

Abstract
In plants, association of O-acetylserine sulfhydrylase (OASS) and Ser acetyltransferase (SAT) into the Cys synthase complex plays a regulatory role in sulfur assimilation and Cys biosynthesis. We determined the crystal structure of Arabidopsis thaliana OASS (At-OASS) bound with a peptide corresponding to the C-terminal 10 residues of Arabidopsis SAT (C10 peptide) at 2.9-Å resolution. Hydrogen bonding interactions with key active site residues (Thr-74, Ser-75, and Gln-147) lock the C10 peptide in the binding site. C10 peptide binding blocks access to OASS catalytic residues, explaining how complex formation downregulates OASS activity. Comparison with bacterial OASS suggests that structural plasticity in the active site allows binding of SAT C termini with dissimilar sequences at structurally similar OASS active sites. Calorimetric analysis of the effect of active site mutations (T74S, S75A, S75T, and Q147A) demonstrates that these residues are important for C10 peptide binding and that changes at these positions disrupt communication between active sites in the homodimeric enzyme. We also demonstrate that the C-terminal Ile of the C10 peptide is required for molecular recognition by At-OASS. These results provide new insights into the molecular mechanism underlying formation of the Cys synthase complex and provide a structural basis for the biochemical regulation of Cys biosynthesis in plants.
INTRODUCTION
Multienzyme complexes play central roles in a variety of cellular processes, including translation, transcription, gene expression, and signal transduction. Association of macromolecules into complexes provides cells with a means of organizing molecular networks and of regulating metabolism by colocalizing key enzymes or channeling metabolites between enzyme active sites (Srere, 1987; Hrazdina and Jensen, 1992; Winkel, 2004; Xia et al., 2004). Although Kredich et al. (1969) identified one of the earliest examples of a macromolecular complex in primary metabolism by isolating the Cys synthase complex, exploration of the molecular basis for assembly of this multienzyme complex is only beginning.
In plants and bacteria, Cys biosynthesis occurs in two steps (Rabeh and Cook, 2004; Wirtz and Droux, 2005; Kopriva, 2006). Ser acetyltransferase (SAT) generates O-acetylserine by transferring acetate from acetyl-CoA to Ser. Next, O-acetylserine sulfhydrylase (OASS) uses pyridoxal phosphate (PLP) as a cofactor to form Cys from O-acetylserine and sulfide. In plants, strict regulation of the pathway by two mechanisms maintains intracellular Cys levels (Saito et al., 1994). First, feedback inhibition of SAT by Cys can control production of the amino acid (Noji et al., 1998). The second mechanism involves association of SAT and OASS to form the Cys synthase complex. The function of the complex is not metabolic channeling, since O-acetylserine freely diffuses out of the complex (Kredich et al., 1969; Cook and Wedding, 1977; Droux et al., 1998). Instead, interaction of SAT and OASS coordinates sulfate assimilation and modulates Cys synthesis at the cellular level in plants (Hell and Hillebrand, 2001) (Figure 1).
Figure 1.

Regulation of Cys Synthesis by Formation of the Cys Synthase Complex.
Active forms of OASS and SAT are indicated by red text; inactive (or less active) forms of either enzyme are indicated by black text. Modified from Hell and Hillebrand (2001). CS, Cys synthase.
Under sulfur-sufficient conditions, the two enzymes form the complex in which SAT activity increases and OASS activity decreases (Saito et al., 1995; Droux et al., 1998). This results in production of O-acetylserine. If intracellular sulfur levels are low, then O-acetylserine accumulates because free OASS is unable to generate Cys due to a lack of sulfide. Elevated O-acetylserine levels dissociate the complex, which downregulates SAT. Meanwhile, the increased O-acetylserine concentration activates expression of genes encoding sulfate transporters, ATP sulfurylase, OASS, and SAT (Smith et al., 1997; Koprivova et al., 2000; Hopkins et al., 2005). This leads to increased sulfur uptake and reduction. As sulfur levels elevate, free OASS catalyzes Cys formation, which reduces O-acetylserine levels. This allows association of SAT and OASS, activation of SAT, and resumption of Cys biosynthesis.
The molecular details of how OASS and SAT form the Cys synthase complex are unclear. One SAT hexamer (Mr of ∼180 kD) and two OASS dimers (each Mr of ∼70 kD) comprise the complex in plants and bacteria (Kredich et al., 1969; Saito et al., 1995; Zhu et al., 1998). Crystallographic information on the proteins in the complex includes individual structures of OASS from bacteria (Burkhard et al., 1998; Claus et al., 2005) and Arabidopsis thaliana (Bonner et al., 2005) and bacterial SAT (Gorman and Shapiro, 2004; Olsen et al., 2004; Pye et al., 2004). Demonstration of the interaction between OASS and SAT in plants and bacteria has employed multiple approaches, including size-exclusion chromatography, yeast two-hybrid analysis, surface plasmon resonance, and fluorescence spectroscopy (Bogdanova and Hell, 1997; Mino et al., 1999, 2000; Wirtz et al., 2001; Berkowitz et al., 2002; Bonner et al., 2005; Campanini et al., 2005; Zhao et al., 2006).
Efforts to map the protein–protein interaction regions in the Cys synthase complex indicate that the C terminus of SAT plays a role in association with OASS in plants and bacteria (Bogdanova and Hell, 1997; Mino et al., 1999, 2000; Wirtz et al., 2001; Zhao et al., 2006). Recently, determination of the structure of Haemophilus influenzae OASS (Hi-OASS) in complex with a peptide corresponding to the C terminus of SAT from the same organism, and protein–protein interaction studies of Arabidopsis OASS (At-OASS) and At-SAT showed that the OASS active site is a key SAT interaction site (Bonner et al., 2005; Huang et al., 2005). Sequence variation in the C termini of SAT (Figure 2A) suggests that differences in the OASS active site may provide specificity for formation of the Cys synthase complex in plants and bacteria.
Figure 2.

Overview of the At-OASS·C10 Peptide Structure.
(A) Comparison of the C-terminal sequences of SAT from Arabidopsis (At-SAT), Glycine max (soybean) (Gm-SAT), Escherichia coli (Ec-SAT), and H. influenzae (Hi-SAT). Portions of the At-SAT and Hi-SAT peptides observed crystallographically in structures of At-OASS and Hi-OASS, respectively, are highlighted in red.
(B) Ribbon diagram of the At-OASS dimer. The monomers of the dimer are colored in light purple and white. The location of the active site is defined by the Schiff base formed between PLP and Lys-46 (yellow). C10 peptides (colored green or rose in each monomer) are bound in the two active sites of the At-OASS homodimer.
(C) Electron density of the C10 peptide is shown as a 2Fo-Fc omit map contoured at 1.2σ. Eight (TEWSDYVI) of 10 residues in the peptide were observable.
(D) Stereo view of the molecular surface of At-OASS forming the peptide binding site. The C10 peptide (rose) is shown as a stick drawing. The surface corresponding to the active site PLP is colored yellow. The surface of residues previously implicated in interaction between At-OASS and At-SAT are shown in green.
To elucidate the structural basis of protein–protein interactions in the plant Cys synthase complex, we determined the x-ray crystal structure of At-OASS bound with a peptide consisting of the 10 residues found at the C terminus of At-SAT (C10 peptide). Examination of specific contacts between the C10 peptide and At-OASS using calorimetry shows that key active site residues have different energetic contributions to binding the C terminus of SAT. These results provide new insights into the molecular mechanism underlying formation of the plant Cys synthase complex.
RESULTS
Overall Structure of the At-OASS·C10 Peptide Complex
At-OASS was crystallized in the presence of At-SAT C10 peptide. Crystals were of the same space group and similar unit cell dimensions as previously described At-OASS structures (Table 1) (Bonner et al., 2005). The overall structure of the At-OASS·C10 peptide complex (Figure 2B) is similar to those of wild-type At-OASS and the K46A At-OASS mutant with root mean square deviations for 320 Cα-atoms of 0.32 and 0.39 Å, respectively (Bonner et al., 2005). In the initial electron density maps, contiguous density for a ligand was observed extending from the active site (Figure 2C). Subsequently, eight of 10 amino acids in the C10 peptide were modeled in the final structure, with the two N-terminal residues disordered. Binding of the C10 peptide blocks entry to the active site, with the C-terminal Ile of the peptide positioned near the PLP and the remaining residues occupying a cleft that forms the active site entrance (Figure 2D).
Table 1.
Crystallographic Data Collection and Refinement Statistics
| Space Group | P43212 |
| Cell dimensions | a = b = 104.9 Å; c = 99.3 Å; α = β = γ = 90° |
| Resolution | 100 to 2.9 Å |
| Reflections (total/unique) | 200,355/18,447 |
| Completeness (highest shell)a | 100% (100%) |
| 〈I/σ〉 (highest shell) | 7.94 (2.60) |
| Rsym (highest shell)b | 10.7% (40.8%) |
| Rcrystc/Rfreed | 18.8%/24.8% |
| No. of protein atoms/peptide atoms/sulfate atoms/water moleculese | 2,374/73/5/112 |
| Average B-factor, protein/peptide/sulfate/water | 21.1 Å2/32.8 Å2/66.7 Å2/24.5 Å2 |
| RMSD, bond lengthsf | 0.006 Å |
| RMSD, bond angles | 1.28° |
Highest-resolution shell: 3.0 to 2.9 Å.
Rsym = Σ|Ih − 〈Ih〉|/ΣIh, where 〈Ih〉 is the average intensity over symmetry.
Rcryst = Σ|Fo − 〈Fc〉|/ΣFo, where summation is over the data used for refinement.
Rfree is defined the same as Rcryst but was calculated using 5% of data excluded from refinement.
Includes 15 PLP atoms.
RMSD, root mean square deviation.
Interactions in the C10 Peptide Binding Site
The C10 peptide binding site is centered on the At-OASS active site (Figure 3). The PLP cofactor, Gln-147, and the active site loop containing Thr-74 and Ser-75 define the bottom of the site, where the C-terminal Ile of the peptide binds. The binding site extends ∼25 Å from the Schiff base formed by Lys-46 and PLP through a surface cleft delineated by the β8A-β9A loop (Lys-217–Phe-230) on one side and an α-helix (α4), which includes Met-125 and Lys-126, on the opposite side.
Figure 3.

The C10 Peptide Binding Site.
(A) Stereo view of interactions in the C10 peptide binding site. All modeled residues of the C10 peptide are shown and labeled (rose). Sidechains of At-OASS residues that interact with the C10 peptide are colored yellow. Residues providing peptide backbone contacts are drawn in white without sidechains. Water molecules are shown as red spheres. Dotted lines indicate hydrogen bonds.
(B) Schematic drawing of interactions between the C10 peptide (blue) and At-OASS (black). Only the five C-terminal residues of the peptide are shown in the scheme. Hydrogen bonds are shown as dotted lines with distances in angstroms. W, water molecule (red).
Multiple sidechain, main chain, and water-mediated interactions, made primarily with the C-terminal half of the C10 peptide, lock the ligand into the binding pocket (Figure 3). Hydrogen bonds from the main-chain nitrogen of Thr-78, the sidechain hydroxyl group of Thr-74, and the amide moiety of Gln-147 anchor the C-terminal carboxylate group of the peptide Ile in the binding site. This orients the Ile sidechain toward the PLP cofactor. Additional water-mediated contacts are made with the peptide backbone of the Val and Tyr residues. The sidechain hydroxyl group of Ser-75 also interacts with the carbonyl of the peptide Tyr residue. The protein backbone nitrogens of Met-125 and Lys-126 form hydrogen bonds with the hydroxyl group of the peptide Ser. In addition, the peptide Trp stacks against Pro-220. The two N-terminal residues of the peptide are solvent-exposed.
Plasticity in the C10 Peptide Binding Site
Because the C10 peptide binds at the At-OASS active site, we compared the current structure with the previously determined structure of the At-OASS K46A mutant, which has the PLP and a Met covalently linked to form an external aldimine in the active site (Figure 4A) (Bonner et al., 2005). The PLP-Met external aldimine structure approximates the position of O-acetylserine after reaction with PLP in the catalytic mechanism (Rabeh and Cook, 2004). Overlaying the two structures reveals that the C-terminal Ile of the peptide and the covalently linked Met occupy the same region of the active site. The carboxylate group of either the Ile or the Met forms hydrogen bonds with Thr-74 and Gln-147. Likewise, their respective sidechains fill the same steric space in the active site. The only difference between the two active site structures is that the sidechain of Ser-75 adopts a different rotamer conformation in the C10 peptide complex, which orients the hydroxyl group toward the bound peptide. Unlike in the Met complex, the amide linkage of the peptide's C-terminal Ile is positioned away from the PLP, thus preventing formation of a covalent adduct with PLP. These structures indicate that O-acetylserine and the C terminus of At-SAT compete for the same binding site and explain how OASS activity is downregulated upon formation of the Cys synthase complex. Moreover, these structures show that At-OASS employs the same promiscuous binding site to accommodate substrate (O-acetylserine) or inhibitors (Ile or Met).
Figure 4.

Comparisons of the At-OASS·C10 Peptide Complex.
(A) Stereo view of the structural overlay of the At-OASS·C10 peptide complex (rose) and the At-OASS K46A mutant structure (green). The last six residues of the C10 peptide and three key active site residues are drawn as sticks. In the K46A mutant structure, Met is covalently linked to the PLP to form an external aldimine in the active site.
(B) Stereo view of the structural overlay of the At-OASS·C10 peptide complex (rose) and the Hi-OASS·peptide complex (green). Amino acid sidechains and residues of each peptide are shown in ball-and-stick representation.
The At-OASS·C10 peptide complex structure also reveals how the structurally similar active sites of the plant and bacterial OASS recognize the dissimilar C-terminal sequences of their cognate SAT partners (Figure 2A). The At-OASS·C10 peptide complex and the Hi-OASS·peptide complex (Huang et al., 2005) share the same overall fold with a root mean square deviation of 1.71 Å (302 Cα-atoms). Although only the four C-terminal residues of the Hi-SAT peptide were crystallographically ordered and visible, comparison with the At-OASS·C10 peptide complex indicates that structural plasticity in the active site plays a role in recognizing different ligands (Figure 4B).
In At-OASS and Hi-OASS, the same set of interactions is formed between residues of the active site (Thr-74 and Gln-147 in At-OASS; Thr-69 and Gln-143 in Hi-OASS) and the invariant Ile of either the At-SAT or Hi-SAT C10 peptide. Interestingly, both the sidechain and protein backbone of Ser-75 in At-OASS are positioned 2.7 Å further into the peptide binding cleft than the corresponding Ser (Ser-70) of Hi-OASS. Repositioning of the Ser accommodates the different sidechains in the At-SAT C10 peptide (Val) versus the Hi-SAT C10 peptide (Arg). In addition, the loop that includes residues 120 to 130 of At-OASS shifts 4 Å into the cleft in the plant enzyme structure relative to its position in the Hi-OASS structure. This comparison implies that repositioning of structural features in the OASS active site plays a role in recognition of the SAT C terminus.
At-OASS·C10 Peptide Binding Studies
To probe the functional role of Thr-74, Ser-75, and Gln-147 in binding the C10 peptide, isothermal titration calorimetry (ITC) was used to determine binding constants of wild-type At-OASS and the T74S, S75A, S75T, and Q147A At-OASS mutants for the C10 peptide (Figure 5, Table 2). ITC directly measures changes in the heat or enthalpy of binding (ΔH), binding energy (ΔG) from fitting the integrated heat to appropriate model, stoichiometry of a given interaction from equivalence points of the titration, and entropy (ΔS) changes that occur upon binding, thereby giving a full thermodynamic characterization of an interaction.
Figure 5.

Calorimetric Titrations of At-OASS with C10 Peptide.
(A) Titration of At-OASS with C10 peptide. ITC data are plotted as heat signal (μcal/s) versus time (min). The experiment consisted of 20 injections of 12 μL each of C10 peptide (23.7 μM) into a solution containing At-OASS (1.5 μM) at 25°C.
(B) Integrated heat responses per injection from (A), plotted as normalized heat per mole of injectant. The solid line represents the best fit to a two-site binding model.
(C) Titration of At-OASS mutants with C10 peptide. The integrated heat responses are plotted as normalized heat per mole of injectant for the T74S (open circles), S75A (closed squares), S75T (open squares), and Q147A (closed circles) At-OASS mutants. The solid line for each mutant represents the best fit to a one-site binding model.
Table 2.
Thermodynamic Parameters of Association for At-OASS and C10 Peptide
| Kd (nM) | 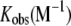 |
ΔH | −TΔS | ΔG | |
|---|---|---|---|---|---|
| Wild type | 5.6 ± 2.0 (37 ± 12) | 1.8 ± 0.8 × 108 (2.7 ± 0.9 × 107) | −9.7 ± 0.3 (−1.3 ± 0.4) | −1.6 (−8.8) | −11.3 ± 0.3 (−10.1 ± 0.2) |
| T74S | 100 ± 11 | 9.4 ± 0.1 × 106 | −5.8 ± 0.17 | −3.6 | −9.5 ± 0.08 |
| S75A | 40 ± 3 | 2.5 ± 0.2 × 107 | −9.2 ± 0.12 | −0.8 | −10.0 ± 0.05 |
| S75T | 1471 ± 50 | 6.8 ± 0.5 × 105 | −5.9 ± 0.3 | −2.0 | −7.8 ± 0.08 |
| Q147A | 56 ± 13 | 1.8 ± 0.3 × 107 | −8.8 ± 0.3 | −0.9 | −9.9 ± 0.08 |
Values of ΔH, TΔS, and ΔG are in kcal mol−1. Wild-type data were fit to the equation describing a two-site binding, with the parameters for the second binding site shown in parentheses. Mutant data were fit to the equation describing a single-site binding model.
Analysis of the interaction between At-OASS and the C10 peptide using ITC (Figures 5A and 5B) shows that two moles of peptide bind per mole of At-OASS homodimer, with the data best fit to a two-site binding model. The calculated change in enthalpy (ΔH) and the association constant (Kobs) are summarized in Table 2. For wild-type At-OASS, negative cooperativity was observed, as binding of the first C10 peptide (Kd = 5 nM) reduced affinity for the second peptide (Kd = 37 nM).
Mutations of Thr-74 (T74S), Ser-75 (S75A and S75T), and Gln-147 (Q147A) reduce binding affinity for the C10 peptide with 6- to 300-fold increases in Kd values (Table 2). Surprisingly, each mutant (T74S, S75A, S75T, and Q147A) displays altered communication between the two active sites of the homodimer, as evidenced by a loss of cooperativity and two-site binding (Figure 5C). In contrast with the wild-type enzyme, the titration data for each mutant are best fit to a single-site binding model (i.e., only one mole of C10 peptide binds per mole of At-OASS homodimer). Fits of the mutant data to a two-site binding model yielded unsatisfactory results. These experiments demonstrate that Thr-74, Ser-75, and Gln-147 energetically contribute to binding of the C10 peptide and play a role in allowing the second active site to bind another C10 peptide, albeit with decreased affinity.
At-OASS·C10 Mutant Peptide Binding
The plant and bacterial SAT contain an invariant Ile at the C terminus (Figure 2A). To examine the role of the Ile as a determinant for At-OASS binding, a mutant C10 peptide containing an Ala substitution at the C terminus was synthesized. ITC analysis of the mutant peptide binding to At-OASS suggested that no interaction occurs (data not shown). To verify this result, fluorescence spectroscopy was used. Binding of C10 peptide to OASS enhances the fluorescence emission signal of the PLP cofactor (Campanini et al., 2005). Figure 6 shows that addition of C10 peptide to At-OASS increases the signal of the cofactor, whereas titration of At-OASS with the mutant peptide does not. The fluorescence emission of At-OASS remained unchanged with addition of up to 3 mM mutant peptide (data not shown), which was the limit due to peptide solubility. This experiment demonstrates that mutation of the C-terminal Ile disrupts binding to At-OASS.
Figure 6.

Comparison of C10 Peptide and Mutant Peptide Interactions with At-OASS.
The fluorescence emission signals of At-OASS (1 μM) without peptide, At-OASS + 3 μM C10 peptide, and At-OASS + 3 μM mutant peptide are shown as solid black, dashed black, and solid gray lines, respectively. a.u., arbitrary units.
DISCUSSION
Cellular processes like signal transduction and gene expression rely on protein–protein interactions, but little structural information for macromolecular assemblies with metabolic functions is available. The plant Cys synthase complex differs from other multienzyme complexes in metabolism because the physical association of OASS and SAT provides a regulatory mechanism for sulfur assimilation and Cys biosynthesis, not a metabolite channeling function (Figure 1) (Droux et al., 1998; Hell and Hillebrand, 2001; Berkowitz et al., 2002).
The structure of the At-OASS·C10 peptide complex provides the molecular basis for how association with At-SAT regulates At-OASS activity and why O-acetylserine binding dissociates the Cys synthase complex (Bogdanova and Hell, 1997; Droux et al., 1998; Wirtz et al., 2001; Berkowitz et al., 2002). Binding of the C10 peptide to At-OASS completely blocks access to the catalytic center of the enzyme (i.e., PLP and Lys-46) (Figures 2 and 3). The interactions between residues of the active site and the C-terminal Ile of the peptide are similar to those formed with the covalently bound Met in the active site of the At-OASS K46A mutant (Figure 4A) (Bonner et al., 2005). Functional studies are also consistent with structural overlap of the C10 peptide and O-acetylserine binding sites. Calorimetric analysis of At-OASS mutants (Table 2) indicates that Thr-74, Ser-75, and Gln-147 are important residues for C10 peptide binding. Previously, we showed that mutations of these residues reduce catalytic efficiency and affinity for substrate (Bonner et al., 2005). Since the two binding sites overlap, increases in intracellular O-acetylserine levels, which occur during sulfur deprivation, will compete with SAT binding to OASS and dissociate the complex, thereby decreasing SAT activity. Evolution of the At-OASS active site architecture has yielded a functionally versatile binding site for both catalysis and regulation of activity.
Formation of the Cys synthase complex raises a molecular recognition issue. Although the plant and bacterial OASS share identical active site residues (Bonner et al., 2005), each recognize the sequentially distinct C terminus of their respective SAT interaction partner (Figure 2A). Comparison of how different C10 peptides bind in At-OASS and Hi-OASS shows the structural conservation of a core set of interactions between the invariant C-terminal Ile and key active site residues (Thr-74 and Gln-147 in At-OASS; Thr-69 and Gln-143 in Hi-OASS) (Figure 4B). Importantly, the C-terminal Ile is a critical feature for interaction with At-OASS, as substitution of an Ala for this residue abolishes binding (Figure 6).
With the C-terminal Ile of SAT serving as an anchor to both the plant and bacterial OASS, structural plasticity elsewhere in the OASS active site allows for binding of divergent C10 peptides. Repositioning of residues in the binding site, in particular Ser-75 and the 120-130 loop, allows At-OASS and Hi-OASS to accommodate different C10 peptides (Figure 4B). As described for Salmonella OASS (Burkhard et al., 1998, 1999), large-scale conformational changes (i.e., transition between open and closed binding conformations) are important in substrate binding and catalysis. Similarly, the structures of the OASS·peptide complexes indicate that structural differences within the active site play a role in recognition of the SAT C terminus.
Multiple approaches demonstrate formation of the Cys synthase complex (Bogdanova and Hell, 1997; Mino et al., 1999, 2000; Wirtz et al., 2001; Bonner et al., 2005; Zhao et al., 2006), but only recently have quantitative studies probed the SAT–OASS interaction. Campanini et al. (2005) used fluorescence spectroscopy to measure binding of Hi-SAT and the Hi-SAT C10 peptide to Hi-OASS. Likewise, Berkowitz et al. (2002) reported a Kd = 25 nM for binding of At-OASS to At-SAT determined by surface plasmon resonance (i.e., BIAcore). Unlike BIAcore technology, in which one binding partner is immobilized on a two-dimensional surface, calorimetry is performed with both molecules in solution and allows determination of the binding constant, stoichiometry, and thermodynamics of a molecular interaction.
Analysis of the interaction between At-OASS and the C10 peptide by calorimetry yielded Kd values comparable to those obtained by surface plasmon resonance for binding of At-SAT (Berkowitz et al., 2002), with fitting of the data showing negative cooperativity between the two binding sites of At-OASS (Figure 5, Table 2). In the plant OASS, reports of positive cooperativity during O-acetylserine binding and catalysis suggest that structural changes upon binding mediate communication between the active sites in the homodimer (Kuske et al., 1994; Rolland et al., 1996; Berkowitz et al., 2002). Binding of the first C10 peptide is accompanied by more favorable enthalpy, which suggests the net formation of noncovalent interactions upon complex formation. This favorable enthalpy effect likely arises from burial of hydrophobic surface area in the binding cleft and formation of noncovalent bonds between C10 peptide and At-OASS binding site (Renzoni et al., 1996). By contrast, entropy changes dominate binding of the second peptide (Table 2). Either removal of ordered water molecules by burial of more hydrophobic surface area in the binding site or major structural changes, such as the transition between open and closed binding conformations of At-OASS, would account for this result (Renzoni et al., 1996).
Additional evidence for communication between the At-OASS active sites is provided by the effect of site-directed mutations. ITC analysis of the T74S, S75A, S75T, and Q147A mutants shows binding of only one peptide per homodimer. The mechanism for the loss of second site binding is unclear but presumably involves perturbation of the interaction network that triggers structural changes. The mutations also alter the energetic contributions of the residues to binding. Entropy changes with the S75A and Q147A mutants are similar to that observed for the binding of the first C10 peptide to At-OASS (Table 2). Thus, Ser-75 and Gln-147 contribute to binding energy by favoring the enthalpy component, which is consistent with hydrogen bond formation by these residues, as observed in the structure (Figure 3). The effects of the T74S and S75T mutants on the C10 peptide binding parameters are larger than anticipated for the change of a single methyl group in the binding site. The T74S mutant should retain the ability to form a hydrogen bond with the correct sidechain rotamer, but the affinity for the C10 peptide is decreased 20-fold. Likewise, the observed rotational flexibility of Ser-75 (Figure 4A) would suggest that the Thr mutation could be accommodated, but the S75T mutant displays a 300-fold increase in Kd value for the C10 peptide. Compared with wild-type At-OASS, the T74S and S75T mutations reduce the enthalpy component and increase the entropic contributions, which suggest structural changes in the active site. Large-scale conformational changes can be ruled out by gel filtration chromatography and circular dichroism spectroscopy (data not shown); however, localized structural differences, such as repositioning of the active site loop containing Thr-74 and Ser-75, that affect peptide binding may occur.
In conclusion, the At-OASS·C10 peptide complex structure offers an initial glimpse of the organization of the plant Cys synthase complex. The structure reveals how interaction with SAT modulates OASS activity and shows that structural plasticity in the plant and bacterial OASS active sites is important for the recognition of dissimilar C termini from plant and bacterial SAT. Analysis of the protein–peptide interaction shows that key active site residues and the C-terminal Ile of SAT are involved in complex formation. Currently, it is unclear how binding of OASS influences SAT activity. Ultimately, a complete understanding of how the plant Cys synthase complex is assembled will require continued structural and functional studies.
METHODS
Protein and Peptide Preparation
Recombinant wild-type, T74S, S75A, S75T, and Q147A mutants were expressed and purified as previously described (Bonner et al., 2005). Peptides corresponding to the 10 C-terminal residues of At-SAT (C10; sequence: YLTEWSDYVI) and the same with a mutation in the C-terminal Ile (sequence: YLTEWSDYVA) were synthesized by Sigma-Genosys.
Protein Crystallography
For cocrystallization, At-OASS and C10 peptide (12.5 mM) were combined to yield a protein/peptide stock solution (12 mg mL−1 At-OASS and 2.5 mM C10 peptide). Crystals were obtained by the vapor diffusion method in 6 μL hanging drops of a 1:1 mixture of protein/peptide solution and crystallization buffer (1.5 M ammonium sulfate and 0.1 M PIPES, pH 6.5) at 4°C over a 0.5-mL reservoir. Crystals were stabilized in cryoprotectant (30% [v/v] glycerol, 1.5 M ammonium sulfate, and 0.1 M PIPES, pH 6.5) before freezing at 100K. All data were collected at 100K using a Proteum-R Smart 6000 CCD detector connected to a Bruker-Nonius FR591 rotating anode generator. Diffraction intensities were integrated, merged, and scaled using the Proteum II software suite (Table 1). The structure of the protein·peptide complex was obtained by difference Fourier methods and was refined using CNS (Brünger et al., 1998). The starting model was the apoenzyme structure of At-OASS lacking PLP (Bonner et al., 2005). After iterative rounds of manual rebuilding in O (Jones et al., 1993) and positional and B-factor refinement in CNS, the R-factors converged with those reported in Table 1. The final model includes residues 3 to 322 of At-OASS, the PLP cofactor, residues 3 to 10 of the C10 peptide, a sulfate molecule, and 112 water molecules. For the structure, 88.6, 11.1, and 0.4% of the amino acids are in the most-favored, additional-allowed, and generously allowed regions of the Ramachandran plot. All structural figures were generated with PyMol (http://www.pymol.org).
Calorimetric Measurements
ITC experiments were performed using a VP-ITC calorimeter (Microcal). Protein and peptide concentrations were determined using molar extinction coefficients for At-OASS homodimer (ɛ280nm = 44,762 M−1 cm−1) and the C10 peptide (ɛ280nm = 8480 M−1 cm−1), as determined by the method of Gill and von Hippel (1989). Extinction coefficients for the At-OASS mutants were assumed to be the same as for wild-type protein. Wild-type and mutant At-OASS and C10 peptide were dialyzed extensively versus 50 mM Tris buffer, pH 7, with 500 mM NaCl at 25°C. All samples and buffer solutions were degassed at room temperature prior to use. Control experiments were done to determine the heat of dilution for each injection by injecting the same volumes of C10 peptide into the sample cell containing only buffer.
Data obtained from the titration of C10 peptide with wild-type and mutant At-OASS were analyzed using either a 1:1 binding model (Equation 1) or a two-site binding model (Equation 2), as described by the following equations:
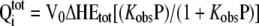 |
(1) |
and
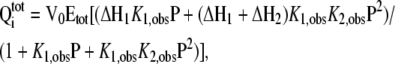 |
(2) |
where Qitot is total heat after the ith injection, V0 is the volume of calorimetric cell, Kobs is the observed equilibrium constant, and ΔH the corresponding enthalpy change. In these equations, P and Etot are the free concentrations of the C10 peptide and total concentration of At-OASS, respectively, in the calorimetric cell after the ith injection. Estimates of Kobs and ΔH were obtained by fitting the experimental data to either one site or two site binding models using software provided by the instrument manufacturer (Microcal). Values for the change in free energy (ΔG) were calculated using ΔG = −RTln(Kobs), where R is the gas constant and T is absolute temperature. Changes in entropy (ΔS) were calculated using ΔG = ΔH − TΔS. Kd was calculated as 1/Kobs.
Fluorescence Measurements
The intrinsic fluorescence emission of At-OASS was monitored using a Cary Eclipse fluormeter (Varian). The excitation wavelength was 412 nm (slit width of 5 nm) with emission signal scanned from 440 to 575 nm (slit width of 5 nm). Scans of At-OASS (1 μM) were performed in 50 mM Tris, pH 7, in the absence or presence of peptide (C10 or mutant) (0.5 mL final volume).
Accession Numbers
Atomic coordinates and structure factors (PDB2ISQ) were deposited in the Protein Data Bank (www.rcsb.org).
Acknowledgments
We thank Eliot Herman for providing support to J.A.F., Xuemin (Sam) Wang for access to the calorimeter in his lab, Tom Smith for advice, and Rebecca Cahoon for all her help. This work was supported by a grant from the USDA (NRI-2005-02518) to J.M.J.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantcell.org) is: Joseph M. Jez (jjez@danforthcenter.org).
References
- Berkowitz, O., Wirtz, M., Wolf, A., Kuhlmann, J., and Hell, R. (2002). Use of biomolecular interaction analysis to elucidate the regulatory mechanism of the cysteine synthase complex from Arabidopsis thaliana. J. Biol. Chem. 277 30629–30634. [DOI] [PubMed] [Google Scholar]
- Bogdanova, N., and Hell, R. (1997). Cysteine synthesis in plants: Protein-protein interactions of serine acetyltransferase from Arabidopsis thaliana. Plant J. 11 251–262. [DOI] [PubMed] [Google Scholar]
- Bonner, E.R., Cahoon, R.E., Knapke, S.M., and Jez, J.M. (2005). Molecular basis of cysteine biosynthesis in plants: Structural and functional analysis of O-acetylserine sulfhydrylase from Arabidopsis thaliana. J. Biol. Chem. 280 38803–38813. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., et al. (1998). Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Rao, G.S., Hohenester, E., Schnackerz, K.D., Cook, P.F., and Jansonius, J.N. (1998). Three-dimensional structure of O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 283 121–133. [DOI] [PubMed] [Google Scholar]
- Burkhard, P., Tai, C.H., Ristroph, C.M., Cook, P.F., and Jansonius, J.N. (1999). Ligand binding induces a large conformational change in O-acetylserine sulfhydrylase from Salmonella typhimurium. J. Mol. Biol. 291 941–953. [DOI] [PubMed] [Google Scholar]
- Campanini, B., Speroni, F., Salsi, E., Cook, P.F., Roderick, S.L., Huang, B., Bettati, S., and Mozzarelli, A. (2005). Interaction of serine acetyltransferase with O- acetylserine sulfhydrylase active site: Evidence from fluorescence spectroscopy. Protein Sci. 14 2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus, M.T., Zocher, G.E., Maier, T.H.P., and Schulz, G.E. (2005). Structure of the O-acetylserine sulfhydrylase isoenzyme CysM from Escherichia coli. Biochemistry 44 8620–8626. [DOI] [PubMed] [Google Scholar]
- Cook, P.F., and Wedding, R.T. (1977). Initial kinetic characterization of the multienzyme complex, cysteine synthetase. Arch. Biochem. Biophys. 178 293–302. [DOI] [PubMed] [Google Scholar]
- Droux, M., Ruffet, M.L., Dounce, R., and Job, B. (1998). Interactions between serine acetyltransferase and O-acetylserine(thiol)lyase in higher plants: Structural and kinetic properties of the free and bound enzymes. Eur. J. Biochem. 255 235–245. [DOI] [PubMed] [Google Scholar]
- Gill, S.C., and von Hippel, P.H. (1989). Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Gorman, J., and Shapiro, L. (2004). Structure of serine acetyltransferase from Haemophilus influenzae. Acta Crystallogr. D Biol. Crystallogr. 60 1600–1605. [DOI] [PubMed] [Google Scholar]
- Hell, R., and Hillebrand, H. (2001). Plant concepts for mineral acquisition and allocation. Curr. Opin. Biotechnol. 12 161–168. [DOI] [PubMed] [Google Scholar]
- Hopkins, L., Parmar, S., Blaszczyk, A., Hesse, H., Hoefgen, R., and Hawkesford, M.J. (2005). O-acetylserine and the regulation of expression of genes encoding components for sulfate uptake and assimilation in potato. Plant Physiol. 138 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrazdina, G., and Jensen, R.A. (1992). Spatial organization of enzymes in plant metabolic pathways. Annu. Rev. Plant Physiol. Plant Mol. Biol. 43 241–267. [Google Scholar]
- Huang, B., Vetting, M.W., and Roderick, S.L. (2005). The active site of O-acetylserine sulfhydrylase is the anchor point for bienzyme complex formation with serine acetyltransferase. J. Bacteriol. 187 3201–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. (1993). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. D Biol. Crystallogr. 49 148–157. [DOI] [PubMed] [Google Scholar]
- Kopriva, S. (2006). Regulation of sulfate assimilation in Arabidopsis and beyond. Ann. Bot. (Lond.) 97 479–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprivova, A., Suter, M., den Camp, R.O., Brunold, C., and Kopriva, S. (2000). Regulation of sulfate assimilation by nitrogen in Arabidopsis. Plant Physiol. 122 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kredich, N.M., Becker, M.A., and Tomkins, G.M. (1969). Purification and characterization of cysteine synthetase, a bifunctional protein complex, from Salmonella typhimurium. J. Biol. Chem. 244 2428–2439. [PubMed] [Google Scholar]
- Kuske, C.R., Ticknor, L.O., Guzman, E., Gurley, L.R., Valdez, J.G., Thompson, M.E., and Jackson, P.J. (1994). Purification and characterization of O-acetylserine sulfhydrylase isoenzymes from Datura innoxia. J. Biol. Chem. 269 6223–6232. [PubMed] [Google Scholar]
- Mino, K., Hiraoka, K., Imamura, K., Sakiyama, T., Eisaki, N., Matsuyama, A., and Nakanishi, K. (2000). Characteristics of serine transacetylase from Escherichia coli deleting different lengths of amino acid residues from the C-terminus. Biosci. Biotechnol. Biochem. 64 1874–1880. [DOI] [PubMed] [Google Scholar]
- Mino, K., Yamanoue, T., Sakiyama, T., Eisaki, N., Matsuyama, A., and Nakanishi, K. (1999). Purification and characterization of serine acetyltransferase from Escherichia coli partially truncated at the C-terminal region. Biosci. Biotechnol. Biochem. 63 168–179. [DOI] [PubMed] [Google Scholar]
- Noji, M., Inoue, K., Kimura, N., Gouda, A., and Saito, K. (1998). Isoform-dependent differences in feedback regulation and subcellular localization of serine acetyltransferase involved in cysteine biosynthesis from Arabidopsis thaliana. J. Biol. Chem. 273 32739–32745. [DOI] [PubMed] [Google Scholar]
- Olsen, L.R., Huang, B., Vetting, M.B., and Roderick, S.L. (2004). Structure of serine acetyltransferase in complexes with CoA and its cysteine feedback inhibitor. Biochemistry 43 6013–6019. [DOI] [PubMed] [Google Scholar]
- Pye, V.E., Tingey, A.P., Robson, R.L., and Moody, P.C.E. (2004). The structure and mechanism of serine acetyltransferase from Escherichia coli. J. Biol. Chem. 279 40729–40736. [DOI] [PubMed] [Google Scholar]
- Rabeh, W.M., and Cook, P.F. (2004). Structure and mechanism of O-acetylserine sulfhydrylase. J. Biol. Chem. 279 26803–26806. [DOI] [PubMed] [Google Scholar]
- Renzoni, D.A., Pugh, D.J., Siligardi, G., Das, P., Morton, C.J., Rossi, C., Waterfield, M.D., Campbell, I.D., and Ladbury, J.E. (1996). Structural and thermodynamic characterization of the interaction of the SH3 domain from Fyn with the proline-rich binding site on the p85 subunit of PI3-kinase. Biochemistry 35 15646–15653. [DOI] [PubMed] [Google Scholar]
- Rolland, N., Ruffet, M.L., Job, D., Douce, R., and Droux, M. (1996). Spinach chloroplast O-acetylserine(thiol)-lyase exhibits two catalytically non-equivalent pyridoxal-5′-phosphate-containing active sites. Eur. J. Biochem. 236 272–282. [DOI] [PubMed] [Google Scholar]
- Saito, K., Kurosawa, M., Tatsuguchi, K., Takagi, Y., and Murakoshi, I. (1994). Modulation of cysteine biosynthesis in chloroplasts of transgenic tobacco overexpressing cysteine synthase (O-acetylserine(thiol)lyase). Plant Physiol. 106 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, K., Yokoyama, H., Noji, M., and Murakoshi, I. (1995). Molecular cloning and characterization of a plant serine acetyltransferase playing a regulatory role in cysteine biosynthesis from watermelon. J. Biol. Chem. 270 16321–16326. [DOI] [PubMed] [Google Scholar]
- Smith, F.W., Hawkesford, M.J., Ealing, P.M., Clarkson, D.T., Vanden Berg, P.J., Belcher, A.R., and Warrilow, A.G. (1997). Regulation of expression of a cDNA from barley roots encoding a high affinity sulphate transporter. Plant J. 12 875–878. [DOI] [PubMed] [Google Scholar]
- Srere, P.A. (1987). Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 56 89–124. [DOI] [PubMed] [Google Scholar]
- Winkel, B.S.J. (2004). Metabolic channeling in plants. Annu. Rev. Plant Biol. 55 85–107. [DOI] [PubMed] [Google Scholar]
- Wirtz, M., Berkowitz, O., Droux, M., and Hell, R. (2001). The cysteine synthase complex in plants: Mitochondrial serine acetyltransferase from Arabidopsis thaliana carries a bifunctional domain for catalysis and protein-protein interaction. Eur. J. Biochem. 268 686–693. [DOI] [PubMed] [Google Scholar]
- Wirtz, M., and Droux, M. (2005). Synthesis of the sulfur amino acids: Cysteine and methionine. Photosynth. Res. 86 345–346. [DOI] [PubMed] [Google Scholar]
- Xia, Y., Yu, H., Jansen, R., Seringhaus, M., Baxter, S., Greenbaum, D., Zhao, H., and Gerstein, M. (2004). Analyzing cellular biochemistry in terms of molecular networks. Annu. Rev. Biochem. 73 1051–1087. [DOI] [PubMed] [Google Scholar]
- Zhao, C., Moriga, Y., Feng, B., Kumada, Y., Imanaka, H., Imamura, K., and Nakanishi, K. (2006). On the interaction site of serine acetyltransferase in the cysteine synthase complex from Escherichia coli. Biochem. Biophys. Res. Commun. 341 911–916. [DOI] [PubMed] [Google Scholar]
- Zhu, X., Yamaguchi, T., and Masada, M. (1998). Complexes of serine acetyltransferase and isozymes of cysteine synthase in spinach leaves. Biosci. Biotechnol. Biochem. 62 947–952. [DOI] [PubMed] [Google Scholar]