

Abstract
Most restriction endonucleases use Mg2+ to hydrolyze phosphodiester bonds at specific DNA sites. We show here that BfiI, a metal-independent restriction enzyme from the phospholipase D superfamily, catalyzes both DNA hydrolysis and transesterification reactions at its recognition site. In the presence of alcohols such as ethanol or glycerol, it attaches the alcohol covalently to the 5′ terminus of the cleaved DNA. Under certain conditions, the terminal 3′-OH of one DNA strand can attack the target phosphodiester bond in the other strand to create a DNA hairpin. Transesterification reactions on DNA with phosphorothioate linkages at the target bond proceed with retention of stereoconfiguration at the phosphorus, indicating, uniquely for a restriction enzyme, a two-step mechanism. We propose that BfiI first makes a covalent enzyme–DNA intermediate, and then it resolves it by a nucleophilic attack of water or an alcohol, to yield hydrolysis or transesterification products, respectively.
Keywords: alcoholysis, BfiI, covalent intermediate, restriction endonuclease, stereochemistry
In the presence of Mg2+, type II restriction endonucleases (REases) hydrolyze specific phosphodiester bonds in DNA to yield 5′-phosphate and 3′-hydroxyl termini (1). The stereochemical pathway for phosphodiester hydrolysis by four such enzymes, EcoRI, EcoRV, SfiI, and HpaII (2–4), have been determined. All proceed with inversion of configuration at the scissile phosphate, which implies an odd number of chemical steps in the hydrolytic reaction, and it is most simply accounted for by a direct in-line displacement of the 3′-leaving group by water (5–7). High-resolution structures are available for many more REases than have been analyzed stereochemically (1). Their active sites are generally similar, with several carboxylates to act as ligands for Mg2+ in spatially equivalent positions (1). All such enzymes probably act in the same way, by using water to attack the scissile phosphate, to invert its configuration.
Endonucleases from the transposase-retroviral integrase family, like MuA or HIV-integrase, catalyze at a single active site two sets of reactions on DNA (4, 8, 9). The first, the endonucleolytic cleavage of the 3′ end of the transposon or viral DNA, is mechanistically similar to the hydrolysis reactions of REases. The second, termed DNA strand transfer, is a single-step transesterification in which the newly liberated 3′-OH group attacks and becomes linked to a phosphate group in the target DNA (9, 10).
We ask here whether the BfiI REase can catalyze transesterification reactions in addition to DNA hydrolysis. BfiI cleaves DNA at specified positions downstream from an asymmetric recognition sequence (11). Unlike other REases, it functions without metal ions (12). The N-terminal half of BfiI (13, 14) is similar to Nuc, an EDTA-resistant nuclease from Salmonella typhimurium (15, 16) that belongs to the phospholipase D (PLD) superfamily. The PLD superfamily is a diverse group of proteins that includes phospholipases, phospholipid synthases, bacterial toxins, phosphodiesterases (PDEs), and nucleases (17, 18). The PLD enzymes catalyze phosphoryl transfer reactions from a donor to an acceptor, to either water or an alcohol (19). The phospholipases and the nucleases in this family use water to hydrolyze phosphodiester bonds, whereas the phospholipid synthases use an alcohol in a transesterification reaction to make a new phosphodiester.
PLD proteins are characterized by a conserved sequence motif called HXK (17, 19), and they use the conserved histidine in the motif for nucleophilic attack on the target phosphorus. They follow a two-step mechanism (18, 20): in the first step, they attack the phosphorus to form a covalent phosphohistidine intermediate and release the alcohol; in the second, the intermediate is attacked by either another alcohol or water to yield, respectively, the transesterification or the hydrolysis product (21, 22).
Transesterification reactions have, however, yet to be observed with any PLD enzyme acting on DNA, either tyrosyl-DNA PDE (18, 23) or the nucleases (15) in this superfamily. Here we show that alcohols can act as nucleophiles in the DNA cleavage reaction catalyzed by BfiI to yield transesterification products. Moreover, we demonstrate that the 3′-hydroxyl group of DNA can act as an intramolecular nucleophile to generate a DNA hairpin. Finally, we show from the stereochemistry of transesterification that, unlike other restriction enzymes, the reaction pathway of BfiI follows a two-step mechanism.
Results
DNA Alcoholysis by BfiI.
BfiI has a single active site, which it uses sequentially to cut both DNA strands (24, 25). It first cuts the bottom strand 4 nt downstream from its recognition sequence, 5′-ACTGGG-3′. Then it slowly cuts the top strand at varied positions 5–7 nt downstream from the site (25). To study the reactions of BfiI, a minimal substrate, 14/15 (Table 1 and Fig. 1A), was constructed by annealing 14- and 15-nt oligonucleotides for its top and bottom strands, respectively. The duplex contains the recognition site for BfiI, but it can only be cleaved in the bottom strand because it lacks the scissile phosphodiester bonds in the top strand. The 15-nt bottom strand was radiolabeled at its 3′ end. In single-turnover reactions in aqueous buffer, BfiI cut this strand at a relatively rapid rate, 2 s−1 (see below and Fig. 4B), to give the 13-nt (radiolabeled) product with a 5′-phosphate and a dinucleotide with a 3′-OH; no other products were detected (Fig. 1B).
Table 1.
Oligonucleotide duplexes and yields of hairpin DNA products in BfiI reactions
| Duplex | Oligonucleotide duplex sequence* | Max. hairpin yield, % |
|---|---|---|
| 14/15 | 5′-AGCACTGGGCTGC–T-3′ | 0 |
| 3′-TCGTGACCCGACGpAC-5′ | ||
| 25C/15 | 5′-AGCACTGGGCTGC–TGAACTAGTTCA-3′ | 38 ± 2 |
| 3′-TCGTGACCCGACGpAC-5′ | ||
| 25CddA/15† | 5′-AGCACTGGGCTGC–TGAACTAGTTCddA-3′ | 0 |
| 3′-TCGTGACCCGACGpAC-5′ | ||
| 15/15 | 5′-AGCACTGGGCTGC–TG-3′ | 0 |
| 3′-TCGTGACCCGACGpAC-5′ | ||
| 16/15 | 5′-AGCACTGGGCTGC–TGA-3′ | 5.5 ± 1 |
| 3′-TCGTGACCCGACGpAC-5′ | ||
| 17/15 | 5′-AGCACTGGGCTGC–TGAG-3′ | 14 ± 1 |
| 3′-TCGTGACCCGACGpAC-5′ | ||
| 25/15 | 5′-AGCACTGGGCTGC–TGAACTGTGCTG-3′ | 28 ± 3 |
| 3′-TCGTGACCCGACGpAC-5′ | ||
| 29/15 | 5′-AGCACTGGGCTGC–TGAACTGTGCTGCGGC-3′ | 1.4 ± 0.5 |
| 3′-TCGTGACCCGACGpAC-5′ | ||
| 25C/15(S)Rp‡ | 5′-AGCACTGGGCTGC–TGAACTAGTTCA-3′ | 36 ± 5 |
| 3′-TCGTGACCCGACGsAC-5′ | ||
| 25C/15(S)Sp§ | 5′-AGCACTGGGCTGC-TGAACTAGTTCA-3′ | 37 ± 5 |
| 3′-TCGTGACCCGACGsAC-5′ |
*The BfiI recognition sequence is underlined, and the scissile phosphodiester bond in the bottom DNA strand is noted as p. Unless noted otherwise, all duplexes carried a radiolabel at the 3′ terminus of the bottom strand.
†The 3′-terminal nucleoside of the top strand of 25CddA/15 is dideoxyadenosine (ddA).
‡The 25C/15(S)Rp duplex contains an Rp–PTO linkage at the scissile position of the bottom strand (marked s).
§The 25C/15(S)Sp duplex contains an Sp–PTO linkage at the scissile position of the bottom strand (marked s).
Fig. 1.

DNA alcoholysis by BfiI. (A) Scheme for the hydrolysis and alcoholysis reactions of BfiI on the 14/15 duplex (for the nucleotide sequence, see Table 1). The star marks the 3′ radiolabel on the bottom strand. (B) Samples from reactions of BfiI on 14/15 in aqueous buffer (lane marked H2O) or in the presence of an alcohol at the concentration indicated above each lane were subjected to denaturing PAGE and analyzed by PhosphorImager. Lanes marked S and P contain the intact substrate and the chemically synthesized 5′-phosphorylated hydrolysis product of 15 and 13 nt, respectively.
Fig. 4.

BfiI hydrolysis of the PTO-modified and unmodified DNA. (A) Representations of the 14/15 substrates (Table 1) with either Rp–or Sp–PTO linkages at the site of bottom-strand cleavage by BfiI are shown. (B) Extents of DNA cleavage were measured in single-turnover reactions of BfiI (see Materials and Methods) on unmodified DNA (open diamonds) or PTO-substituted DNA (open circles for the Rp and crosses for the Sp derivatives). The solid lines are the best fits to single exponentials to the data, which gave rate constants of 2.0 ± 0.3 s−1 for the unmodified DNA and 0.0034 ± 0.0003 s−1 for the Rp and the Sp substrates.
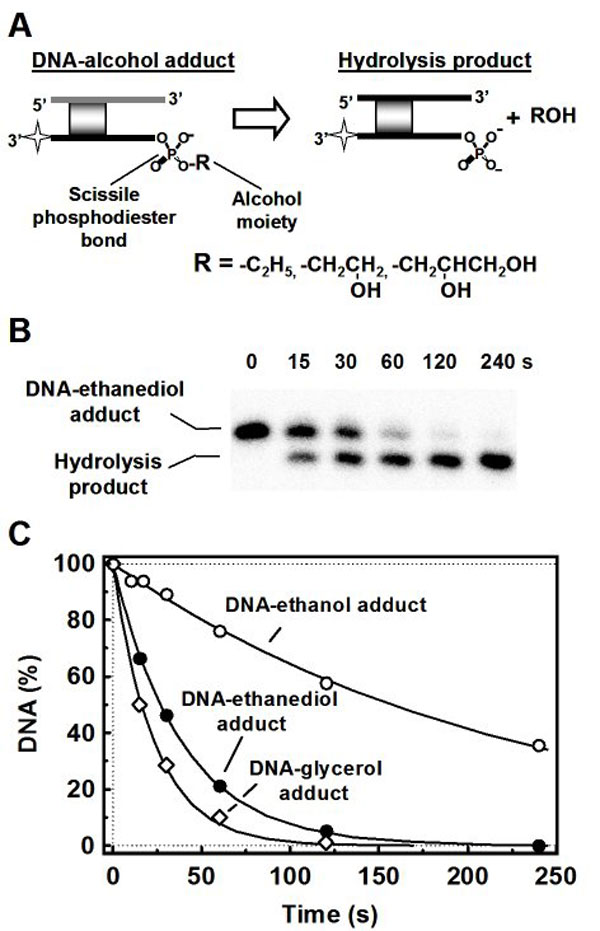
When BfiI reactions on 14/15 were conducted in the presence of various alcohols, additional products were observed by gel electrophoresis, with mobilities between those of the 15-nt substrate and the 13-nt product (Fig. 1B). The mobilities of the extra bands varied inversely with the molecular mass of the alcohol: Tris gave the slowest extra band and ethanol the fastest, with glycerol and 1,2-ethanediol at intermediate values. In all cases, the yield of extra product increased proportionally with the alcohol concentration (Fig. 1B). The additional species generated during DNA cleavage in the presence of alcohols may therefore be transesterification products formed by the transfer of the scissile phosphate to the alcohol, rather than to water, to leave the alcohol attached to the 5′-phosphate of the 13-nt product. This reaction was confirmed by mass spectrometry, which revealed two 13-nt products from the reaction with each alcohol: one with the same mass as the hydrolysis product and one with an increased mass. In each case, the increase in mass matched the alcohol in question [see supporting information (SI) Fig. 7].
The additional products were also tested with calf intestine alkaline phosphatase (CIAP), which cleaves phosphomonoesters, and with polynucleotide kinase (PNK), which uses ATP to phosphorylate 5′-OH groups. The hydrolysis product was dephosphorylated by CIAP, as expected for a DNA with a 5′-phosphate, but the extra products formed in the presence of the alcohols were all resistant to CIAP, so they lack the 5′-monoester (see SI Fig. 7). Neither the hydrolysis product nor the adduct formed with ethanol was phosphorylated by PNK, which shows that they lack free 5′-OH groups. In contrast, the glycerol, the 1,2-ethanediol, and the Tris adducts were all phosphorylated by PNK (see SI Fig. 7). These three alcohols possess two or more OH groups, so they can presumably be modified by PNK at their free OH group(s) in the corresponding DNA–alcohol adduct, as noted in ref. 9. Subsequent treatment of the phosphorylated adducts with CIAP removed the phosphate group.
A transesterification reaction that leaves an alcohol on the 5′-phosphate of the cleaved DNA creates a novel phosphodiester bond, which may in turn be cleaved by BfiI. When challenged with BfiI, the additional products were cleaved to the normal product with a 5′-phosphate, albeit at rates that were ≥40-fold slower than the native phosphodiester in DNA (see SI Fig. 8).
Taken together, the above data show that BfiI generates transesterification products during DNA hydrolysis in the presence of alcohols.
Hairpin Synthesis by BfiI.
The BfiI REase may therefore be able to catalyze transesterification reactions between two DNA molecules, resulting in strand transfer. It is possible that if the top strand carries a protruding OH group at its 3′ terminus, that OH group could act as a nucleophile to attack the scissile phosphodiester in the bottom strand to create a DNA hairpin (Fig. 2A). To test this possibility, the reactions of BfiI were studied on an oligoduplex substrate, 25C/15, that has in its top strand a 10-nt self-complementary extension terminating with a 3′-OH group (Table 1).
Fig. 2.

DNA hairpin formation. (A) A scheme for the transesterification and hydrolysis reactions of BfiI on the 3′-tailed duplex, 25C/15 (Table 1) is shown. The star marks the 3′ radiolabel on the bottom strand. (B) Samples from reactions of BfiI on 25C/15 were subjected to denaturing PAGE and analyzed by PhosphorImager. Lanes marked M contain the intact 15-nt substrate (left) or chemically synthesized hydrolysis and hairpin products (right). (C) Quantitative data from the gel in B: open circles, intact substrate; filled circles, hydrolysis product; open diamonds, hairpin DNA.
The reaction of BfiI on 25C/15, 3′-radiolabled in the bottom strand as before, yielded two radiolabeled products (Fig. 2B). In addition to the expected 13-nt product from the cleavage of the bottom strand, an extra band was observed by denaturing PAGE that migrated much more slowly than the 15-nt substrate (Fig. 2B). The extra band was unaffected by proteolysis (data not shown), so the low mobility of this DNA is not the result of covalent attachment of BfiI protein.
The low-mobility band could be the DNA hairpin that would be generated by joining the 3′-OH of the top strand of the DNA to the scissile phosphate in the bottom strand (Fig. 2A). Because hairpin formation requires a 3′-OH in the top strand, a substrate containing a dideoxynucleoside at the 3′ end of the top strand should be unable to support hairpin formation. When the 3′-terminal dA on the top strand of 25C/15 was replaced by ddA (the substrate 25CddA/15), none of the low-mobility product was formed (Table 1). Maxam–Gilbert sequencing verified that the low-mobility species is indeed the postulated hairpin (Fig. 3). The sequence of the low-mobility product matched the bottom strand of the DNA substrate from its 3′ end up to the scissile phosphate; thereafter, it matched the top strand of the substrate from its 3′ to its 5′ terminus. Furthermore, the sequencing pattern of the BfiI-derived hairpin was identical to that of the chemically synthesized hairpin (Fig. 3).
Fig. 3.

Maxam–Gilbert sequencing. (A) A scheme for hairpin formation in the reaction of BfiI on 25C/15 is shown. The asterisk (∗) marks the 33P label at the 5′ end of the top strand. (B) Maxam–Gilbert sequencing of the low-mobility product from the reaction of BfiI on 25C/15 and of the chemically synthesized hairpin DNA. The G, A+G, C+T, and A>C chemical sequencing reactions were performed as in Materials and Methods. The junction positions between the top and the bottom strands of the 25C/15 substrate and the BfiI recognition sequence are indicated.
Kinetic analysis of the cleavage of the 3′-tailed oligoduplex by BfiI reveals that both DNA hydrolysis and transesterification reactions proceed concurrently, resulting initially in the parallel accumulation of both hydrolysis and hairpin products (Fig. 2C). The amount of hairpin reached a peak after ≈3 s, and then it decreased upon cleavage to the hydrolysis product (Fig. 2C).
To characterize the processes involved in hairpin formation, reactions were carried out on a series of DNA duplexes that had in their top strands 3′ extensions of varied lengths and sequences: the 15/15, 16/15, 17/15, 25/15, and 29/15 duplexes (Table 1). Hairpin formation was not observed with the blunt-ended substrate, 15/15, but it was detected in increasing amounts as the length of the 3′-extension was increased from 1 to 10 nt (Table 1), from a low level with a 1-nt tail (16/15), an intermediate level with 2 nt (17/15), and a higher level with 10 nt (25/15). Even so, less of the hairpin was formed from the 25/15 substrate (Table 1) than from the 25C/15 substrate used previously (Fig. 2). Both 25/15 and 25C/15 have 10-nt extensions, but only that in 25C/15 has a self-complementary sequence. When the length of the 3′ extension was increased further, to 14 nt of noncomplementary sequence (the duplex 29/15), only trace amounts of hairpin were formed.
Hence, during the reaction of BfiI on the bottom strand of a DNA that has a free OH group at the 3′ end of its top strand, that 3′-OH can compete effectively with water at the enzyme active site, to yield a DNA hairpin. This REase can therefore not only cleave DNA at specific phosphodiester bonds, but it can also join together two DNA segments by transferring the 5′-phosphate at the site of cleavage to a 3′-OH from elsewhere in the DNA.
Stereochemical Course of the BfiI Reaction.
Analysis of the stereochemical course of a phosphoryl transfer reaction can reveal whether the pathway includes a covalent intermediate. A single-step substitution reaction without a covalent intermediate should lead to inversion of configuration, whereas a reaction involving a covalent intermediate should proceed with two inversions and thus result in overall retention of configuration (5, 6).
The BfiI REase belongs to the PLD superfamily of enzymes (12), and covalent phosphohistidine intermediates have been demonstrated previously for a number of PLD enzymes, including PLD itself, tyrosyl-DNA PDE, and Nuc (18, 20, 26). Stereochemical pathways for many endonucleases acting at specific DNA sites have been determined by using DNA substrates in which the scissile phosphate is replaced with a chiral phosphorothioate (PTO) of known configuration (27). This strategy was followed here to determine the stereochemical path of DNA transesterification by BfiI.
The 15-nt bottom strand for the 14/15 (and the 25C/15; Table 1) substrate was synthesized with a PTO linkage in place of the target phosphodiester and the Rp and Sp diastereoisomers purified by HPLC (see SI Methods). The ability of BfiI to hydrolyze PTO linkages was tested on 14/15-nt duplexes with either an Rp– or an Sp–PTO at the site of bottom-strand cleavage (Fig. 4A). BfiI hydrolyzed both Rp and Sp PTO derivatives at equal rates, both ≈500-fold slower than the phosphoester substrate (Fig. 4B). Cleavage occurred only at the PTO bonds (data not shown).
Hairpin formation by BfiI was examined on substrates with either an Rp– or Sp–PTO at the site of bottom-strand cleavage but whose top strands carried 10-nt extensions of self-complementary sequence at their 3′ ends: duplexes 25C/15(S)Rp and 25C/15(S)Sp, respectively (Table 1). In both cases, the hairpin DNA was formed in parallel to the hydrolysis product (data not shown) in the same manner as the native substrate (Fig. 2). The yield of hairpin from both PTO-containing duplexes was similar to that for the phosphoester (Table 1).
To monitor the stereochemical course of the transesterification reaction, 32P radiolabels were placed between the 3′-terminal and penultimate nucleosides in the top strands of the 25C/15(S)Rp and 25C/15(S)Sp duplexes (Fig. 5A). The hairpin products formed from these duplexes were then isolated and analyzed with two nonspecific nucleases that have complementary specificities for PTO stereoisomers (Fig. 5A): snake venom PDE cleaves Rp– but not Sp–PTO bonds (28), whereas nuclease P1 cleaves Sp– but not Rp–PTO linkages (29).
Fig. 5.

Stereochemical course of DNA transesterification by BfiI. (A) Experimental strategy: the 25C/15(S)Rp duplex contains an Rp–PTO linkage at the site of bottom-strand DNA cleavage and a 32P label [indicated by an asterisk (∗)] immediately upstream from the 3′-terminal nucleoside of the top strand (for nucleotide sequence, see Table 1). After incubation with BfiI to form hairpin DNA and subsequent purification (Fig. 2), aliquots were digested with either nuclease P1 or snake venom PDE. (B) Denaturing PAGE analysis of the nuclease P1 and the snake venom PDE digests of the hairpins formed from 25C/15(S)Rp and 25C/15(S)Sp (Table 1). The two substrates contain, respectively, Rp– and Sp–PTO linkages, as indicated above the gel. Lane M contains the mononucleotide marker dAMP.
The digests of the hairpin DNA obtained from 25C/15(S)Rp duplex (Fig. 5A) yielded as the end products a radiolabeled dinucleotide with nuclease P1 and a labeled mononucleotide with PDE (Fig. 5B). This hairpin therefore contains an Rp–PTO linkage adjacent to the radiolabel, the same configuration as in the substrate. Conversely, the hairpin from 25C/15(S)Sp gave mainly a radiolabeled mononucleotide when digested with P1 and a dinucleotide with PDE (Fig. 5B), and so it contains the Sp linkage. Thus, on both Rp and Sp substrates, the configuration of the scissile phosphate remained unchanged during the transesterification reaction. The retention of stereoconfiguration suggests that transesterification by BfiI occurs with two inversions and involves a covalent intermediate.
DNA hydrolysis by BfiI probably follows the same pathway as transesterification. For example, ribonuclease A (30, 31) and MuA transposase (4, 8) catalyze both transesterification and hydrolysis with inversion of configuration, implying the same mechanism for both reactions. BfiI is the only REase known to date that seems to employ a covalent intermediate.
Discussion
Most enzymes that hydrolyze phosphodiester bonds use water to attack the scissile phosphate directly, in a single-step mechanism. Relatively few enzymes catalyze phosphodiester hydrolysis by two-step mechanisms, where a nucleophile from the enzyme first attacks the phosphate to form a covalent intermediate that is then hydrolyzed by water (5–7). All of the REases whose stereochemistries had been analyzed before cleave their target phosphodiester bonds with inversion of configuration at the phosphorus, indicating single-step mechanisms (1). We have shown here that the BfiI endonuclease catalyzes not only the hydrolysis of phosphodiester bonds at specific DNA sites but also site-specific DNA transesterification reactions, a process never seen before with a restriction enzyme. By using DNA substrates with either Rp or Sp PTO groups at the target phosphodiester, the transesterification reaction of BfiI was shown to proceed with retention of configuration (Fig. 5), indicating a two-step mechanism.
Rp and Sp PTOs.
The ability of BfiI to hydrolyze both Rp and Sp stereoisomers at equal rates (Fig. 4) is unusual; most nucleases show a very marked preference for one isomer. For example, the EcoRI, EcoRV, SfiI, and Vsr nucleases can cleave substrates with Rp–PTO linkages at their target sites, albeit more slowly than the phosphoester, but they are inactive against substrates with Sp bonds (4, 32). Consequently, whereas the stereochemical paths of most nucleases can be determined on only one stereoisomer, it was possible to identify the path for BfiI on both Rp and Sp substrates.
The discrimination between Rp and Sp substrates has been examined fully with the 3′–5′ exonuclease of Escherichia coli DNA polymerase I (33). The Rp–PTO was cleaved at a slightly reduced rate, and it caused only minor perturbations to the active site. In contrast, the Sp–PTO was not hydrolyzed at all; in the structure, its proS sulfur occupied the position that would otherwise be occupied by the active-site metal ions (33). The Sp–PTO thus excludes the catalytic metal ions from the active site. Likewise, PTO derivatives of ATP often prevent ATP-dependent enzymes from binding Mg2+ (27). BfiI, on the other hand, does not use metal ions in its reactions (12), so factors that affect metal ion binding will not affect its activity.
Covalent Intermediate in a Restriction Enzyme.
BfiI belongs to the PLD family of enzymes, which catalyze many different reactions on many different phosphodiester substrates, although perhaps by a common mechanism. The reactions of cabbage PLD (26) and of E. coli phosphatidylserine synthase (34) occur with retention of configuration, indicating a covalent phosphoenzyme intermediate. In both S. typhimurium Nuc (15) and Yersinia pestis toxin (35), an active-site histidine can form a covalent complex with inorganic phosphate at acidic pH.
In BfiI, the equivalent active-site residue in the conserved HKD motif is His-105 (24). We suggest that His-105 first attacks the phosphorus at the scissile bond to form a covalent intermediate and release the 3′-DNA (Fig. 6). In a second step, the presumed phosphohistidine intermediate is attacked by water, to liberate the 5′-phosphorylated DNA, or by an alcohol, to give the transesterification product carrying the alcohol on the 5′-phosphate (Fig. 6).
Fig. 6.

Putative reaction mechanism of REase BfiI.
The hydrolysis and transesterification reactions occur concurrently, indicating direct competition between water and the alcohol for the putative phosphohistidine intermediate (Fig. 6). The relative yields of the two products vary with the alcohol concentration. In reactions containing 10% vol/vol glycerol or 1,2-ethanediol (1.4 and 1.8 M, respectively), most of the DNA was converted to transesterification products, and only a small fraction was hydrolyzed (Fig. 1B), indicating perhaps preferential binding of hydrophobic moieties at the active site. The high yields of DNA–alcohol adducts formed by BfiI raise the possibility of exploiting this reaction as a tool for the site-specific labeling of DNA; its transesterification reactions appear to be able to use a wide range of OH-containing compounds.
Strand Transfer by a Restriction Enzyme.
The ability of BfiI to catalyze an intramolecular DNA transesterification reaction, to form a hairpin, is a novel reaction for a restriction enzyme. Nevertheless, hairpin intermediates are central to V(D)J recombination (36) and to many transposition reactions (10, 37, 38) because they allow a single active site to make a double-stranded break. These systems first cut one DNA strand to leave a 3′-OH, which then attacks the other strand to generate the hairpin. The hairpin is an obligatory intermediate, and it is resolved before transfer to the target DNA.
In contrast to enzymes mediating DNA rearrangements, hairpin formation is not obligatory during DNA cleavage by BfiI. Rather, BfiI produces hairpins as a result of its ability to catalyze transesterifications to an appropriately positioned OH group. It produced none of the hairpin from the blunt-ended duplex 15/15 and very little from a duplex with a 1-nt 3′ extension, 16/15 (Table 1). In this respect, BfiI differs from the Tn5 (38) and Tn10 transposases (10) and the RAG1/RAG2 proteins (36), which do not require any protruding DNA for hairpin formation and which use the new 3′-OH group to attack the opposite phosphodiester bond on the complementary strand.
BfiI generated higher yields of hairpin DNA from duplexes with 2- and 10-nt 3′-tails, 17/15 and 25/15, respectively, although a further increase to a 14-nt tail (in 29/15) virtually abolished hairpin formation (Table 1). However, a duplex with a 10-nt extension of self-complementary sequence, 25C/15, gave more of the hairpin than a duplex with a 10-nt extension of noncomplementary sequence, 25/15. Hairpin formation by BfiI presumably requires the terminal 3′-OH of the top strand to be positioned in line with the histidine in the postulated phosphohistidine intermediate (Fig. 6) so as to displace the histidine and transfer the 5′-phosphate onto the 3′-OH group. The frequency with which the 3′-OH occupies the correct position will be affected by both the length and the sequence of the extension. A self-complementary sequence may form a snap-back structure that holds the 3′-OH group in position to attack the intermediate. In contrast, on a short extension or on a long extension of noncomplementary sequence, the 3′-OH will only rarely be in position to attack the intermediate.
Recombinases, integrases, transposases, and topoisomerases have all been shown previously to catalyze DNA strand transfer reactions (8, 37, 39–42). We have identified here a restriction enzyme that cannot only cleave DNA but can also catalyze strand transfer reactions, which possibly implies novel functions for these enzymes. The REase in question, BfiI, is from the PLD superfamily, and other members of this family are also likely to be capable of catalyzing transesterification reactions on DNA.
Materials and Methods
Hydrolysis and Transesterification Reactions.
Oligonucleotide duplexes (Table 1) were obtained by annealing synthetic oligonucleotides (Metabion, Martinsried, Germany). Oligonucleotides were 5′-labeled with [γ-33P]ATP and PNK or 3′-labeled with [α-32P]ddATP and terminal deoxynucleotidyl transferase [TdT (from Fermentas UAB, Vilnius, Lithuania)] (25) and gel-purified. The 25-nt top strand of the 25CddA/15 oligoduplex (Table 1) was obtained by incubating the 24-nt oligonucleotide 5′-AGCACTGGGCTGCTGAACTAGTTC-3′ with ddATP and TdT.
DNA-cleavage reactions were performed by mixing manually radiolabeled oligonucleotides (1–10 nM) with BfiI enzyme (10–100 nM dimer) in 20 mM potassium phosphate, pH 7.4/120 mM KCl/1 mM EDTA/0.01 mg/ml BSA at 25°C. For some experiments, the above buffer was supplemented with glycerol, 1,2-ethanediol, or ethanol (1–10%, vol/vol). Buffers for reactions in Tris were: 0.3 M Tris-acetate, pH 8.3/0.3 M KCl/8 mM EDTA or 0.1 M Tris/0.1 M KCl/2.5 mM EDTA. The samples were collected at timed intervals and quenched with phenol/chloroform or loading dye solution (25). Rapid DNA hydrolysis and transesterification reactions were studied in a quench-flow device (KinTek, Austin, TX): equal volumes of radiolabeled oligonucleotide (1 nM) and BfiI enzyme (50–100 nM dimer) were mixed and quenched with 1.0 M NaOH. The samples were neutralized by adding 20% CH3COOH and 2.4% SDS solution. Separation of DNA hydrolysis and transesterification products was performed by denaturing PAGE as described in ref. 25. Radiolabeled DNA was detected and quantified by PhosphorImager (PerkinElmer, Wellesley, MA) (25).
Maxam–Gilbert Sequencing of Hairpin DNA.
Oligonucleotide duplex 25C/15 (Table 1), with a 33P radiolabel at the 5′ terminus of the top strand was incubated with BfiI for 10 s at 25°C. The reactions were stopped with chloroform, and the hairpin product was gel-purified. The G (dimethyl sulfate), A+G (formic acid), C+T (hydrazine), and A>C (NaOH) sequencing reactions were performed as described in ref. 43. In parallel, the sequencing reactions were performed on a chemically synthesized hairpin oligonucleotide.
Stereochemical Studies.
To place a 32P radiolabel immediately upstream from the 3′-terminal dA of the top strand of the oligonucleotide duplexes 25C/15(S)Sp and 25C/15(S)Rp (see Table 1 and SI Methods), the 24-nt oligonucleotide 5′-AGCACTGGGCTGCTGAACTAGTTC-3′ was annealed to the oligonucleotide 5′-ATGAACTAGTTCAGCA-3′ and extended by incubation with [α-32P]dATP and exo− Klenow fragment. The resulting 25-nt reaction product was gel-purified and annealed to the 15-nt bottom DNA strand with either an Rp– or Sp–PTO modification. After incubation with BfiI for 15 min, the reactions were quenched with CHCl3, and the DNA was precipitated with ethanol. The hairpin products were gel-purified and digested with either nuclease P1 or snake venom PDE. The resulting 32P-labeled mono- and dinucleotides were analyzed by denaturing PAGE and PhosphorImager as above.
Supplementary Material
Acknowledgments
We thank A. Lagunavicius and S. Klimasauskas for discussions, E. Merkiene for the help with the quench-flow equipment, V. Shaw for the mass spectrometry, and E. Manakova for the BfiI sample. We also thank the Marie Curie Research Training Network on DNA Enzymes. This work was supported by Collaborative Research Initiative Grant 074498/Z/04 from the Wellcome Trust (to V.S. and S.E.H.), the Howard Hughes Medical Institute Grant 55000336 (to V.S.), and the Lithuania Science and Studies Foundation Grant T-09106 (to G.S.).
Abbreviations
- CIAP
calf intestine alkaline phosphatase
- ddA
dideoxyadenosine
- PDE
phosphodiesterase
- PLD
phospholipase D
- PNK
polynucleotide kinase
- PTO
phosphorothioate
- REase
restriction endonuclease.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission. L.J.-J. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0608689104/DC1.
References
- 1.Pingoud A, Fuxreiter M, Pingoud V, Wende W. Cell Mol Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PubMed] [Google Scholar]
- 2.Connolly BA, Eckstein F, Pingoud A. J Biol Chem. 1984;259:10760–10763. [PubMed] [Google Scholar]
- 3.Grasby JA, Connolly BA. Biochemistry. 1992;31:7855–7861. doi: 10.1021/bi00149a016. [DOI] [PubMed] [Google Scholar]
- 4.Mizuuchi K, Nobbs TJ, Halford SE, Adzuma K, Qin J. Biochemistry. 1999;38:4640–4648. doi: 10.1021/bi990054p. [DOI] [PubMed] [Google Scholar]
- 5.Knowles JR. Annu Rev Biochem. 1980;49:877–919. doi: 10.1146/annurev.bi.49.070180.004305. [DOI] [PubMed] [Google Scholar]
- 6.Frey PA. Adv Enzymol Relat Areas Mol Biol. 1989;62:119–201. doi: 10.1002/9780470123089.ch4. [DOI] [PubMed] [Google Scholar]
- 7.Gerlt JA. In: Nucleases. 2nd Ed. Linn SM, Lloyd RS, Roberts RJ, editors. Plainview, NY: Cold Spring Harbor Lab Press; 1993. pp. 1–35. [Google Scholar]
- 8.Mizuuchi K, Adzuma K. Cell. 1991;66:129–140. doi: 10.1016/0092-8674(91)90145-o. [DOI] [PubMed] [Google Scholar]
- 9.Engelman A, Mizuuchi K, Craigie R. Cell. 1991;67:1211–1221. doi: 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- 10.Kennedy AK, Haniford DB, Mizuuchi K. Cell. 2000;101:295–305. doi: 10.1016/s0092-8674(00)80839-9. [DOI] [PubMed] [Google Scholar]
- 11.Vitkute J, Maneliene Z, Petrusyte M, Janulaitis A. Nucleic Acids Res. 1998;26:3348–3349. doi: 10.1093/nar/26.14.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sapranauskas R, Sasnauskas G, Lagunavicius A, Vilkaitis G, Lubys A, Siksnys V. J Biol Chem. 2000;275:30878–30885. doi: 10.1074/jbc.M003350200. [DOI] [PubMed] [Google Scholar]
- 13.Grazulis S, Manakova E, Roessle M, Bochtler M, Tamulaitiene G, Huber R, Siksnys V. Proc Natl Acad Sci USA. 2005;102:15797–15802. doi: 10.1073/pnas.0507949102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaremba M, Urbanke C, Halford SE, Siksnys V. J Mol Biol. 2004;336:81–92. doi: 10.1016/j.jmb.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 15.Stuckey JA, Dixon JE. Nat Struct Biol. 1999;6:278–284. doi: 10.1038/6716. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Stuckey JA, Lohse DL, Dixon JE. Protein Sci. 1997;6:2655–2658. doi: 10.1002/pro.5560061221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponting CP, Kerr ID. Protein Sci. 1996;5:914–922. doi: 10.1002/pro.5560050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Interthal H, Pouliot JJ, Champoux JJ. Proc Natl Acad Sci USA. 2001;98:12009–12014. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waite M. Biochim Biophys Acta. 1999;1439:187–197. doi: 10.1016/s1388-1981(99)00094-3. [DOI] [PubMed] [Google Scholar]
- 20.Gottlin EB, Rudolph AE, Zhao Y, Matthews HR, Dixon JE. Proc Natl Acad Sci USA. 1998;95:9202–9207. doi: 10.1073/pnas.95.16.9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang SF, Freer S, Benson AA. J Biol Chem. 1967;242:477–484. [PubMed] [Google Scholar]
- 22.Eibl H, Kovatchev S. Methods Enzymol. 1981;72:632–639. doi: 10.1016/s0076-6879(81)72055-x. [DOI] [PubMed] [Google Scholar]
- 23.Interthal H, Chen HJ, Champoux JJ. J Biol Chem. 2005;280:36518–36528. doi: 10.1074/jbc.M508898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagunavicius A, Sasnauskas G, Halford SE, Siksnys V. J Mol Biol. 2003;326:1051–1064. doi: 10.1016/s0022-2836(03)00020-2. [DOI] [PubMed] [Google Scholar]
- 25.Sasnauskas G, Halford SE, Siksnys V. Proc Natl Acad Sci USA. 2003;100:6410–6415. doi: 10.1073/pnas.1131003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruzik K, Tsai MD. Biochemistry. 1984;23:1656–1661. doi: 10.1021/bi00303a012. [DOI] [PubMed] [Google Scholar]
- 27.Eckstein F. Annu Rev Biochem. 1985;54:367–402. doi: 10.1146/annurev.bi.54.070185.002055. [DOI] [PubMed] [Google Scholar]
- 28.Burgers PM, Eckstein F, Hunneman DH. J Biol Chem. 1979;254:7476–7478. [PubMed] [Google Scholar]
- 29.Potter BV, Romaniuk PJ, Eckstein F. J Biol Chem. 1983;258:1758–1760. [PubMed] [Google Scholar]
- 30.Usher DA, Erenrich ES, Eckstein F. Proc Natl Acad Sci USA. 1972;69:115–118. doi: 10.1073/pnas.69.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Usher DA, Richardson DI, Jr, Eckstein F. Nature. 1970;228:663–665. doi: 10.1038/228663a0. [DOI] [PubMed] [Google Scholar]
- 32.Elliott SL, Brazier J, Cosstick R, Connolly BA. J Mol Biol. 2005;353:692–703. doi: 10.1016/j.jmb.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 33.Brautigam CA, Steitz TA. J Mol Biol. 1998;277:363–377. doi: 10.1006/jmbi.1997.1586. [DOI] [PubMed] [Google Scholar]
- 34.Raetz CR, Carman GM, Dowhan W, Jiang RT, Waszkuc W, Loffredo W, Tsai MD. Biochemistry. 1987;26:4022–4027. doi: 10.1021/bi00387a042. [DOI] [PubMed] [Google Scholar]
- 35.Rudolph AE, Stuckey JA, Zhao Y, Matthews HR, Patton WA, Moss J, Dixon JE. J Biol Chem. 1999;274:11824–11831. doi: 10.1074/jbc.274.17.11824. [DOI] [PubMed] [Google Scholar]
- 36.van Gent DC, Mizuuchi K, Gellert M. Science. 1996;271:1592–1594. doi: 10.1126/science.271.5255.1592. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy AK, Guhathakurta A, Kleckner N, Haniford DB. Cell. 1998;95:125–134. doi: 10.1016/s0092-8674(00)81788-2. [DOI] [PubMed] [Google Scholar]
- 38.Lovell S, Goryshin IY, Reznikoff WR, Rayment I. Nat Struct Biol. 2002;9:278–281. doi: 10.1038/nsb778. [DOI] [PubMed] [Google Scholar]
- 39.Knudsen BR, Dahlstrom K, Westergaard O, Jayaram M. J Mol Biol. 1997;266:93–107. doi: 10.1006/jmbi.1996.0767. [DOI] [PubMed] [Google Scholar]
- 40.Bhasin A, Goryshin IY, Reznikoff WS. J Biol Chem. 1999;274:37021–37029. doi: 10.1074/jbc.274.52.37021. [DOI] [PubMed] [Google Scholar]
- 41.McBlane JF, van Gent DC, Ramsden DA, Romeo C, Cuomo CA, Gellert M, Oettinger MA. Cell. 1995;83:387–395. doi: 10.1016/0092-8674(95)90116-7. [DOI] [PubMed] [Google Scholar]
- 42.Shuman S. J Biol Chem. 1992;267:8620–8627. [PubMed] [Google Scholar]
- 43.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab Press; 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}