L. monocytogenes Intercellular Spread
Since any pathogenic relationship requires contributions from both the pathogen and the host, selection of a model host system is important. We chose to study intercellular spread of L. monocytogenes in MDCK cells, whose columnar epithelial morphology makes them ideal subjects both practically (given their extensive cell-cell contacts) and physiologically (since L. monocytogenes initially invades its human host across the columnar epithelium of the intestine). However, bacteria inside these cells are difficult to observe by phase-contrast microscopy, since the cells are tall and contain many intracellular phase-dense structures.
To solve this problem, an MDCK cell line expressing GFP-actin was selected based on its normal cytoskeletal organization and gross morphology (Fig. 1). Examination by Western blot revealed that the amount of GFP-actin expressed was <2% of total monomeric actin (Fig. 1 a), although expression levels varied from cell to cell. Upon cell extraction in buffer containing Triton X-100 (Fig. 1 a, lower blot), GFP-actin was more abundant in the Triton X-100 soluble fraction than the pellet (Fig. 1 a, upper blot) compared with endogenous actin. Nevertheless, we detected GFP-actin incorporation into actin filaments that stained with phalloidin. The level of GFP-actin incorporated into filaments was low enough that it did not appear to disrupt the structure of the filament, but was sufficient to observe actin filament structure and dynamics in living cells (Fig. 1 b).
The GFP-actin enabled simultaneous phase-contrast and fluorescence video microscopic observation of L. monocytogenes–induced actin comet tail formation and movement (Fig. 1 c), and actin-dependent bacterial motility in these cells was similar to that previously described in other cells not expressing GFP-actin (Sanger et al. 1992) (Fig. 1 d) with movement speed averaging 0.137 μm/s (SD = 0.065, n = 48). Using this system, we were able to observe many incidences of intercellular spread, and thereby analyze, for the first time, stages in transfer of bacteria and membrane protrusions between neighboring cells.
Formation of an Intercellular Membrane-bound Protrusion Depends on Permissive Host Cell Structure
The first hypothesized step in intercellular spread is formation of a membrane-bound protrusion into a neighboring cell. We frequently observed this step in infected MDCK cells. Here, L. monocytogenes–containing membrane protrusions are defined as extracellular if formed from a host surface free of cell-cell contacts, and intercellular if formed from one cell and protruding into an adjacent cell. Intercellular protrusions must therefore distend two membranes from the point of their inception, one from the recipient and the other from the donor cell. Only these protrusions permitted the ultimate release of a bacterium into another cell. Intercellular protrusions ranged in length from 3 to 18 μm (average length: 8.5 μm, SD = 4.3, n = 16). Since the host cells were many focal planes deep, intercellular protrusions into neighboring cells were easy to distinguish from those protruding apically into the extracellular medium.
In making a protrusion, the bacterium did not simply distend the lipid bilayer: it pulled the associated cellular structure along with it (Fig. 2, a and b). Adherens junctions, mediated by the transmembrane protein, E-cadherin, line the lateral plasma membrane in the form of puncta in MDCK cells partially polarized by growth on coverslips (Shore and Nelson 1991; Adams et al. 1996). We noted that intercellular protrusions induced by bacteria were lined by E-cadherin puncta with a distribution similar to that on the lateral membranes of cell-cell contacts. This suggests that adherens junctions maintain their geometry in the plasma membrane with respect to one another despite the distension of the membrane during protrusive activity. Some extracellular protrusions displayed similar E-cadherin staining (Fig. 2c and Fig. d); however, extracellular protrusions that did not contain detectable levels of E-cadherin were morphologically indistinguishable from those that did (Fig. 2e and Fig. f), suggesting that E-cadherin on noncontacting membranes does not play an appreciable role in creating the protrusion structure.
Figure 2.
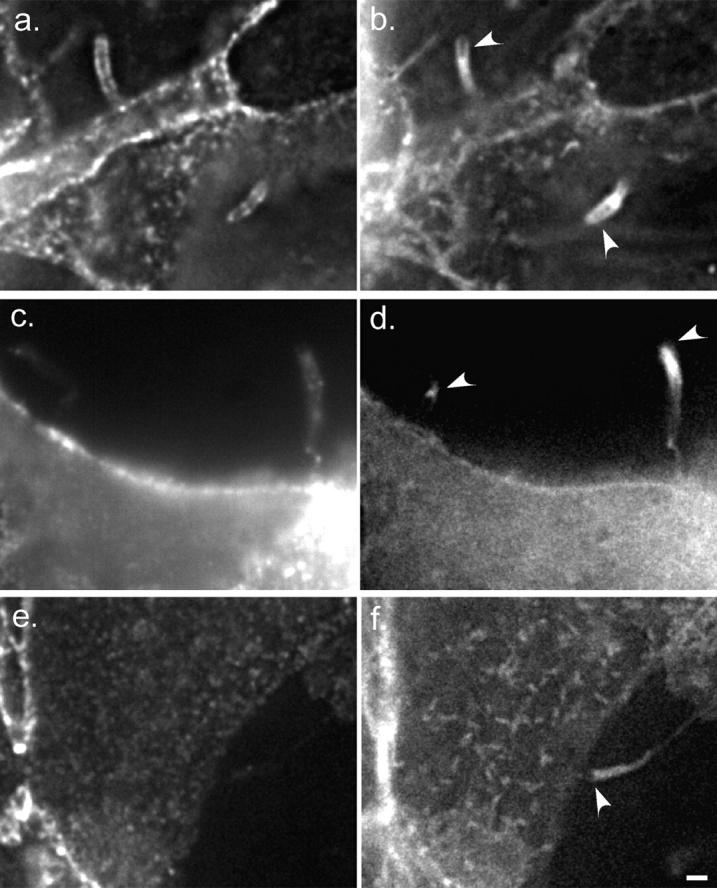
E-cadherin localizes to protrusions but is not required for protrusion structure. Deconvolved sections of fixed cells stained for E-cadherin (a, c, and e) and GFP-actin (b, d, and f). White arrowheads indicate positions of bacteria. (a and b) Two intercellular protrusions. (c and d) Two extracellular protrusions which contain a few E-cadherin puncta. The left-hand protrusion, which contains fewer puncta, passes through several focal planes; only one plane is shown. (e and f) Extracellular protrusions without E-cadherin are morphologically indistinguishable from those with E-cadherin. Bar, 2 μm.
Surprisingly, a protrusion did not form on every occasion that a bacterium was propelled by its actin tail into the plasma membrane (Fig. 3 a). Instead, the bacterium occasionally ricocheted off the membrane and continued to race around the cell. A single bacterium might ricochet off one face of the plasma membrane and immediately form a membrane-bound protrusion from another, demonstrating that the ricochet events were dependent on properties of the host cell membrane, rather than a bacterial deficiency.
We examined cells to learn more about the basis for ricochet versus membrane protrusion events. There did not appear to be particular regions of the cell membrane that exhibited a predilection for a ricochet or protrusion event, and neither the instantaneous velocity nor angle with which the bacterium struck the membrane was predictive of which fate would result. Bacteria moving at rates >0.02 μm/s and striking the membrane at an angle ≥30° were equally likely to have been members of the protrusion or ricochet subsets.
However, the probability of a ricochet was highly dependent upon the age of the MDCK monolayer (Fig. 3 b). The percentage of membrane contacts resulting in ricochets increased with monolayer age. A possible cause of this change could be structural alterations commensurate with development of cell polarity. MDCK cells are known to undergo structural changes during maturation of the monolayer on solid or permeable substrates (Rodriguez-Boulan and Nelson 1989). These include extensive cell-cell contact formation as cells increase their height, and formation of a microtubule lattice oriented vertically with respect to the substrate along basolateral membranes (Grindstaff et al. 1998; Bacallao et al. 1989). It is possible that the change in susceptibility of the plasma membrane to bacteria-induced protrusions is due to changes in the extent of cell-cell adhesion and/or the development of an organized submembranous cytoskeleton.
Intercellular Protrusion Dynamics Are Biphasic
We observed that membrane-bound intercellular protrusions frequently underwent a period of erratic motility (which we termed fitful movement) followed by a period of stillness before their uptake (see next section). 62% (n = 29) of intercellular protrusions exhibited fitful movement in the recipient cell's cytoplasm (Fig. 4, a and b). The remaining 38% exhibited little or no movement throughout their duration. Of the protrusions that underwent fitful movement, 78% moved erratically for 7–15 min (average = 10.3 min, SD = 2.3, n = 15) followed by an additional 10–25 min of stillness. 60% of that population was stalled for 2–3 min before beginning fitful movement (Fig. 4). The remaining protrusions which moved (n = 4) exhibited fitful movement throughout the duration of their time in the protrusion.
This fitful movement is consistent with continued polymerization of actin at the bacterial surface, as most of the motion occurs in a vector along the protrusion's axis of formation (Fig. 4). We plotted the bacterial path as a function of time with respect to axes defined by the three time points which constituted the initial direction of the intrusive pathway as it crossed the cell border (x axis) (Fig. 4 a). We found that 73% (SD = 9, n = 17) of the motility was along the protrusion axis (Fig. 4 c), strongly suggesting that the bacterium was largely responsible for fitful movement of the protrusion.
Extracellular membrane-bound protrusions into free space exhibit rapid erratic movement (Fig. 5 a). When we inhibited the Brownian component of that movement by increasing extracellular fluid viscosity, we observed fitful movement of the extracellular protrusions similar to that of intercellular protrusions (Fig. 5 b). Under these experimental conditions, bacteria moved more slowly and in a directed fashion, occasionally taking steps backward, as intercellular protrusions do, but did not stop moving completely. We monitored these protrusions for motility and found that they displaced an average of 11.1 μm over 10 min (SD = 8.1, n = 10) with an average instantaneous velocity of 0.021 μm/s (SD = 0.018). When host cell ATP production (and therefore actin polymerization) was inhibited by addition of 50 μM 2,4-dinitrophenol and 10 mM 1-deoxyglucose, cytoplasmic bacteria stopped moving in <90 s. Likewise, extracellular protrusions stopped virtually all movement, displacing an average of 1.1 μm over 10 min (SD = 1.5) with an average instantaneous velocity of 0.0018 μm/s (SD = 0.0022; video available at http://www.jcb.org/cgi/content/full/146/6/1333/DC2). 5 min after washing out these inhibitors, the average displacement of the protrusions returned to 10.0 μm over 10 min (SD = 9.6), with average instantaneous velocity 0.016 μm/s (SD = 0.015), which is similar to movements of protrusions in untreated cells (see above).
Figure 5.

Extracellular protrusions exhibit fitful movement. Paths are traced in the adjacent boxes for 5–10 min (each dot = 10 s). (a) Infected cell incubated in the absence of methyl cellulose. Extracellular protrusions appear stubby and thick and move rapidly through many focal planes. (b) Infected cell incubated in 0.5% methyl cellulose, which inhibits Brownian motion. Protrusions are long and slender and are less likely to move out of focus, exhibiting directed motility along the protrusion axis. No new protrusions were formed during either observation. Bar, 10 μm.
Taken together, these experiments demonstrate that the fitful movement of membrane-bound protrusions is most likely due to bacterially directed actin polymerization, though it is possible that the recipient cell may make a minor contribution by tugging on the protrusion. Since polymerization-based movement of protrusions requires ATP, it is probable that cessation of that movement following entry of the protrusion into a recipient cell corresponds to the pinching off and sealing of the donor cell's protrusion membrane. This would prevent further generation of actin-based force as it denies bacterial access to the cellular pool of ATP.
Recipient Cell Dictates L. monocytogenes Protrusion Uptake
Extracellular protrusions were not taken up by nearby cells, even when constrained by them. We captured occasional (n = 4) video sequences of membrane-bound protrusions that lay confined in the intercellular space between two neighboring cells. After >1.5 h observation, they were never taken up by these cells, though they occasionally twitched back and forth along the protrusion axis. Furthermore, we never observed a protrusion that was initially extracellular taken up by a neighboring cell, indicating that classical, actin-based phagocytosis of membrane-bound protrusions does not occur frequently, if at all, in these cells. Since we never observed bacterial escape from extracellular protrusions (n > 300), we concluded that the receiving cell must actively pinch off a protrusion that has intruded into its cytoplasmic space. In other words, the bacterium initially plays the active role, pushing the protrusion directly into the cytoplasm of the neighboring cell, which then takes over the active role to engulf the protrusion.
Approximately 30–40 min (average = 34 min, SD = 7, n = 14) after the start of membrane-bound protrusion into the recipient cell (corresponding to 15–25 min after the cessation of fitful movement), a distinct morphological change in protrusion geometry was apparent in all but one case (Fig. 6). Initially, membrane protrusions in recipient cells were an elongated ovoid in shape. Subsequently the protrusion appeared to shrink, and then snap to a roughly spherical morphology over a period of 30–150 s. Visually, the protrusion appeared to have been released from tension, as if it had been pinched off.
Of the four protrusions that exhibited constant fitful movement, two underwent this geometric collapse after >1 h of observation. The other two failed to collapse even after 90 min.
We did not observe any correlation between entry speed and protrusion length, or entry speed and time before the change in geometry, suggesting that these variables did not depend on the activity of the bacterium (Fig. 7). These results, combined with the properties of fitful movement (see above) lead us to conclude that protrusion uptake occurs in two steps, the timing of which are primarily determined by the recipient host cell. In the first, the supply of ATP to the bacterium is cut off, as the bacterium becomes sealed in a single membrane structure. In the second, which occurs ∼20 min later, tension is released as the second membrane closes off and the protrusion snaps to a spherical shape. Shortly after this second step, the vacuole lyses.
Figure 7.
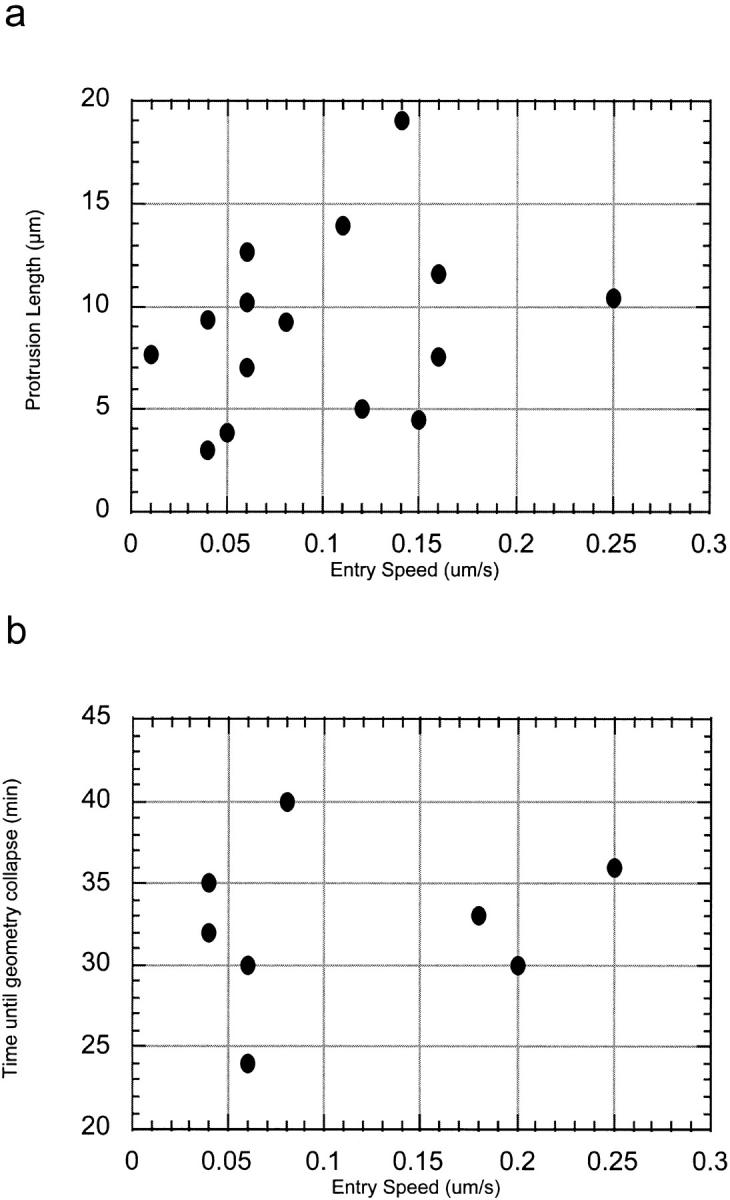
Protrusion length and time to collapse are host dependent. No correlation exists between entry speed and (a) maximum protrusion length and (b) time until protrusion collapse, suggesting that these variables are dependent on the recipient host cell rather than bacterial activity.
Efficient Vacuolar Lysis by the Bacterium Depends on Acidification by the Recipient Cell
Interactions between the membranes supplied by the donor and recipient cells and bacterial lysis of the double membrane vacuole are important steps in intercellular spread, but difficult to observe using the heretofore described system of GFP-actin expressing MDCK cells. Therefore, we added a plasma membrane label to the donor cell. Cells were incubated with TRITC, which covalently and nonspecifically labels membrane proteins on lysine residues, and were then coplated with unlabeled cells. Protrusions in the act of spreading from labeled into unlabeled cells were selected for observation. Simultaneous time–lapse recording of phase contrast and the two fluorescent channels, GFP-actin and TRITC, was performed.
In every case examined (n = 9), the membrane TRITC signal vanished 1–5 min (average = 2.4, SD = 1.4) after the change in geometry, always fading over a time interval <50 s (Fig. 8, a–c). In the presence of bafilomycin A1, which inhibits vacuolar acidification (Bowman et al. 1988), the geometric collapse of the protrusion occurred in 9 out of 10 cases examined, but the membrane TRITC signal remained detectable for at least 1 h in 8 of those 9 cases, at which time we stopped recording. In the 9th case, the signal disappeared after 45 min of observation. These manipulations and observations lead us to conclude that: (a) The internalized membrane protrusion becomes a sealed vacuole; (b) Subsequent acidification of the vacuole is not required for the abrupt change in the geometric shape of the protrusion; and (c) Vacuolar acidification dramatically increases the efficiency of bacterial escape from the double membrane.
Throughout the period after cessation of fitful movement, but preceding the abrupt loss of the membrane TRITC signal, we noticed that the strength of the GFP-actin signal declined (Fig. 8 d). This could represent photobleaching, but could also in part be due to vacuolar acidification, since GFP intensity declines with decreasing pH (Haupts et al. 1998). Coincident with or one frame (20–30 s) before vacuolar lysis, a sudden increase in GFP-actin fluorescence intensity (flash), followed by a significant decrease in intensity, was observed in 37% of cases examined (Fig. 8 d). This flash might be expected if the GFP-actin surrounding the bacterium was freed from the acidified vacuole and returned to a neutral pH, at which time a fraction of it is free to diffuse away into the recipient cell's cytoplasm.
Association of the L. monocytogenes Comet Tail with the Plasma Membrane Stabilizes Actin Filaments
Since we detected a GFP signal for up to 30 min after intercellular spread around the bacterium in recipient cells that did not express detectable levels of GFP-actin, we conclude that a fraction of the GFP-actin from the donor cell depolymerized very slowly after escape from the secondary vacuole. This was unexpected, as actin turnover in L. monocytogenes tails is rapid, with an average half-life of 33 s in PtK2 cells (Theriot et al. 1992). Furthermore, even in highly organized actin structures such as stress fibers in PtK2 cells, individual actin filaments have a half-life of <4 min (Theriot and Mitchison 1991).
We hypothesized that the stabilization of actin around the bacterium is the legacy of the lysed protrusion. Therefore, we examined actin turnover in extracellular and intercellular membrane protrusions. The rate of actin turnover in intercellular and extracellular protrusions was examined by FRAP. For these measurements, cells were grown at medium density in the presence of 0.5% methyl cellulose, which permitted formation of extracellular membrane-bound protrusions and actin polymerization-dependent movement, but inhibited Brownian motion (see above). Neither extracellular (n = 5) nor intercellular protrusions (n = 10) exhibited any measurable recovery of fluorescent actin along the length of the protrusion >10 min of observation after complete photobleaching (27% of photobleached protrusions exhibited some very gradual recovery within 1 μm around the bacterial surface, particularly the tail-associated end, where new polymerization is expected to occur). These results indicate that actin filaments in protrusions, like those in clouds surrounding bacteria that have just escaped from a secondary vacuole, are highly stabilized, with a half-life significantly >10 min. This is consistent with our unpublished observations that bacteria-containing extracellular membrane-bound protrusions persist for >30 min and continue to react with phalloidin along their length, demonstrating the presence of persistent actin filaments, even when the protrusion is extending very slowly.
Bacteria associated with uniform actin clouds in the cytoplasm were also photobleached and their fluorescence recovery measured. Unbleached clouds were observed to be at steady state (their fluorescence intensity and area neither increased nor decreased), demonstrating that the rate of actin polymerization must equal that of depolymerization. Hence FRAP, which measures polymerization rates, allowed us to calculate filament half-life. Unlike clouds surrounding bacteria which have just escaped from the secondary vacuole, filaments in ordinary cytoplasmic clouds turn over at rates close to that in actin tails (average = 45 s, SD = 15, n = 8).
Recovery of Bacterial Motility Is Delayed after Escape from the Secondary Vacuole
Bacteria that had escaped from a secondary vacuole did not recover motility over a period of >30 min of observation (n = 13). After initial invasion, bacteria require 1–3 h to acquire tails and begin movement (Tilney and Portnoy 1989; Theriot, J.A., unpublished observations). This is due in part to the fact that actA expression is not induced until after the bacteria escape from the primary vacuole (Freitag and Jacobs 1999). Therefore, we hypothesized that the delay in the recovery of bacterial movement might be due to inhibition or destruction of existing ActA protein by acidification of the secondary vacuole.
To observe effects of pH on ActA stability, we turned to the well-characterized model of J774 cells (Sun et al. 1990; Smith et al. 1995a; Wadsworth et al., 1999). In these cells (unlike MDCK cells), L. monocytogenes infects with very high efficiency, so newly synthesized ActA protein can be readily detected. Using pulse–chase experiments we found that the ActA protein is cleaved in a pH-dependent manner (Fig. 9) reminiscent of the acid-dependent cleavage of proPC-PLC (see introduction), which is proteolytically activated in the secondary vacuole by the metalloprotease Mpl (Marquis et al. 1997). Cells were pulse-labeled with 35S-methionine for 10 min, followed by a 15-min chase. After the chase, the medium was replaced with a buffer at pH 6.5 (see Materials and Methods). In lanes 3, 6, and 9, 10 μM nigericin was added to allow pH to equilibrate across all membranes. Three bands corresponding to phosphorylated forms of ActA were observed. The ActA was cleaved in the wild-type strain in a pH-dependent manner (lanes 2 and 3), but not in the mpl mutant (lanes 5 and 6). In an Mpl-deficient strain, no cleavage of ActA was observed (lanes 8 and 9). This strongly suggests that Mpl, which is activated upon acidification, mediates ActA proteolysis in the secondary vacuole. This destruction of ActA explains why newly escaped bacteria do not immediately recruit actin from the recipient cell and reinitiate movement in the cytoplasm.
Figure 9.

ActA is proteolytically cleaved upon acidification in an Mpl-dependent manner. J774 cells were infected with the wild-type strain 10403S (lanes 1–3), a mpl mutant (lanes 4–6) and an actA mutant (lanes 7–9). Cells were pulse-labeled with 35S-methionine and chased, and the medium was replaced with a pH 6.5 buffer (for more details see Materials and Methods). In some cases nigericin was added (lanes 3, 6, and 9) to equilibrate pH across all membranes. Position of molecular mass markers is indicated at the left.
To further examine the kinetics of the recovery of motility in epithelial cells, we examined individual infectious foci in MDCK cells fixed at different time points after infection (Fig. 10; see Materials and Methods). Most intercellular spread into secondary cells occurred between 4 and 5 h after infection, with tertiary infections following 2.5 h later at a similar rate (Fig. 10 a). The initial baseline percentage of foci with secondary infections (2–3 h) is probably due to primary infections occurring simultaneously in neighboring cells, as insufficient numbers of bacteria have acquired tails at this time for intercellular spread to have occurred (Fig. 10 b). Interestingly, as only 6.9% of cells are infected at t = 2 h, it is highly improbable (P < 0.01 by chi-squared test) that primary infections of neighboring cells (apparent in 12% of foci) are independent events. This may be due to a sensitization effect. It has been observed that transient calcium spikes occur during L. monocytogenes invasion (Wadsworth et al., 1999) and also during invasion by at least one other invasive bacterial species (Pace et al. 1993). This ion flux could be translated to adjacent cells via gap junctions, thereby sensitizing the cells and increasing the likelihood of bacterial invasion in neighbors.
Figure 10.
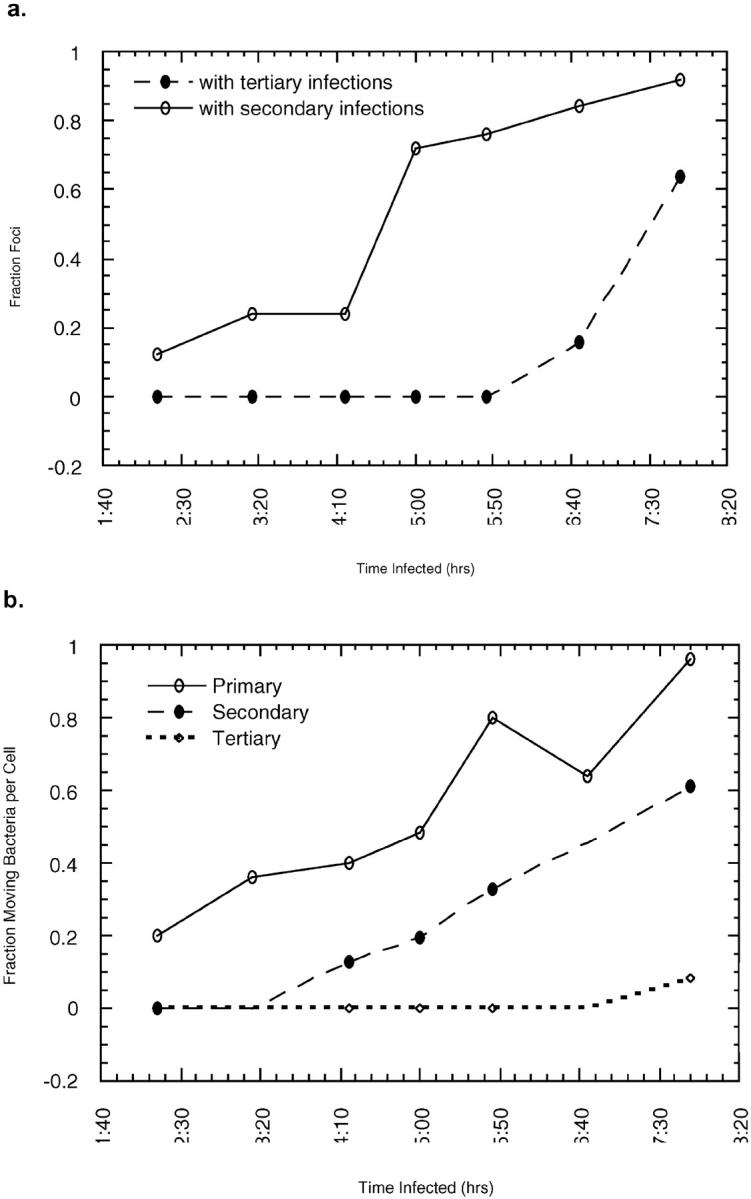
Infectious foci growth and acquisition of motility. (a) Analysis of infectious foci over time reveals that significant intercellular spread to secondary cells occurs between 4 and 5 h. Tertiary cells (two cells removed from the primary cell) begin to be infected at ∼7 h p.i. (b) Increase in the number of motile bacteria occurs before intercellular spread in both primary and secondary cells.
The increase in secondary infections was not followed by immediate recovery of bacterial motility (Fig. 10 b). The fraction of moving bacteria in secondary cells followed a curve almost identical to that obtained from primarily infected cells, but with a shorter lag time and slightly steeper slope. To determine how many bacterial divisions were required before initiation of motility, we counted the total number of bacteria in cells that contained 1–2 bacteria with tails. To exclude the possibility that other, motile bacteria had already left the cell, primary cells with infected neighbors were not counted. For secondarily infected cells, spreading to a tertiary cell or back to the primary cell are equally probable; thus, we excluded secondary cells after 7 h infection, when tertiary infections were first observed. Primary cells contained an average of 6.3 bacteria per cell when 1–2 tails were observed (SD = 2.6, n = 18), suggesting approximately three divisions were required for acquisition of motility. Long-term video observation in PtK2 cells also indicates that an average of three divisions are required before bacterial motility is initiated (Theriot, J.A., unpublished results). Secondarily infected cells, however, required only two bacterial divisions, with an average bacterial population of 4.0 per cell hosting 1–2 bacterial tails (SD = 1.5, n = 28). This difference may reflect the time required to upregulate transcription of ActA after initial infection (Freitag and Jacobs 1999) versus the shorter time required to merely translate and polarize ActA after its proteolytic destruction in the secondary vacuole.
Out of 711 cells observed, only one was inhabited by a single, moving bacterium, and it was unclear whether this bacterium's sister could have recently spread to a neighboring cell. All other cells with tail-associated bacteria possessed at least one other bacterium, strongly suggesting that motility cannot initiate except after at least one bacterial division. The near-absolute dependence of motility on bacterial division is consistent with the model that division is responsible for the establishment of ActA polarity and therefore actin polarity (Tilney et al., 1992; Smith et al. 1996).
Membrane Engulfment by Neighboring Cells Occurs in the Absence of Bacteria
We were surprised to observe that when TRITC-labeled cells were incubated with unlabeled cells in the absence of L. monocytogenes, 5–18% of the unlabeled cells adjacent to labeled cells contained 1–2 TRITC-labeled vacuoles (Fig. 11). Percentages varied greatly according to plating density and seed ratio, and did not appear to increase over time (20–72 h). Vacuoles ranged in diameter up to ∼0.5 μm, and exhibited some saltatory movement inside cells.
Figure 11.
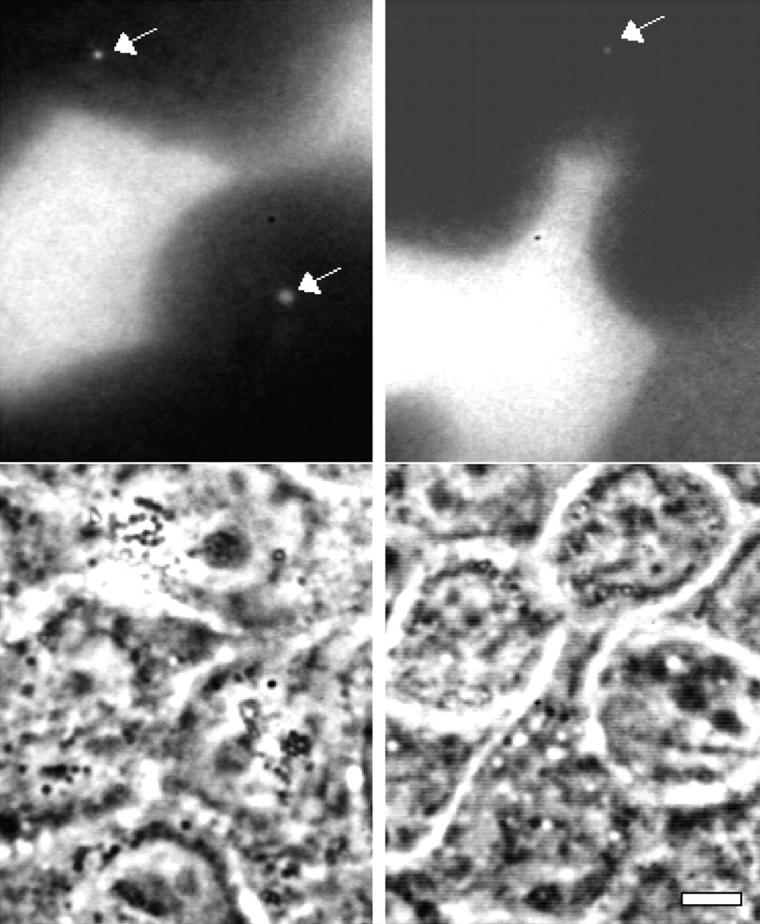
Two examples of TRITC-labeled vacuoles in unlabeled cells adjacent to TRITC-labeled cells. The fluorescent image shows the vacuoles (white arrows), and the corresponding phase contrast image is shown below. Bar, 10 μm.
To confirm that these vacuoles did not merely contain labeled extracellular matrix proteins or products of trypsinization, we colabeled TRITC-dyed cells with the dialkylcarbocyanine DiO, a lipophilic tracer. Multiple DiO-containing vesicles were observed inside unlabeled cells, and a subset of these, usually those largest in size, were also TRITC-labeled.
To our knowledge, this is the first demonstration that MDCK cells engulf portions of their neighbors' membranes. L. monocytogenes intercellular spread may capitalize on this natural paracytophagic behavior.
Discussion
Temporal and Physical Kinetics of Intercellular Spread
Since the columnar epithelium of the intestine is the initial target of L. monocytogenes infection, monolayers of MDCK columnar epithelial cells (albeit derived from kidney) provide a useful model system in which to study pathogenicity at the cellular level. The extensive characterization of this cell line as a polarized epithelium increases its utility. It is clear from our work that the host cell is not merely a passive victim of L. monocytogenes infection, but extensively cooperates in perpetuating the infection as an accomplice in intercellular spread.
Intercellular spread by L. monocytogenes can be described by a stereotyped sequence of contingent events that are highly reproducible. Fig. 12 a shows the timeline for spread of a typical bacterium, using average times for each event. First, the bacterium is propelled by actin-based motility into a membrane-bound protrusion, which undergoes bacterially directed fitful movement for ∼15 min. Cessation of this movement represents cutoff of the bacterium from the donor cell's pool of ATP or possibly other factors. The protrusion's morphology is maintained for some time, then is suddenly released from tension. Within minutes, the vacuole is lysed in an acidification-dependent manner, leaving the bacterium free in the cytoplasm of the recipient cell. Recovery of motility occurs much later, after at least one bacterial division. The process requires approximately one bacterial generation time (∼40–50 min; Brundage et al. 1993); however, bacteria were rarely observed to divide during intercellular spread (in the membrane-bound protrusion or vacuole), so the process may have an inhibitory effect on division. This is consistent with the observation that bacteria deficient in PC-PLC have slower growth unless intercellular spread is blocked by addition of cytochalasin B (Smith et al. 1995a).
Figure 12.

Timeline and model for protrusion uptake. (a) Timeline of a typical bacterium spreading from one cell to another. The initial period of bacterially directed fitful movement lasts ∼15 min and is followed by ∼20 min of stillness, ended by the sudden collapse of the protrusion to a roughly spherical geometry. 3–5 min later, the membrane signal is abrogated, marking vacuole lysis, which is sometimes preceded by a flash of intensity in GFP-actin. (b, 1) The bacterial protrusion, stabilized by E-cadherin, has access to donor cell ATP, and undergoes fitful movement. (2) Movement stops. Tail filaments are partially stabilized by contact with plasma membrane-cytoskeletal linking proteins. (3) Vacuole is formed and acidified, activating PC-PLC and LLO. (4) Membrane is lysed, immediately preceded by actin flash. Remaining filaments are stable and division must occur before movement can resume.
A Two-Step Model for Uptake of Bacterium-containing Membrane-bound Protrusions
Uptake of membrane-bound protrusions has been described as being phagocytic in nature by analogy to phagocytic uptake of bacteria by macrophages (Tilney and Portnoy 1989). However, intercellular spread is fundamentally different, requiring that the cell not only physically enclose the bacterium but also pinch off the donor cell's membrane. Our data suggest that protrusion uptake requires two steps: first, the donor membrane is closed off and second (15–25 min later) the recipient cell's membrane seals (Fig. 12 b).
Since bacteria are never observed to escape from or lyse extracellular protrusions despite many hours of observation, the recipient cell must play an active role in pinching off that protrusion and permitting vacuolar lysis. In this model, the first host-dependent step is the closure of the donor membrane, which corresponds to the cessation of fitful movement (Fig. 12 b, panel 2). This is analogous to the inhibition of fitful movement in extracellular protrusions by host cell ATP depletion.
After the donor membrane is sealed, the protrusion's morphology is maintained under tension by existing cell-cell adherens junctions (Fig. 12 b, panel 2). The host proteasomes, which are known to degrade LLO (Villanueva et al. 1995), will no longer have access to bacterially secreted proteins. We speculate that the recipient cell does not recognize the open-ended distension of its membrane as an endosome, the ovoid protrusion will not be acidified, and the acid-dependent proteins LLO (Geoffroy et al. 1987; Jones et al. 1996) and PC-PLC (Marquis et al. 1997) will not be activated.
Collapse of the protrusion to a roughly spherical geometry marks the closure of the recipient cell membrane (Fig. 12 b, panel 3). Now the cell recognizes the endocytosed vacuole as a target for acidification. How this recognition occurs remains an open question. The sudden drop in pH activates LLO and Mpl; the latter cleaves proPC-PLC to its active form and also cleaves ActA. Vacuolar lysis occurs rapidly (Fig. 12 b, panel 4).
The temporal coupling between protrusion collapse and vacuolar lysis is striking in that they are consistently separated by <5 min. This timing is consistent with biochemical observations that proteolytic activation of PC-PLC occurs very rapidly, with maximal amounts accumulated within 5 min of acidification (Marquis, H., unpublished observations). The observation that inhibition of acidification decreases the efficiency of vacuolar lysis is consistent with the phenotype of bacteria deficient for the acid-activated PC-PLC which exhibit decreased intercellular spread (Vazquez-Boland et al. 1992; Smith et al. 1995a).
It has been shown that cadherin-cadherin interactions are required for intercellular spread of Shigella flexneri (Sansonetti et al. 1994), a Gram-negative pathogen that also undergoes actin-based motility (Bernardini et al. 1989; Pal et al. 1989) and may spread from cell to cell by similar mechanisms. Our observations suggest that this is not a molecular requirement, but rather a need for membrane adhesion that creates a cellular architecture that permits intrusion of a bacterial membrane-bound protrusion into the cytoplasmic space of a neighboring recipient cell.
The two-step model presented above suggests a plausible resolution to the biochemical problem of regulating two separate membrane fusion events. The model is also consistent with the observation that protrusions which exhibit fitful movement throughout their duration are taken up only after extremely long periods of time or not at all, as it posits their donor membrane was not sealed. Further studies will elucidate the mechanism by which this occurs.
Actin Tail Structure and Dynamics Are Modified by Close Association with Host Cell Plasma Membrane
After escape into the cytoplasm of the recipient cell, actin from the donor cell remains associated with the bacterium for up to 30 min or more, indicating that is not turned over. This is in contrast to cytoplasmic bacteria surrounded by uniform actin clouds in which the actin is rapidly turned over at a rate similar to that in comet tails (half-lives of ∼30–45 s). However, slow actin turnover around secondarily spread bacteria is consistent with the actin turnover rate in protrusions, which is much slower than that in comet tails. We suggest that intimate association of the bacterial comet tail with the host's membrane in these protrusions forces interactions between actin and other membrane-associated host proteins that stabilize the actin filaments associated with the bacterium.
Characterization of such interactions is beyond the scope of this report, but this hypothesis is supported by previous data. Ultrastructural examination of cytoplasmic comet tails reveals short, randomly oriented actin filaments that are 0.2–0.3 μm long (Tilney and Portnoy 1989; Tilney et al. 1990). However, tails in protrusions possess two filament populations; one is comprised of short, random filaments, the other is comprised of long (≥1 μm) filaments that are axially oriented along the long axis of the protrusion (Sechi et al. 1997). This second population, which is not found in cytoplasmic tails, may be stabilized by its proximity to membrane-localized proteins responsible for forming cytoskeleton-membrane associations. Ezrin and radixin, members of a family of proteins responsible for stabilizing actin-membrane associations, have been localized to a subset of tails that may correspond to bacteria-containing protrusions (Temm-Grove et al. 1994).
This may also explain the tendency of L. monocytogenes to occasionally slide back out of short extracellular protrusions in PtK2 cells (Sanger et al. 1992; our unpublished data) which do not posses microvillus-like structures. We have never observed this behavior in MDCK cells, which exhibit many microvilli on their apical surface. It is possible that the presence of microvillus-stabilizing proteins in MDCK cells cause them to recognize the bacterial actin tail in close association with the membrane as a structure worthy of stabilization. PtK2 cells are less likely to express or positively regulate the proteins required for such stabilization. The composition and behavior of L. monocytogenes-containing protrusions could prove a useful probe of host actin-membrane interactions.
Membrane-dependent stabilization of actin filaments along the length of the protrusion does not prevent further polymerization at the tip, which is apparently responsible for the fitful movement of extracellular (Fig. 5) and intercellular (Fig. 4 b) protrusions. Similar actin-dependent fitful movement has been described for dendritic spines in neurons (Fischer et al. 1998).
Bacterial Division Is Required before Initiation of Motility
Recovery of motility after intercellular spread is not immediate for two reasons. First, the actin turnover rate is very low in clouds surrounding bacteria that have just spread into a neighboring cell (see above). More importantly, new actin polymerization is inhibited by the acid-dependent cleavage of ActA. Not only does the new bacterium need to replace its ActA, but it must polarize it.
Other studies have reported that ActA is asymmetrically distributed in a gradient along the bacterial surface of bacteria (Kocks et al. 1993). The simplest explanation is that this gradient is generated by bacterial elongation during division combined with a slow turnover rate of ActA of ∼3 h (Moors, M.A., and D.A. Portnoy, manuscript in preparation) that leaves the zone of septation relatively free of ActA. This explanation is consistent with our observations that in cells bacteria only begin moving immediately after division and that stopped bacteria will not start moving again unless they divide (n > 100).
Our data also suggest an explanation for the difference in tail acquisition time between primarily and secondarily infecting bacteria. Transcription of the actA gene followed by translation requires more time than just translation to recover sufficient surface ActA density for motility. However, this alone cannot account for the lag in tail recovery, since bacteria begin to nucleate actin at least one generation before they acquire tails (Tilney and Portnoy 1989). Thus we conclude that bacterial division acts to reorganize the symmetric actin cloud into an asymmetric structure capable of generating unidirectional force.
Who Calls the Shots in Protrusion Uptake: Bacterium or Host?
Our work shows that epithelial cells do not actively internalize extracellular protruding membranes containing bacteria. Instead, cell to cell spread requires that a bacterium actively drive a membrane protrusion to intrude into the cytoplasm of the adjacent cell in order to trigger uptake. After that, the recipient cell takes over, with uptake occurring in two distinct phases, each considerably longer than classical mechanisms of phagocytosis, which normally requires only a few minutes.
It is surprising that an epithelial cell should ingest a membrane-bound protrusion, considering that the only thing that the recipient cells should recognize is the surface of its neighbor. We cannot exclude the possibility of a bacterially produced presentation molecule that lines the external surface of the protrusion and directs the recipient cell to engulf it; however, there is no evidence for such a signal. If such a molecule existed, it would either require posttranslational modification and plasma membrane targeting by the host cell or a multicomponent bacterial secretory system which could insert the signal directly into the host cell membrane during bacterium-membrane contact. The former is unlikely, since it requires secretion in the protrusion but modification and targeting in the cell body, though it is possible the signal is produced in the cytoplasm and then targeted ubiquitously to the host membrane. The latter is unlikely since genetic screens have failed to find a signal, upstream regulatory components, or components of a secretory system with the required properties in L. monocytogenes. It is also possible that the bacterium modifies the interactions of host membrane proteins at the protrusion tip, however, this is unlikely since extracellular membrane-bound protrusions which are confined between cells are not taken up by their neighbors. Finally, unrelated bacterial pathogens, such as S. flexneri, also undergo actin-based motility to spread from cell to cell (Bernardini et al. 1989) and no signal molecule has been found. It is possible that both bacteria evolved independent mechanisms for intercellular spread; however, the simpler explanation is that they are both taking advantage of the same natural host process.
The engulfment and uptake of membrane from one cell into another, as described here for the lateral spread of bacteria, also occurs in several natural processes. One little-understood but well-known example is the formation of dendrites by melanophores to deliver melanin-containing vesicles to numerous nearby keratinocytes (Fitzpatrick et al. 1967). Dendrite formation is actin-dependent (Hirobe 1978; Lacuor et al., 1992), and the required molecules for melanin transfer are unknown. It is possible that this protrusive activity triggers uptake of proffered vesicles by keratinocytes much as L. monocytogenes protrusion triggers uptake by recipient cells.
Another example of plasma membrane exchange includes a set of several cell signaling events in which a transmembrane ligand on the surface of one cell binds to a cognate receptor on an adjacent cell and is internalized. The Drosophila proteins boss and sevenless (Cagan et al. 1992) are internalized in this manner. The uptake of boss–sevenless complexes can be inhibited by mutations in the hook gene, a factor required by the recipient R7 cell of Drosophila ommatidium for such endocytosis (Krämer and Phistry 1996), whose mutation also results in sharply bent bristles (Mohr 1927), a potential cytoskeletal defect. That gene also inhibits endocytosis of the transmembrane ligand delta (Krämer and Phistry 1996), which binds notch (Kooh et al. 1993; Parks et al. 1995). Multivesicular bodies in R7 cells costain for delta and boss (Krämer and Phistry 1996), suggesting that this endocytic mechanism may depend only on either the boss/sevenless or delta/notch interaction, or neither. Similarly, the C. elegans Lag-2 transmembrane ligand, a homologue of delta (Henderson et al. 1994; Tax et al. 1994), can be internalized into adjacent cells via interaction with its receptor Glp-1 (Henderson et al. 1994). Although none of these studies has demonstrated uptake of membrane phospholipids across cell borders, we have shown that both membrane proteins labeled on the surface with TRITC and the lipophilic dye DiO are found inside adjacent, unlabeled cells in membrane-bounded compartments indicating constitutive, albeit rare, engulfment of membranes by adjacent cells.
We also note that epithelial cells frequently take on the role of semi-professional phagocytes to digest their apoptosing neighbors, thereby eliminating the need for macrophages to perform this function (Sulston and Horvitz 1977; Sulston et al., 1983). Very little is known about semi-professional phagocytosis by epithelial cells (for a review, see Platt et al. 1998). At least six genes critical for this process have been identified in C. elegans; three of these mutants also manifest defects in cell migration (Hedgecock et al. 1983; Ellis et al. 1991). One of these genes, ced-5, has been demonstrated to function in extending the membrane of the engulfing cell over the apoptotic cell (Wu and Horvitz 1998), and it is a homologue of DOCK180, a Drosophila melanogaster protein that has been implicated in cell motility and integrin-mediated signaling. When recruited to the membrane, DOCK180 induces cell spreading and, in the presence of EGF, causes cells to take on a highly protrusive, dendritic morphology (Hasegawa et al. 1996). This link between protrusive cytoskeletal activity and the natural process of neighbor consumption of apoptotic cells underscores the possibility that L. monocytogenes intercellular spread could co-opt a natural host process, inducing uptake simply by initiating an actin-stabilized protrusion that intrudes into the cytoplasmic space of a neighboring cell.
The type of membrane engulfment observed in these genetically characterized pigment delivery, signaling, and apoptotic systems is a normal feature of epithelial cell biology, and we suggest the descriptive term paracytophagy (eating of nearby cells) for this widespread behavior. L. monocytogenes, an excellent bacterial cell biologist, appears to have exploited the tendency of epithelial cells to engulf bits of their neighbors to enable its efficient intercellular spread. Video microscopy has enabled us to observe the dynamic physical and temporal constraints under which this host-pathogen relationship has evolved.
Acknowledgments
We gratefully acknowledge Daniel Portnoy for communication of unpublished results and for providing us with bacterial strains. We also thank Sandra McCallum and Raka Mustaphi for helpful comments on the manuscript and thank members of the Theriot and Nelson labs for stimulating discussions throughout the course of this work.
J.R. Robbins was supported by a National Science Foundation Graduate Research Fellowship. This work was supported by National Institutes of Health grants AI3629 (to J.A. Theriot), GM3527 (to W.J. Nelson), and AI42800 (to H. Marquis), and by a fellowship from the David and Lucile Packard Foundation (to J.A. Theriot).
Footnotes
1.used in this paper: CCD, charge-coupled device; DiO, dioctadecyloxacarbocyanine perchlorate; DMF, dimethylformamide; LLO, listeriolysin O; Mpl, metalloprotease; p.i., post-infection; PLC, phospholipase C
✪
The online version of this article contains supplemental material.
References
- Adams C.L., Nelson W.J., Smith S.J. Quantitative analysis of cadherin-catenin-actin reorganization during development of cell-cell adhesion. J. Cell Biol. 1996;135:1899–1911. doi: 10.1083/jcb.135.6.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpuche Aranda C.M., Swanson J.A., Loomis W.P., Miller S.I. Salmonella typhimurium activates virulence gene transcription within acidified macrophage phagosomes. Proc. Natl. Acad. Sci. USA. 1992;89:10079–10083. doi: 10.1073/pnas.89.21.10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacallao R., Antony C., Dotti C., Karsenti E., Stelzer E.H., Simons K. The subcellular organization of Madin-Darby canine kidney cells during the formation of a polarized epithelium. J. Cell Biol. 1989;109:2817–2832. doi: 10.1083/jcb.109.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth A.I., Pollack A.L., Altschuler Y., Mostov K.E., Nelson W.J. NH2-terminal deletion of β-catenin results in stable colocalization of mutant β-catenin with adenomatous polyposis coli protein and altered MDCK cell adhesion. J. Cell Biol. 1997;136:693–706. doi: 10.1083/jcb.136.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini M.L., Mounier J., d'Hauteville H., Coquis-Rondon M., Sansonetti P.J. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. USA. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielecki J., Youngman P., Connelly P., Portnoy D.A. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature. 1990;345:175–176. doi: 10.1038/345175a0. [DOI] [PubMed] [Google Scholar]
- Bishop D.K., Hinrichs D.J. Adoptive transfer of immunity to Listeria monocytogenes. The influence of in vitro stimulation on lymphocyte subset requirements. J. Immunol. 1987;139:2005–2009. [PubMed] [Google Scholar]
- Bowman E.J., Siebers A., Altendorf K. Bafilomycinsa class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage R.A., Smith G.A., Camilli A., Theriot J.A., Portnoy D.A. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc. Natl. Acad. Sci. USA. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagan R.L., Kramer H., Hart A.C., Zipursky S.L. The bride of sevenless and sevenless interactioninternalization of a transmembrane ligand. Cell. 1992;69:393–399. doi: 10.1016/0092-8674(92)90442-f. [DOI] [PubMed] [Google Scholar]
- Cameron L.A., Footer M.J., van Oudenaarden A., Theriot J.A. Motility of ActA protein-coated microspheres driven by actin polymerization. Proc. Natl. Acad. Sci. USA. 1999;96:4908–4913. doi: 10.1073/pnas.96.9.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier M.F., Laurent V., Santolini J., Melki R., Didry D., Xia G.X., Hong Y., Chua N.H., Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnoverimplication in actin-based motility. J. Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabiri G.A., Sanger J.M., Portnoy D.A., Southwick F.S. Listeria monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc. Natl. Acad. Sci. USA. 1990;87:6068–6072. doi: 10.1073/pnas.87.16.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R.E., Jacobson D.M., Horvitz H.R. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans . Genetics. 1991;129:79–94. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M., Kaech S., Knutti D., Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick T.B., Miyamoto M., Ishikawa K. The evolution of concepts of melanin biology. Arch. Dermatol. 1967;96:305–323. [PubMed] [Google Scholar]
- Freitag N.E., Jacobs K.E. Examination of Listeria monocytogenes intracellular gene expression by using the green fluorescent protein of Aequorea victoria . Infect. Immun. 1999;67:1844–1852. doi: 10.1128/iai.67.4.1844-1852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard J.L., Berche P., Mounier J., Richard S., Sansonetti P. In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect. Immun. 1987;55:2822–2829. doi: 10.1128/iai.55.11.2822-2829.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy C., Gaillard J.L., Alouf J.E., Berche P. Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes . Infect. Immun. 1987;55:1641–1646. doi: 10.1128/iai.55.7.1641-1646.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiffenberg L., Goebel W., Kim K.S., Weiglein I., Bubert A., Engelbrecht F., Stins M., Kuhn M. Interaction of Listeria monocytogenes with human brain microvascular endothelial cellsInlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect. Immun. 1998;66:5260–5267. doi: 10.1128/iai.66.11.5260-5267.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindstaff K.K., Bacallao R.L., Nelson W.J. Apiconuclear organization of microtubules does not specify protein delivery from the trans-Golgi network to different membrane domains in polarized epithelial cells. Mol. Biol. Cell. 1998;9:685–699. doi: 10.1091/mbc.9.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H., Kiyokawa E., Tanaka S., Nagashima K., Gotoh N., Shibuya M., Kurata T., Matsuda M. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol. Cell. Biol. 1996;16:1770–1776. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupts U., Maiti S., Schwille P., Webb W.W. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 1998;95:13573–13578. doi: 10.1073/pnas.95.23.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock E.M., Sulston J.E., Thomson J.N. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans . Science. 1983;220:1277–1279. doi: 10.1126/science.6857247. [DOI] [PubMed] [Google Scholar]
- Henderson S.T., Gao D., Lambie E.J., Kimble J. lag-2 may encode a signaling ligand for the GLP-1 and LIN-12 receptors of C. elegans . Development. 1994;120:2913–2924. doi: 10.1242/dev.120.10.2913. [DOI] [PubMed] [Google Scholar]
- Hirobe T. Stimulation of dendritogenesis in the epidermal melanocytes of newborn mice by melanocyte-stimulating hormone. J. Cell Sci. 1978;33:371–383. doi: 10.1242/jcs.33.1.371. [DOI] [PubMed] [Google Scholar]
- Jones S., Preiter K., Portnoy D.A. Conversion of an extracellular cytolysin into a phagosome-specific lysin which supports the growth of an intracellular pathogen. Mol. Microbiol. 1996;21:1219–1225. doi: 10.1046/j.1365-2958.1996.00074.x. [DOI] [PubMed] [Google Scholar]
- Kocks C., Hellio R., Gounon P., Ohayon H., Cossart P. Polarized distribution of Listeria monocytogenes surface protein ActA at the site of directional actin assembly. J. Cell Sci. 1993;105:699–710. doi: 10.1242/jcs.105.3.699. [DOI] [PubMed] [Google Scholar]
- Kocks C., Marchand J.B., Gouin E., d'Hauteville H., Sansonetti P.J., Carlier M.F., Cossart P. The unrelated surface proteins ActA of Listeria monocytogenes and IcsA of Shigella flexneri are sufficient to confer actin-based motility on Listeria innocua and Escherichia coli respectively. Mol. Microbiol. 1995;18:413–423. doi: 10.1111/j.1365-2958.1995.mmi_18030413.x. [DOI] [PubMed] [Google Scholar]
- Kooh P.J., Fehon R.G., Muskavitch M.A. Implications of dynamic patterns of Delta and Notch expression for cellular interactions during Drosophila development. Development. 1993;117:493–507. doi: 10.1242/dev.117.2.493. [DOI] [PubMed] [Google Scholar]
- Krämer H., Phistry M. Mutations in the Drosophila hook gene inhibit endocytosis of the boss transmembrane ligand into multivesicular bodies. J. Cell Biol. 1996;133:1205–1215. doi: 10.1083/jcb.133.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacour J.P., Gordon P.R., Eller M., Bhawan J., Gilchrest B.A. Cytoskeletal events underlying dendrite formation by cultured pigment cells. J. Cell. Physiol. 1992;151:287–299. doi: 10.1002/jcp.1041510210. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Marquis H., Doshi V., Portnoy D.A. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect. Immun. 1995;63:4531–4534. doi: 10.1128/iai.63.11.4531-4534.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis H., Goldfine H., Portnoy D.A. Proteolytic pathways of activation and degradation of a bacterial phospholipase C during intracellular infection by Listeria monocytogenes . J. Cell Biol. 1997;137:1381–1392. doi: 10.1083/jcb.137.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs, J.A., and W.J. Nelson. 1998. Epithelial cell polarity development: roles for the membrane-cytoskeleton and cell adhesion. In Principles of Medical Biology. Vol. 11. JAI Press. 69–87.
- Mohr O. The second chromosome recessive hook bristles in Drosophila melanogaster . Hereditas. 1927;9:169–179. [Google Scholar]
- Mounier J., Ryter A., Coquis-Rondon M., Sansonetti P.J. Intracellular and cell-to-cell spread of Listeria monocytogenes involves interaction with F-actin in the enterocyte-like cell line Caco-2. Infect. Immun. 1990;58:1048–1058. doi: 10.1128/iai.58.4.1048-1058.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace J., Hayman M.J., Galan J.E. Signal transduction and invasion of epithelial cells by S. typhimurium . Cell. 1993;72:505–514. doi: 10.1016/0092-8674(93)90070-7. [DOI] [PubMed] [Google Scholar]
- Pal T., Newland J.W., Tall B.D., Formal S.B., Hale T.L. Intracellular spread of Shigella flexneri associated with the kcpA locus and a 140-kilodalton protein. Infect. Immun. 1989;57:477–486. doi: 10.1128/iai.57.2.477-486.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parida S.K., Domann E., Rohde M., Muller S., Darji A., Hain T., Wehland J., Chakraborty T. Internalin B is essential for adhesion and mediates the invasion of Listeria monocytogenes into human endothelial cells. Mol. Microbiol. 1998;28:81–93. doi: 10.1046/j.1365-2958.1998.00776.x. [DOI] [PubMed] [Google Scholar]
- Parks A.L., Turner F.R., Muskavitch M.A. Relationships between complex Delta expression and the specification of retinal cell fates during Drosophila eye development. Mech. Dev. 1995;50:201–216. doi: 10.1016/0925-4773(94)00336-l. [DOI] [PubMed] [Google Scholar]
- Platt N., da Silva R.P., Gordon S. Recognizing deaththe phagocytosis of apoptotic cells. Trends Cell Biol. 1998;8:365–372. doi: 10.1016/s0962-8924(98)01329-4. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Boulan E., Nelson W.J. Morphogenesis of the polarized epithelial cell phenotype. Science. 1989;245:718–725. doi: 10.1126/science.2672330. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J., Agnew B.J., Abe H., Bamburg J.R., Mitchison T.J. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J. Cell Biol. 1997;136:1323–1332. doi: 10.1083/jcb.136.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon E.D., Leslie R.J., Saxton W.M., Karow M.L., McIntosh J.R. Spindle microtubule dynamics in sea urchin embryosanalysis using a fluorescein-labeled tubulin and measurements of fluorescence redistribution after laser photobleaching. J. Cell Biol. 1984;99:2165–2174. doi: 10.1083/jcb.99.6.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger J.M., Sanger J.W., Southwick F.S. Host cell actin assembly is necessary and likely to provide the propulsive force for intracellular movement of Listeria monocytogenes . Infect. Immun. 1992;60:3609–3619. doi: 10.1128/iai.60.9.3609-3619.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansonetti P.J., Mounier J., Prevost M.C., Mege R.M. Cadherin expression is required for the spread of Shigella flexneri between epithelial cells. Cell. 1994;76:829–839. doi: 10.1016/0092-8674(94)90358-1. [DOI] [PubMed] [Google Scholar]
- Sechi A.S., Wehland J., Small J.V. The isolated comet tail pseudopodium of Listeria monocytogenesa tail of two actin filament populations, long and axial and short and random. J. Cell Biol. 1997;137:155–167. doi: 10.1083/jcb.137.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore E.M., Nelson W.J. Biosynthesis of the cell adhesion molecule uvomorulin (E-cadherin) in Madin-Darby canine kidney epithelial cells. J. Biol. Chem. 1991;266:19672–19680. [PubMed] [Google Scholar]
- Smith G.A., Marquis H., Jones S., Johnston N.C., Portnoy D.A., Goldfine H. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread Infect. Immun. 63 1995. 4231 4237a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G.A., Portnoy D.A., Theriot J.A. Asymmetric distribution of the Listeria monocytogenes ActA protein is required and sufficient to direct actin-based motility Mol. Microbiol. 17 1995. 945 951b [DOI] [PubMed] [Google Scholar]
- Smith G.A., Theriot J.A., Portnoy D.A. The tandem repeat domain in the Listeria monocytogenes ActA protein controls the rate of actin-based motility, the percentage of moving bacteria, and the localization of vasodilator-stimulated phosphoprotein and profilin. J. Cell Biol. 1996;135:647–660. doi: 10.1083/jcb.135.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm A.M., Dismukes W.E., Simmons B.P., Cobbs C.G., Elliott A., Budrich P., Harmon J. Listeriosis in renal transplant recipientsreport of an outbreak and review of 102 cases. Rev. Infect. Dis. 1982;4:665–682. doi: 10.1093/clinids/4.3.665. [DOI] [PubMed] [Google Scholar]
- Sulston J.E., Horvitz H.R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans . Dev. Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Sun A.N., Camilli A., Portnoy D.A. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect. Immun. 1990;58:3770–3778. doi: 10.1128/iai.58.11.3770-3778.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tax F.E., Yeargers J.J., Thomas J.H. Sequence of C. elegans lag-2 reveals a cell-signalling domain shared with Delta and Serrate of Drosophila . Nature. 1994;368:150–154. doi: 10.1038/368150a0. [DOI] [PubMed] [Google Scholar]
- Temm-Grove C.J., Jockusch B.M., Rohde M., Niebuhr K., Chakraborty T., Wehland J. Exploitation of microfilament proteins by Listeria monocytogenesmicrovillus-like composition of the comet tails and vectorial spreading in polarized epithelial sheets. J. Cell Sci. 1994;107:2951–2960. doi: 10.1242/jcs.107.10.2951. [DOI] [PubMed] [Google Scholar]
- Theriot J.A., Mitchison T.J. Actin microfilament dynamics in locomoting cells. Nature. 1991;352:126–131. doi: 10.1038/352126a0. [DOI] [PubMed] [Google Scholar]
- Theriot J.A., Mitchison T.J., Tilney L.G., Portnoy D.A. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature. 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- Theriot J.A., Rosenblatt J., Portnoy D.A., Goldschmidt-Clermont P.J., Mitchison T.J. Involvement of profilin in the actin-based motility of L. monocytogenes in cells and in cell-free extracts. Cell. 1994;76:505–517. doi: 10.1016/0092-8674(94)90114-7. [DOI] [PubMed] [Google Scholar]
- Tilney L.G., Portnoy D.A. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes . J. Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney L.G., Connelly P.S., Portnoy D.A. Actin filament nucleation by the bacterial pathogen, Listeria monocytogenes . J. Cell Biol. 1990;111:2979–2988. doi: 10.1083/jcb.111.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Boland J.A., Kocks C., Dramsi S., Ohayon H., Geoffroy C., Mengaud J., Cossart P. Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread. Infect. Immun. 1992;60:219–230. doi: 10.1128/iai.60.1.219-230.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva M.S., Sijts A.J., Pamer E.G. Listeriolysin is processed efficiently into an MHC class I-associated epitope in Listeria monocytogenes-infected cells. J. Immunol. 1995;155:5227–5233. [PubMed] [Google Scholar]
- Wadsworth S.J., Goldfine H. Listeria monocytogenes phospholipase C-dependent calcium signaling modulates bacterial entry into J774 macrophage-like cells. Infect. Immun. 1999;67:1770–1778. doi: 10.1128/iai.67.4.1770-1778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.C., Horvitz H.R. C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature. 1998;392:501–504. doi: 10.1038/33163. [DOI] [PubMed] [Google Scholar]
- Yoshimori T., Yamamoto A., Moriyama Y., Futai M., Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- Zhukarev V., Ashton F., Sanger J.M., Sanger J.W., Shuman H. Organization and structure of actin filament bundles in Listeria-infected cells. Cell Motil. Cytoskeleton. 1995;30:229–246. doi: 10.1002/cm.970300307. [DOI] [PubMed] [Google Scholar]