

Abstract
Over 110 structurally diverse missense mutations in the superoxide dismutase (SOD1) gene have been linked to the pathogenesis of familial amyotrophic lateral sclerosis (FALS), yet the mechanism by which these lead to cytotoxicity still remains unknown. We have synthesized wild-type and mutant SOD1 in synchronized cell-free reticulocyte extracts replete with the full complement of molecular chaperones and folding facilitators that are normally required to fold this metalloenzyme. Here, we report that, despite being a small, single-domain protein, human SOD1 folds post-translationally to a hyperstable native-like conformation without a requirement for ATP-dependent molecular chaperones. SOD1 folding requires tight Zn but not Cu binding and proceeds through at least three kinetically and biochemically distinct states. We find that all 11 FALS-associated SOD1 mutants examined using this system delay the kinetics of folding, but do not necessarily preclude the formation of native-like states. These data suggest a model whereby impaired post-translational folding increases the population of on- and off-pathway folding intermediates that could provide an important source of proto-toxic protein, and suggest a unifying mechanism for SOD1-linked FALS pathogenesis.
Keywords: Amyotrophic lateral sclerosis, cell-free protein synthesis, protein folding, superoxide dismutase
Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive, adult-onset neurodegenerative disease that selectively targets motor neurons (Pasinelli and Brown, 2006). Although the majority of ALS cases are sporadic (SALS), approximately 5–10% of patients have a genetically inherited form known as familial ALS (FALS) (Pasinelli and Brown, 2006). Nearly 20% of FALS cases are linked to dominantly inherited missense mutations in SOD1, which encodes the abundant and ubiquitously expressed 32 kDa homodimeric cytoplasmic metalloenzyme Cu, Zn superoxide dismutase (SOD1) (Rosen, 1993). Although there is no compelling evidence implicating SOD1 dysfunction in the vastly more prevalent sporadic SALS, the similarities in clinical presentation and histopathology among SOD1-linked FALS, SALS and animal models expressing human mutant SOD1 transgenes have focused intense attention on SOD1-linked FALS as a genetically and biochemically tractable model for this devastating disease. However, despite intense scrutiny and the identification of over 110 FALS-linked mutations in SOD1 (http://alsod.org), elucidation of a common mechanism linking motor neuron death to structural alterations of the SOD1 protein remains elusive.
It is now widely accepted that the genetic mechanism by which mutations in the SOD1 gene cause FALS pathogenesis is via a toxic gain of function, although the biochemical nature of this toxic property (or whether there is indeed a single common property) is controversial (Pasinelli and Brown, 2006). As the vast preponderance of FALS-linked SOD1 mutations are missense—as opposed to null—it has been logically assumed that disease-causing mutations in SOD1 should give rise to toxic or proto-toxic non-native SOD1 molecules (Hart, 2006). This assumption is supported by the presence of aggregated SOD1 in inclusion bodies in spinal cords from FALS patients harboring SOD1 mutations (Shibata et al, 1996; Nishiyama et al, 1997; Ince et al, 1998; Kato et al, 1999; Kokubo et al, 1999) and from SOD1 transgenic mice (Wong et al, 1995; Bruijn et al, 1997; Dal Canto and Gurney, 1997; Shibata et al, 1998; Watanabe et al, 2001) as well as in cell culture models (Durham et al, 1997; Koide et al, 1998; Johnston et al, 2000). Moreover, the appearance of SDS-insoluble high molecular weight human SOD1-containing complexes in spinal cord extracts from transgenic mice (Johnston et al, 2000; Wang et al, 2003) preceding the onset of neurological symptoms (Johnston et al, 2000) supports a linkage between ‘misfolding' of mutant SOD1 and motor neuron degeneration. Such non-native SOD1 protomers could lead to toxicity in a variety of ways: mislocalization to organelles such as mitochondria (Liu et al, 2004) or peroxisomes (Higgins et al, 2003), aberrant catalytic production of oxygen or peroxide radicals (Pasinelli and Brown, 2006), aggregation into insoluble complexes that could deplete soluble cellular factors (Hart, 2006) or interference with axonal transport (Boillee et al, 2006) or proteasome function (Kabashi et al, 2004).
Additional support for SOD1 misfolding comes from the observation that some SOD1 mutants are incapable of producing fully metallated, native, enzymatically active protein when expressed recombinantly (Hayward et al, 2002). Unsurprisingly, most of these mutations correspond to amino acids that directly or indirectly ligand the bound Zn and Cu atoms that are required for stability and catalysis (Hayward et al, 2002). Nevertheless, many of the FALS-linked SOD1 mutants produce SOD1 molecules that exhibit full enzymatic activity in vivo (Bowling et al, 1995; Ceroni et al, 1999) and in vitro (Hayward et al, 2002) along with tertiary and quaternary structures that are virtually indistinguishable from native wild-type protein (Valentine et al, 2005). These ‘native-like' mutations are scattered throughout the eight-stranded flattened Greek key β-barrel that comprises the bulk of the SOD1 structure and do not appear to impair metal binding. One way to reconcile the synthesis of native, enzymatically active SOD1 mutants with the production of toxic, misfolded SOD1 conformers is if the FALS mutations decrease the stability of the folded state, resulting in an increased propensity to unfold. Although several FALS mutants have been reported to exhibit reduced thermal stability (Rodriguez et al, 2002), analysis of the metal-free states of 20 FALS-linked SOD1 mutants by differential scanning calorimetry revealed a wide range of stabilities that overlap with the wild-type protein (Rodriguez et al, 2005). Therefore, decreased stability is not a universal property of the metal-free state of disease-linked SOD1 mutants, indicating that the toxic, non-native forms of the protein must have an additional source.
A second way that the population of aggregation-prone partially folded states might be increased would be if mutations were to alter the energy landscape upon which SOD1 folding proceeds leading to increased latency of partially folded intermediates. This hypothesis is supported by recent in vitro chemical denaturation/refolding studies that describe SOD1 folding by a three-state model with transitions proceeding from metal-free unfolded monomer to metallated folded monomer and subsequent dimerization (Lindberg et al, 2005; Rumfeldt et al, 2006). It is not clear, however, to what extent these in vitro studies using pure recombinant protein truly reflect the situation in vivo. Physiological folding of SOD1 in the cell takes place in the reducing environment of the cytoplasm, where protein concentrations are in excess of 50 mg/ml and nascent polypeptides are synthesized in an ordered, N-terminal-first vectorial fashion and accessory factors are required to facilitate metal loading (Culotta et al, 2006). It is thus of central importance to understand the pathway of SOD1 folding under these physiological conditions and the effects of FALS mutations thereupon.
In this study, we examined the folding of wild-type and FALS-associated mutants of SOD1 in a synchronized cell-free rabbit reticulocyte lysate (RRL) translation system. These extracts contain physiological complements of molecular chaperones and total protein. As such, they have been extensively exploited to study protein folding under experimentally tractable conditions that approximate those found in the mammalian cytosol (Merrick, 1990; Frydman et al, 1994; Endo and Sawasaki, 2006). Moreover, as reticulocytes normally produce high levels of SOD1, it is highly probable that this system contains all possible factors required for synthesizing and folding native SOD1. We developed several assays to probe the folding state of newly synthesized SOD1, exploiting distinctive characteristics of the native enzyme such as hyper-resistance to proteolysis, dimer formation and intramolecular disulfide bond formation. The data reveal that nascent SOD1 molecules fold post-translationally via a sequence of temporally ordered monomeric intermediates that do not require ATP-dependent chaperones. We find that all 11 FALS-associated SOD1 mutants examined using this system delay the kinetics of folding, but do not necessarily preclude the formation of native-like states. These data suggest a model whereby impaired post-translational folding increases the population of on- and off-pathway folding intermediates that provide an important source of proto-toxic protein, and may provide a unifying mechanism for SOD1-linked FALS pathogenesis.
Results
Post-translational folding of SOD1 in synchronized reticulocyte lysates
To monitor the kinetics and molecular requirements of SOD1 folding, in vitro-transcribed, m7G-capped mRNA encoding WT-SOD1 was used to initiate protein synthesis in a RRL translation system. To create a homogeneous pool of newly synthesized WT-SOD1, translation was synchronized by the addition of aurintricarboxylic acid (ATCA), an inhibitor of translation initiation (Figure 1A) (Stewart et al, 1971; Frydman et al, 1994). 35S-labeled full-length WT-SOD1 was first detected ∼4 min following mRNA addition and plateaued by 8 min, indicating that WT-SOD1 synthesis was tightly synchronized in this system (data not shown). To monitor the kinetics of WT-SOD1 folding, lysates were digested with proteinase K (0.01 mg/ml) at various times following initiation of protein synthesis (Figure 1B). Initially, nascent WT-SOD1 was mostly (∼85% at 4.5 min) susceptible to proteolysis, but became progressively resistant (t1/2∼8 min) to digestion. As translation of WT-SOD1 is completed by ∼8 min, these data indicate that the bulk of newly synthesized WT-SOD1 folds post-translationally. No proteolytic fragments from proteinase K digestion were resolved by SDS–PAGE (data not shown), suggesting that folding of WT-SOD1, as expected for a single-domain protein, did not proceed through any stable protease-resistant intermediates.
Figure 1.
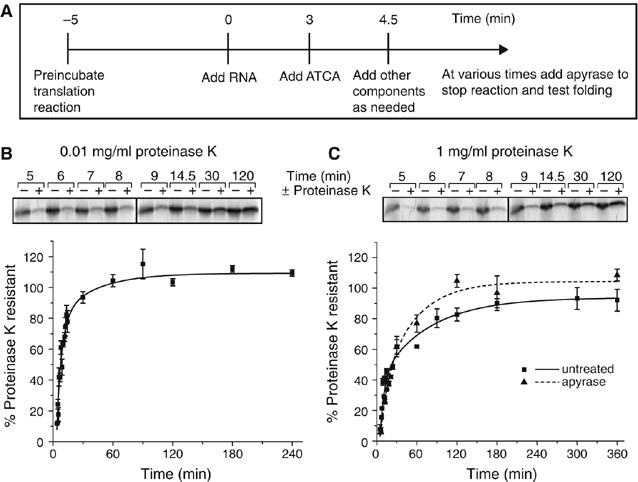
Post-translational acquisition of WT-SOD1 resistance to proteinase K does not require ATP. (A) Schematic diagram of synchronized cell-free translation assay. (B) Kinetics of acquisition of resistance to 0.01 mg/ml proteinase K. Translations were terminated at the indicated times and radioactivity incorporated into SOD1 was assessed by SDS–PAGE/phosphorimage analysis before (−) or after (+) digestion with 0.01 mg/ml proteinase K (top panel). Normalized data (mean±s.e.m.) from multiple experiments (n=3–12) were fitted using least squares regression (bottom panel). (C) Kinetics of acquisition of resistance to 1 mg/ml proteinase K. Samples were analyzed and presented as in (B).
Native WT-SOD1 belongs to a class of hyperstable proteins (Forman and Fridovich, 1973); it is resistant to proteinase K concentrations of up to 1 mg/ml for 30 min at 37°C (Ratovitski et al, 1999). WT-SOD1 chains synthesized in RRL became fully resistant to high (1 mg/ml) proteinase K concentrations, with distinctly slower (t1/2∼19 min) kinetics compared to the lower protease concentration (Figure 1C), indicating that RRL is capable of synthesizing and folding WT-SOD1 to a highly protease-resistant conformation. Thus, the folding of WT-SOD1 in RRL proceeds through at least two steps that can be distinguished by differing levels of protease resistance. Significantly, the folding kinetics of WT-SOD1, assessed by acquisition of resistance to high concentrations of proteinase K, were unaffected by ATP depletion (Figure 1C), suggesting that ATP-dependent molecular chaperones such as Hsc/p70 are not required for its folding.
WT-SOD1 is a homodimeric metalloenzyme that binds a single atom each of copper and zinc per monomer. To examine the requirement for these metals for WT-SOD1 folding, metal chelators were added to the translation reaction before initiating protein synthesis with mRNA. Tetraethylenepentamine pentahydrochloride (TEPA), a copper chelator, at 100 μM had no effect on the kinetics or extent of acquisition of protease resistance of WT-SOD1 (Figure 2A and B). By contrast, addition of N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN), a zinc chelator, at 5 μM completely abrogated the ability of WT-SOD1 to gain detectable resistance to high (Figure 2A) but not low (Figure 2B) concentrations of proteinase K. Neither chelator affected the kinetics or efficiency of WT-SOD1 translation (data not shown), indicating that these metals are not required for protein synthesis in RRL. The inhibitory effect of TPEN on the acquisition of high-level protease resistance was prevented by preincubating TPEN with 1.5 M equivalents of zinc sulfate (Figure 2A) confirming that the TPEN effect was specifically due to chelation of zinc and not other metals. Addition of TPEN to the reaction at 4.5 min, a time point at which translation but not folding of a majority of WT-SOD1 chains was complete, also eliminated the ability of WT-SOD1 to acquire protease resistance (data not shown). By contrast, TPEN treatment of lysates immediately before addition of proteinase K had no effect on the extent of resistance to high proteinase K concentrations (data not shown), suggesting that Zn is extremely tightly bound by the protease-resistant form (Kd TPEN for Zn=10−15 M), as expected for correctly folded, native WT-SOD1. Interestingly, TPEN, even at 100 μM, did not interfere with the ability of WT-SOD1 to become resistant to low (0.01 mg/ml) concentrations of proteinase K (Figure 2B), indicating that the WT-SOD1 conformation detected by this assay does not require zinc binding. To further explore the role of Zn binding in WT-SOD1 folding, we assessed the kinetics of acquisition of proteinase K resistance in a SOD1 mutant lacking one of the triad of imidazole rings, His80, that comprises the Zn-binding site. Like TPEN-treated WT-SOD1, H80R was completely incapable of acquiring resistance to high concentrations of proteinase K even in the absence of chelator (Figure 2A). This mutant was able to become resistant to low concentrations of protease (Figure 2B), albeit with considerably delayed kinetics compared to TPEN-treated WT-SOD1. Together, these data establish that post-translational Zn binding is essential for WT-SOD1 to fold to a native-like hyperstable conformation, but not for an earlier folding intermediate distinguished by resistance to low but not high levels of protease. The fact that H80R, a Zn-binding mutation, is linked to ALS (Alexander et al, 2002) suggests that one way that mutations could give rise to the formation of toxic species is to delay or prevent folding of SOD1 to a hyperstable, native state.
Figure 2.
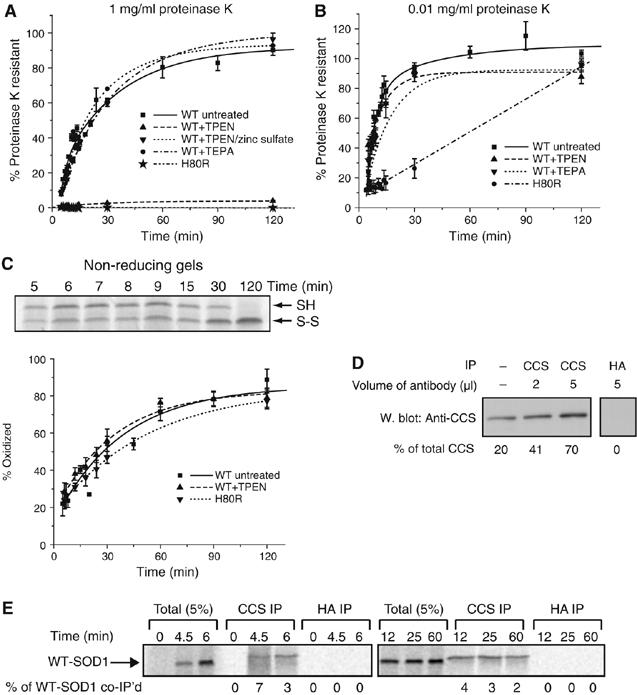
Zinc is required for folding to a compact state but not for disulfide bond formation. Effect of metal chelators on the kinetics of acquisition of resistance of WT-SOD1 to 1 mg/ml (A) or 0.01 mg/ml (B) proteinase K. Samples were prepared and analyzed as in Figure 1B. Proteinase K resistance of zinc-binding mutant H80R is also shown. (C) Top panel, representative non-reducing SDS–PAGE gel of WT-SOD1 showing maturation from mostly reduced to fully oxidized state. Bottom panel, quantification of oxidized fraction of H80R and of WT-SOD1 with or without treatment with 100 μM TPEN, as indicated. Graphical data are mean±s.e.m. from a minimum of three independent experiments per time point. (D) Immunoprecipitation of endogenous CCS from RRL. RRL was immunoprecipitated with human CCS mAb or with control HA mAb, as indicated. Immune complexes were subjected to immunoblotting with a CCS polyclonal antibody and band intensities relative to total CCS were quantified by densitometry, indicated below each lane. Far left lane, loading control of 20% of total RRL in the immunoprecipitations. (E) Co-immunoprecipitation of newly synthesized WT-SOD1 with CCS. Endogenous CCS was immunoprecipitated using the CCS mAb, as in (D), at various times following the translation of newly synthesized WT-SOD1. Anti-HA was used as an immunoprecipitation control. The percentage of newly synthesized WT-SOD1 co-immunoprecipitated with the monoclonal antibodies was quantified via SDS–PAGE/phosphorimage analysis and noted below each lane.
Folding of SOD1 in mammalian cells and yeast normally occurs in the presence of the copper chaperone for SOD1 (CCS), which facilitates copper loading via formation of a transient heterodimer with SOD1. Although the TEPA experiment demonstrated that WT-SOD1 does not require copper binding, it is possible that CCS could facilitate SOD1 folding by a process independent of copper delivery. For example, CCS has been reported to facilitate formation of the intramolecular disulfide bond in yeast (Furukawa et al, 2004). We used quantitative immunoblot analysis with a standard of highly purified CCS to estimate that the RRL contains CCS at a concentration of approximately 200 μg/ml (data not shown). To assess a possible role for CCS in WT-SOD1 folding, we quantified the extent and kinetics of association of newly synthesized WT-SOD1 and CCS by co-immunoprecipitation using a mAb to human CCS. A control experiment indicated that the human CCS mAb was capable of precipitating at least 70% of endogenous rabbit CCS from RRL (Figure 2D). However, only a small (<10%) fraction of nascent WT-SOD1 chains could be co-precipitated with this antibody at any time point following translation (Figure 2E). This fraction remained relatively constant over the course of WT-SOD1 folding for at least 60 min. These results suggest that the WT-SOD1-CCS interaction is either too transient to capture efficiently in this type of experiment, or that it forms asynchronously during the course of post-translational folding.
Intramolecular disulfide bond formation
Formation of the single intramolecular disulfide bond in WT-SOD1 was assessed by quantifying the relative amounts of oxidized and reduced nascent [35S]SOD1 using non-reducing SDS–PAGE of chemically alkylated RRL reactions (Figure 2C). At the earliest time point examined (5 min), approximately 20% of WT-SOD1 was already in the oxidized form (compared to >80% chain completion). As the protein resolved to a monomer-sized band on the gel, this represented an intramolecular and not intermolecular disulfide bond. The kinetics for disulfide bond formation (t1/2∼20 min) were similar to the time required for acquisition of resistance to high levels of proteinase K (Figure 1C). Treatment of folding reactions with TPEN did not appear to influence the rate of disulfide formation. Similarly, the extent of disulfide bond formation was not altered by mutation of the zinc ligand His80. Therefore, zinc binding is not essential for disulfide bond formation in WT-SOD1.
Formation of WT-SOD1 dimers
To assess the oligomeric state of newly synthesized WT-SOD1, folding reactions were separated by gel filtration at various times and fractions were analyzed by SDS–PAGE. The retention time for WT-SOD1 dimers was determined by analysis of WT-SOD1 purified from erythrocytes. The retention time for SOD1 monomers was determined by analysis of an engineered SOD1 variant that contains two mutations (F50E/G51E) in the dimer interface that have been previously shown to prevent dimerization (Bertini et al, 1994). By gel filtration (Figure 3A) and by native gel electrophoresis (data not shown), newly synthesized F50E/G51E remained exclusively monomeric, even upon prolonged incubation. By contrast, newly synthesized WT-SOD1 eluted largely in a monomer-sized peak that progressively shifted to a dimer-sized peak with increasing time (Figure 3A). These peaks were deconvolved and the area under the dimer-sized peak was fit to a single exponential equation yielding a t1/2 for dimer formation of 30 min (Figure 3B), suggesting that WT-SOD1 dimer formation occurs after the protein folds to a proteinase K-resistant, oxidized state.
Figure 3.
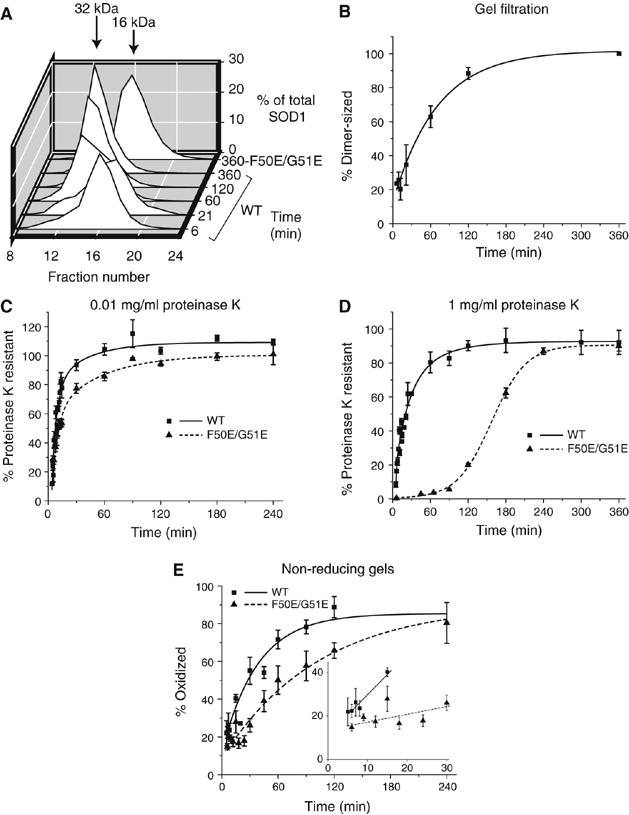
WT-SOD1 dimerization is not required for proteinase K resistance. (A) Representative gel filtration profiles for WT-SOD1 and the monomeric variant F50E/G51E. (B) Kinetics of WT-SOD1 dimerization. (C, D) Kinetics of acquisition of resistance to 0.01 mg/ml (C) and 1 mg/ml (D) proteinase K for the F50E/G51E variant compared to WT-SOD1 (from Figure 1). Samples were prepared and analyzed as in Figure 1B. (E) Quantification of the oxidized fraction of F50E/G51E as a function of time. Inset, initial rates of disulfide bond formation for WT-SOD1 and F50E/G51E fitted to linear least-squares regression model. Graphical data are mean±s.e.m. from a minimum of three independent experiments per time point.
To further examine the relationship between dimerization and folding, monomeric F50E/G51E was subjected to protease digestion. This variant exhibited similar kinetics of acquisition of resistance to low (0.01 mg/ml) concentrations of proteinase K compared to WT-SOD1 (Figure 3C). F50E/G51E was also able to acquire resistance to the higher concentration of proteinase K, albeit with significantly slower kinetics compared to WT-SOD1 (Figure 3D). Likewise, although delayed, F50E/G51E was able to become fully oxidized (Figure 3E).
Together, these data suggest that RRL is capable of supporting the synthesis of WT-SOD1 molecules that resemble native SOD1 in their ability to resist digestion with high concentrations of proteinase K and to form homodimers containing an intrachain disulfide bond. These data also show that WT-SOD1 folding requires the presence of zinc and proceeds through a set of at least three kinetically distinct intermediates that can be probed with simple assays that are suitable to the radiochemical levels of protein produced in this complex milieu that closely mimics the conditions in erythrocytic precursor cells that normally synthesize this protein.
Folding of FALS-associated SOD1 mutants
The inability of the FALS-linked zinc-binding mutant, H80R, to fold in our assay suggested the possibility that defective folding might be a feature common to other FALS mutations. To test this hypothesis, we subjected a subset of FALS mutants, selected to span the major structural and functional domains of the protein, to detailed analysis using the tools described above. The copper ligand mutants, H46R and H48Q, were both slow to acquire resistance to the low concentration of proteinase K (Figure 4A) and were severely defective in gaining resistance to 1 mg/ml proteinase K (Figure 4B). These mutants also exhibited delayed kinetics of disulfide bond formation (Figure 4C). As chelation of copper does not appear to affect WT-SOD1 folding (Figure 2), these data are consistent with previous studies suggesting that mutation of these copper ligand residues influences the geometry of the metal-binding loop (Hayward et al, 2002; Antonyuk et al, 2005). Additionally, H46R shows significantly slower dimerization kinetics (t1/2∼90 min) compared to WT-SOD1 (Figure 4D and E).
Figure 4.
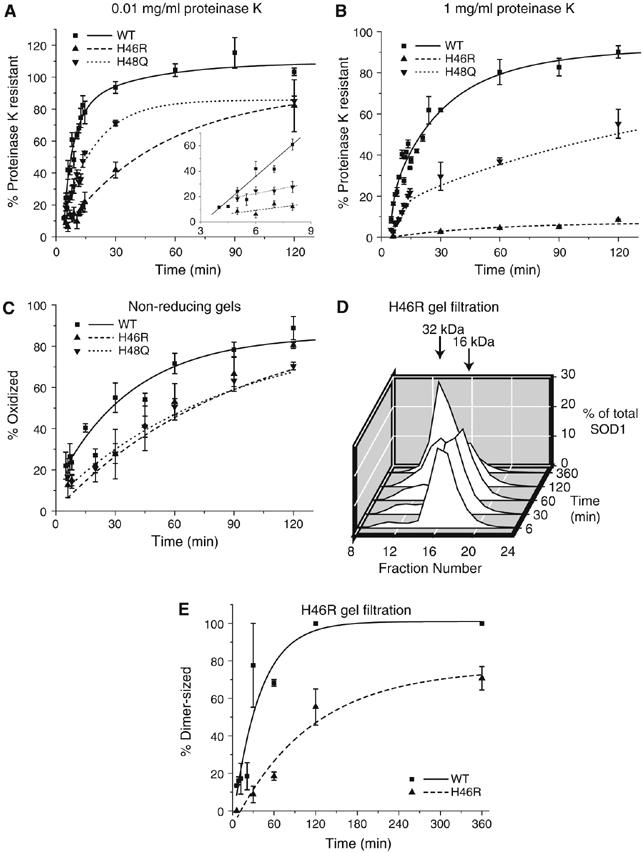
Effects of FALS-linked metal-binding loop mutations on SOD1 folding. Kinetics of acquisition of resistance of metal-binding FALS mutants to 0.01 mg/ml (A) and 1 mg/ml (B) proteinase K. Inset, initial rates of protease resistance for WT-SOD1 and mutants fitted to a linear least-squares regression model. (C) Kinetics of disulfide bond formation as described in Figure 2C. (D) Gel filtration profile for H46R as in Figure 3A. (E) Quantification of H46R dimerization kinetics compared with WT-SOD1 data from Figure 3B. Graphical data are mean±s.e.m. from a minimum of three independent experiments per time point.
SOD1 contains four cysteine residues of which two, Cys57 and Cys146, participate in the formation of the intramolecular disulfide bond. We selected three FALS-linked cysteine mutations for analysis. C6F exhibited a complete inability to fold as assessed by both the protease resistance assay and the disulfide bond formation assay (Figure 5A and C). As expected, the two disulfide bond mutants failed to form a disulfide bond (Figure 5C). Although both disulfide bond mutants exhibited impaired acquisition of proteinase K resistance (Figure 5A and B), mutation of Cys57 was considerably more severe than its counterpart.
Figure 5.
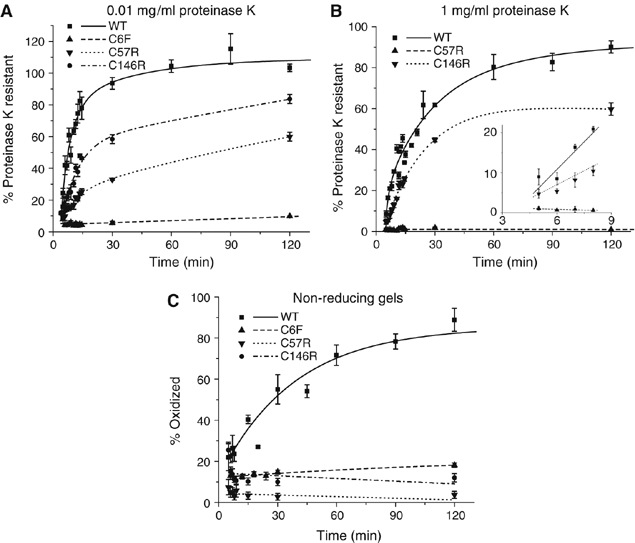
Effects of FALS-linked cysteine mutations on SOD1 folding. Kinetics of acquisition of resistance of FALS-linked Cys mutants to 0.01 mg/ml (A) and 1 mg/ml (B) proteinase K. Inset, initial rates of protease resistance for WT-SOD1 and mutants fitted to a linear least-squares regression model. (C) Kinetics of disulfide bond formation as described in Figure 2C. Graphical data are mean±s.e.m. from a minimum of three independent experiments per time point.
A subset of SOD1 mutants, including E100K, D101N and N139K, have been reported to exhibit wild-type thermal stability and hydrogen–deuterium exchange kinetics, despite being linked to the pathogenesis of autosomal dominant FALS (Rodriguez et al, 2005). We found that all three of these mutants demonstrated significantly slower initial rates of folding as measured by both of the protease resistance assays, although all were able to achieve greater than 50% resistance to both high and low concentrations of proteinase K within the 2 h assay period (Figure 6A and B). Additionally, both D101N and N139K displayed a distinct delay in their initial rate that was not observed with any of the other mutants (Figure 6B). Substitution of Glu100 with Gly (E100G), one of the more common FALS-linked mutations, was slightly more impaired in the rate of acquisition of resistance to proteinase K (Figure 7A and B) in comparison to the more conservative Lys substitution. These four mutants (E100K, E100G, D101N and N139K) also behaved identically to WT-SOD1 in the disulfide bond formation assay (Figure 7C; data not shown). Also, E100G showed similar kinetics to WT-SOD1 in the dimerization assay (Figure 7D). Similar to E100G, A4V, the most common FALS mutation in North America, displayed significantly slower kinetics in the proteolysis assays (Figure 7A and B) with no delay in disulfide bond formation (Figure 7C). Unlike E100G, A4V, which is located along the dimer interface, showed a significant retardation in dimer formation (t1/2∼80 min) (Figure 7D).
Figure 6.
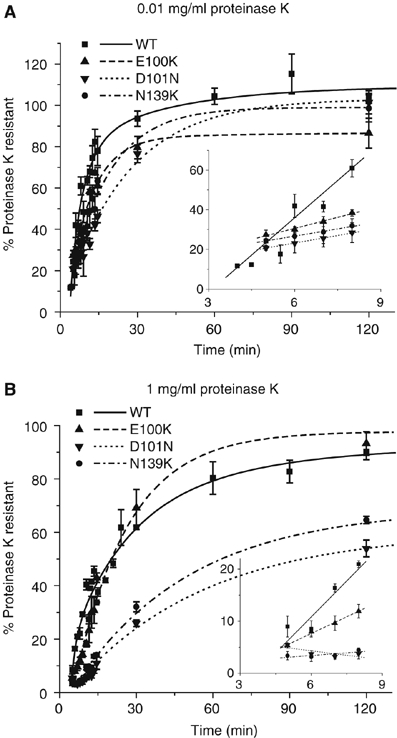
Effects of ‘wild-type like' FALS-linked mutations on SOD1 folding. Kinetics of acquisition of resistance of FALS-linked ‘wild-type-like' mutants to 0.01 mg/ml (A) and 1 mg/ml (B) proteinase K. Inset, initial rates of protease resistance for WT-SOD1 and mutants fitted to a linear least-squares regression model. Graphical data are mean±s.e.m. from a minimum of three independent experiments per time point.
Figure 7.
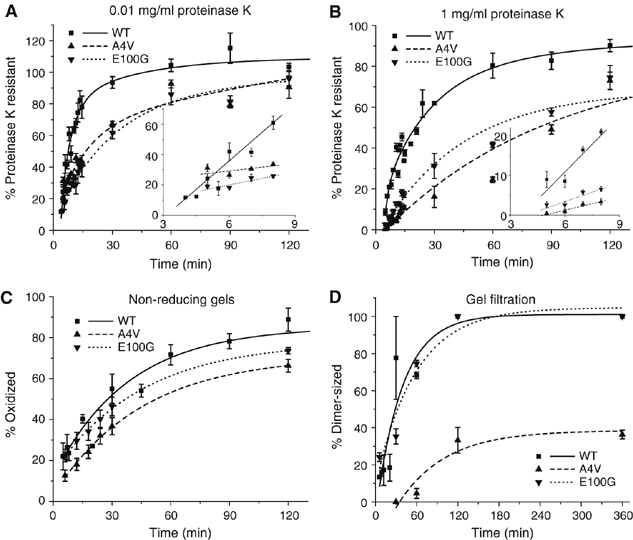
Folding and dimerization kinetics of A4V and E100G. Kinetics of acquisition of resistance of FALS mutants to 0.01 mg/ml (A) and 1 mg/ml (B) proteinase K. Inset, initial rates of protease resistance for WT-SOD1 and mutants fitted to a linear least-squares regression model. (C) Kinetics of disulfide bond formation as described in Figure 2C. (D) Quantification of A4V and E100G dimerization kinetics compared with WT-SOD1 data from Figure 3B. Graphical data are mean±s.e.m. from a minimum of three independent experiments per time point.
Discussion
Although the genetic linkage between SOD1 and FALS pathogenesis has been recognized for over a decade, the mechanism by which missense mutations in this gene give rise to the toxic conformers of the SOD1 protein—the presumed consequence of such gain-of-function mutations—remains unsolved. In this study, we have used cell-free synthesis in reticulocyte lysates to probe the pathway by which WT-SOD1 folds and the effects of FALS-linked mutations on the kinetics and yield of this process. Our data suggest that WT-SOD1 folds post-translationally and requires high-affinity zinc binding. The folding rates of all 11, structurally diverse, FALS mutants examined in this study were significantly impaired compared with WT-SOD1. We propose that FALS mutations alter SOD1's folding landscape, leading to increased population of intermediates that are prone to becoming trapped in inappropriate, potentially toxic aggregates.
Small, single-domain cytoplasmic proteins like SOD1 typically fold rapidly (<1 s) in vitro (Fersht, 2000), by a process best described by a two-state model (Daggett and Fersht, 2003). Surprisingly, in vitro refolding of pure WT-SOD1 is extremely inefficient and slow (>24 h) (Rumfeldt et al, 2006), suggesting that its folding must require factors present within the cytoplasm of cells in which it is normally synthesized. In accord with this view, we find that folding of WT-SOD1 in cell-free synchronized RRL translation extracts proceeds with quantitative efficiency and considerably accelerated kinetics (t1/2=20–30 min). Our data also establish that WT-SOD1 folding in RRL occurs post-translationally and proceeds through a set of biochemically and kinetically distinct intermediates that can be distinguished by resistance to different concentrations of proteinase K, oxidation of Cys57 and Cys146, and assembly into homodimers.
Nascent WT-SOD1 rapidly (t1/2∼8 min) forms a monomeric intermediate that is resistant to 10 μg/ml proteinase K—a concentration often used to define folded states of proteins (Frydman et al, 1994; Manning and Colon, 2004; Hoffmann et al, 2006). Formation of this intermediate, which lacks a disulfide bond, does not require the presence of Zn or Cu. It is possible that this intermediate corresponds to the Zn-free state in which, from solution NMR studies, the eight-stranded β-barrel is opened up, clam-like, where each shell is comprised of two four-stranded antiparallel β-sheets and the metal-binding and electrostatic loops are highly disordered (Banci et al, 2003). This intermediate compacts with time (t1/2∼19 min) into a second intermediate that more closely resembles the native protein in that it is resistant to unusually high concentrations of proteinase K, requires Zn (but not Cu) and contains the correct intrachain disulfide bond. Dimerization proceeds with slower (t1/2∼30 min) kinetics and is not essential for formation of the second compact intermediate, as the engineered monomeric mutant, F50E/G51E, was also capable of becoming resistant to high proteinase K concentrations.
Our data suggest that WT-SOD1 folding does not require the activity of ATP-dependent molecular chaperones as ATP depletion has no effect on its protease resistance, although they do not rule out the possibility that ATP-dependent chaperones are required for dimer formation. The absence of an ATP requirement for WT-SOD1 folding is not surprising as most single-domain proteins are able to fold efficiently without the facilitation of ATP-dependent molecular chaperones (Dobson and Karplus, 1999; Feldman and Frydman, 2000). However, the long delay between chain completion (4–8 min) and formation of the native-like protease-resistant intermediate (t1/2∼19 min) is not consistent with such a spontaneous folding process. By comparison, firefly luciferase, a two-domain protein and obligate chaperone substrate, synthesized in a very similar synchronized RRL exhibits only a short (∼2 min) lag between completion of full-length chains and acquisition of enzymatic activity (Frydman et al, 1994). Although SOD1 is a single-domain protein, it is also a metalloenzyme and exhibits an absolute requirement for Zn in forming a native-like compact intermediate.
Our data show that Cu binding is not required for conformational maturation of WT-SOD1 in RRL. Because of its inherent toxicity, free Cu in cells is maintained at vanishingly low concentrations (Rae et al, 1999); this metal must be delivered to nascent SOD1 molecules by carriers such as CCS. Although we find that conformational maturation of WT-SOD1 does not require copper, our data do not strictly exclude the possibility of a Cu-independent role for CCS in this process. For example, CCS has been implicated in facilitating O2-dependent disulfide formation in yeast SOD1 (Furukawa et al, 2004). Additionally, human CCS can bind the Zn(II) ion and has been proposed to function as a Zn chaperone for SOD1 (Furukawa and O'Halloran, 2006). CCS is highly homologous to SOD1 and the yeast homologs of CCS and SOD1 have been shown to form a transient heterodimer using the same interface normally used for SOD1 homodimer formation. Notably, this heterodimer does not require Cu to be bound to CCS (Lamb et al, 2001). Although CCS can be readily detected by immunoblot in RRL (data not shown), our gel filtration HPLC system does not have the resolution to discriminate between SOD1 homodimers and SOD1-CCS heterodimers. Nevertheless, only a small fraction (<10%) of nascent WT-SOD1 chains could be co-immunoprecipitated with CCS antibodies at any time point following synthesis (Figure 2E), despite near-quantitative pull-down of CCS (Figure 2D, data not shown). Similarly, translation and precipitation of a C-terminal S-tagged form of WT-SOD1 co-precipitated only a small amount (<10%) of endogenous CCS at the folding times examined (data not shown). Thus, although we cannot rule out a possible role for CCS in SOD1 folding in RRL, possible CCS heterodimers must be either very short-lived, uncoupled to the formation of discrete folding intermediates or both.
It is somewhat paradoxical that seemingly subtle missense mutations in SOD1, one of the most stable proteins known, should give rise to polypeptides that share the tendency to form toxic aggregation-prone conformers. The mutants investigated in this study, although far from comprehensive, were selected to be representative of the functional phenotypes and structural domains that characterize the range and diversity of FALS mutations. Three mutants, which each display <7% of wild-type SOD1 levels of activity in vitro (Valentine et al, 2005), are in the metal-binding ligands of SOD1 (H46R, H48Q and H80R). Although both Cu-binding mutants, H46R and H48Q, demonstrate retarded folding, they also behave quite differently from each other. Whereas H48Q is capable of slowly becoming protease resistant, H46R fails to fold to a compact intermediate. This is not surprising as H48Q is known to behave quite differently from H46R in vitro (Hayward et al, 2002; Rodriguez et al, 2002). In fact, unlike H46R, it can still bind copper, albeit in an altered geometry (Hayward et al, 2002). Additionally, recombinant H46R but not H48Q was found to be severely deficient in Zn binding, providing a plausible explanation for the greater effect of this substitution on SOD1 folding.
Mutation of either of the two cysteines, Cys57 and Cys146, that participate in the intrachain disulfide bond also significantly delays SOD1 folding, although C57R is far more impaired than C146R. The solution NMR structure of reduced WT-SOD1 reveals only local conformational disorder around the reduced cysteines, although upon reduction Cys57 becomes highly solvent exposed (Banci et al, 2006). Our data suggest that introduction of a guanidino group at position 57 probably interferes with a critical step in conformational maturation. Interestingly, C6F is also incapable of forming a disulfide bond, even though this residue is not oxidized in the native protein. Not surprisingly, replacement of the relatively buried thiol with a bulky hydrophobic aromatic residue completely abrogates SOD1 folding, although more conservative mutations at this site appear to be tolerated (Stathopulos et al, 2003). Although the enzymatic activities of C6F and C57R have not been examined, C146R displays <10% of wild-type levels of activity in vitro (Valentine et al, 2005).
All of the FALS-associated SOD1 mutants in this study are significantly impaired in their ability to fold to a native-like dimeric, hyperstable state, even though several of the mutants examined herein are capable of eventually reaching a native-like state and have also been previously shown to be enzymatically active in vitro (e.g. A4V and E100G; Hayward et al, 2002). This delayed folding would be expected to increase the fraction of completed SOD1 chains that populate on-pathway folding intermediates or off-pathway states that are prone to becoming mistargeted to organelles such as mitochondria or to form toxic aggregates. We propose that retarded folding kinetics is one potentially unifying mechanism by which to reconcile the diversity of functional and structural alterations imparted on the SOD1 protein by FALS mutations with the toxic gain of function that underlies ALS pathogenesis.
Materials and methods
Reagents
Luciferase and luciferin were a generous gift from J Frydman (Stanford University). Apyrase, TPEN, TEPA and all other chemicals were purchased from Sigma-Aldrich company.
Plasmids
Human WT-SOD1 in pcDNA3.1(−) was constructed via a three-step process. Using the previously published SOD1-HA plasmids (Johnston et al, 2000) as templates, SOD1 was PCR amplified to create an untagged version. All mutants were constructed using the QuikChange mutagenesis kit (Stratagene) with WT-SOD1 in pcDNA3.1(−) as a template. All products were confirmed by sequencing.
In vitro transcription
Plasmid DNA was linearized by digestion with HindIII and purified with the QiaQuick PCR Purification kit (Qiagen). Capped SOD1 RNA was then transcribed using Ambion's MegaScript kit and Promega's Ribo m7G cap analog according to the MegaScript kit's protocol. RNA was purified by twice precipitating with 70 mM sodium acetate and 75% ice-cold ethanol with 70% ice-cold ethanol washes. The final product was resuspended in RNase-free water to a concentration of 7–10 mg/ml as determined by absorbance at 260 nm.
In vitro translation
Translation reactions were performed using Promega's Flexi Rabbit Reticulocyte Lysate kit according to the directions with 70 mM potassium chloride and 0.8 mCi/ml [35S]cysteine. The kit's protocol was modified to include a 5-min incubation step at 30°C before addition of the RNA (0.2 mg/ml). Three minutes after RNA addition, 75 μM ATCA was added to inhibit re-initiation of translation. At various times, 50 mU/μl apyrase was added to halt ATP-dependent processes and reactions were then analyzed. For the ATP depletion study, 50 mU/ μl apyrase was added at 1.5 min after ATCA addition. Depletion was confirmed via a luciferase activity assay (Frydman and Hartl, 1996). For the chelation study, TPEN or TEPA was added during the initial 5-min incubation at 30°C before addition of the RNA. For the quenching of TPEN, 7.5 μM zinc sulfate was added to 5 μM TPEN on ice for 15 min before addition to the translation reaction.
Proteinase K digestion
Halted translations were digested with 1 or 0.01 mg/ml proteinase K for 10 min on ice. Phenylmethylsulfonylfluoride (5 mM) was then added to halt digestion. Samples were boiled for 5 min after addition of Laemmli sample buffer and then separated by 15% SDS–PAGE.
Disulfide bond formation assay
Halted translations were incubated with 6 M guanidine hydrochloride, 50 mM fresh iodoacetamide and 100 mM Tris (pH 8.0) for 2 h at room temperature in the dark. Samples were then precipitated with addition of 0.04% deoxycholic acid and 4 mM trichloroacetic acid. Precipitates were washed twice with 0.6 ml ice-cold acetone. After resuspension in Laemmli sample buffer without reducing agent and 5 min of boiling, samples were separated by 15% non-reducing SDS–PAGE.
Dimerization assay
Gel filtration was performed using a Zorbax GF-250 4.6 × 250 mm gel filtration column (Agilent) on a Dionex DX500 HPLC. The column was equilibrated in 50 mM Tris (pH 7.5) and 150 mM sodium chloride. Molecular weight calibration was performed with Biorad's gel filtration standards and SOD1 (Sigma-Aldrich Company). Halted translations were separated on the column and fractions were collected. After addition of Laemmli sample buffer, the fractions were boiled for 5 min and separated by 15% SDS–PAGE.
Immunoprecipitation
RRL was diluted to the concentration used for the in vitro translations (66%) and 5 μl was incubated with 2 or 5 μl CCS monoclonal antibody (LF-MA0042, Labfrontier) or 5 μl HA.11 antibody (BAbCo) and brought to 100 μl with buffer A (0.1% NP-40, 10 mM Tris, pH 7.5) for 1 h on ice. Reactions were then rotated for 1 h at 4°C after addition of 20 μl of 50% protein G Sepharose in phosphate-buffered saline and 100 mg/ml bovine serum albumin. The resulting bead precipitates were washed five times with 0.5 ml buffer A and once with 0.5 ml buffer B (1% NP-40, 10 mM Tris, pH 7.5). Beads were eluted with the addition of Laemmli sample buffer and boiling for 5 min. They were then separated by 15% SDS–PAGE and transferred to nitrocellulose whereupon they were probed with a CCS polyclonal antibody (Santa Cruz Biotechnology Inc.) and detected with an HRP-conjugated goat anti-rabbit secondary antibody. Immunoblots were then quantified by densitometry using ImageJ (http://rsb.info.nih.gov/ij/). For the co-immunoprecipitation from WT-SOD1 translations, using equal volumes of the stopped translation reaction and the CCS or HA monoclonal antibody, CCS was immunoprecipitated, as described above. Gels were then analyzed for co-immunoprecipitation of translated WT-SOD1, as described below.
Gel analysis and statistics
Gels were briefly rinsed in 10% glycerol and dried. SOD1 bands were imaged by Biorad's Typhoon phosphorimager and quantified using ImageQuant software (Molecular Dynamics). Data fitting and curve deconvolution were performed using OriginPro 7.5 software (OriginLab). All experiments were performed at least three times. Statistical analysis was performed using regression line comparison with Lab Stat (Jim Codde).
Acknowledgments
We are grateful to S Long, P Harbury, G Fang, J Frydman, M Rexach and R Simoni and members of the Kopito laboratory for helpful advice. This work was supported by a grant from NINDS (NS42842) and by a Smith Stanford Graduate Fellowship to CKB.
References
- Alexander MD, Traynor BJ, Miller N, Corr B, Frost E, McQuaid S, Brett FM, Green A, Hardiman O (2002) ‘True' sporadic ALS associated with a novel SOD-1 mutation. Ann Neurol 52: 680–683 [DOI] [PubMed] [Google Scholar]
- Antonyuk S, Elam JS, Hough MA, Strange RW, Doucette PA, Rodriguez JA, Hayward LJ, Valentine JS, Hart PJ, Hasnain SS (2005) Structural consequences of the familial amyotrophic lateral sclerosis SOD1 mutant His46Arg. Protein Sci 14: 1201–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, Bertini I, Cantini F, D'Amelio N, Gaggelli E (2006) Human SOD1 before harboring the catalytic metal: solution structure of copper-depleted, disulfide-reduced form. J Biol Chem 281: 2333–2337 [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Cramaro F, Del Conte R, Viezzoli MS (2003) Solution structure of Apo Cu, Zn superoxide dismutase: role of metal ions in protein folding. Biochemistry 42: 9543–9553 [DOI] [PubMed] [Google Scholar]
- Bertini I, Piccioli M, Viezzoli MS, Chiu CY, Mullenbach GT (1994) A spectroscopic characterization of a monomeric analog of copper, zinc superoxide dismutase. Eur Biophys J 23: 167–176 [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW (2006) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52: 39–59 [DOI] [PubMed] [Google Scholar]
- Bowling AC, Barkowski EE, McKenna-Yasek D, Sapp P, Horvitz HR, Beal MF, Brown RH Jr (1995) Superoxide dismutase concentration and activity in familial amyotrophic lateral sclerosis. J Neurochem 64: 2366–2369 [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18: 327–338 [DOI] [PubMed] [Google Scholar]
- Ceroni M, Malaspina A, Poloni TE, Alimonti D, Rognoni F, Habgood J, Imbesi F, Antonelli P, Alfonsi E, Curti D, deBelleroche J (1999) Clustering of ALS patients in central Italy due to the occurrence of the L84F SOD1 gene mutation. Neurology 53: 1064–1071 [DOI] [PubMed] [Google Scholar]
- Culotta VC, Yang M, O'Halloran TV (2006) Activation of superoxide dismutases: putting the metal to the pedal. Biochim Biophys Acta 1763: 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daggett V, Fersht AR (2003) Is there a unifying mechanism for protein folding? Trends Biochem Sci 28: 18–25 [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME (1997) A low expressor line of transgenic mice carrying a mutant human Cu, Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 93: 537–550 [DOI] [PubMed] [Google Scholar]
- Dobson CM, Karplus M (1999) The fundamentals of protein folding: bringing together theory and experiment. Curr Opin Struct Biol 9: 92–101 [DOI] [PubMed] [Google Scholar]
- Durham HD, Roy J, Dong L, Figlewicz DA (1997) Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol 56: 523–530 [DOI] [PubMed] [Google Scholar]
- Endo Y, Sawasaki T (2006) Cell-free expression systems for eukaryotic protein production. Curr Opin Biotechnol 17: 373–380 [DOI] [PubMed] [Google Scholar]
- Feldman DE, Frydman J (2000) Protein folding in vivo: the importance of molecular chaperones. Curr Opin Struct Biol 10: 26–33 [DOI] [PubMed] [Google Scholar]
- Fersht AR (2000) Transition-state structure as a unifying basis in protein-folding mechanisms: contact order, chain topology, stability, and the extended nucleus mechanism. Proc Natl Acad Sci USA 97: 1525–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman HJ, Fridovich I (1973) On the stability of bovine superoxide dismutase. The effects of metals. J Biol Chem 248: 2645–2649 [PubMed] [Google Scholar]
- Frydman J, Hartl FU (1996) Principles of chaperone-assisted protein folding: differences between in vitro and in vivo mechanisms. Science 272: 1497–1502 [DOI] [PubMed] [Google Scholar]
- Frydman J, Nimmesgern E, Ohtsuka K, Hartl FU (1994) Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature 370: 111–117 [DOI] [PubMed] [Google Scholar]
- Furukawa Y, O'Halloran TV (2006) Posttranslational modifications in Cu, Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal 8: 847–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa Y, Torres AS, O'Halloran TV (2004) Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J 23: 2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PJ (2006) Pathogenic superoxide dismutase structure, folding, aggregation and turnover. Curr Opin Chem Biol 10: 131–138 [DOI] [PubMed] [Google Scholar]
- Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown RH Jr (2002) Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem 277: 15923–15931 [DOI] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Xu Z (2003) ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci 4: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Merz F, Rutkowska A, Zachmann-Brand B, Deuerling E, Bukau B (2006) Trigger factor forms a protective shield for nascent polypeptides at the ribosome. J Biol Chem 281: 6539–6545 [DOI] [PubMed] [Google Scholar]
- Ince PG, Tomkins J, Slade JY, Thatcher NM, Shaw PJ (1998) Amyotrophic lateral sclerosis associated with genetic abnormalities in the gene encoding Cu/Zn superoxide dismutase: molecular pathology of five new cases, and comparison with previous reports and 73 sporadic cases of ALS. J Neuropathol Exp Neurol 57: 895–904 [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR (2000) Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 97: 12571–12576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Agar JN, Taylor DM, Minotti S, Durham HD (2004) Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J Neurochem 89: 1325–1335 [DOI] [PubMed] [Google Scholar]
- Kato S, Horiuchi S, Nakashima K, Hirano A, Shibata N, Nakano I, Saito M, Kato M, Asayama K, Ohama E (1999) Astrocytic hyaline inclusions contain advanced glycation end products in familial amyotrophic lateral sclerosis with superoxide dismutase 1 gene mutation: immunohistochemical and immunoelectron microscopical analyses. Acta Neuropathol (Berl) 97: 260–266 [DOI] [PubMed] [Google Scholar]
- Koide T, Igarashi S, Kikugawa K, Nakano R, Inuzuka T, Yamada M, Takahashi H, Tsuji S (1998) Formation of granular cytoplasmic aggregates in COS7 cells expressing mutant Cu/Zn superoxide dismutase associated with familial amyotrophic lateral sclerosis. Neurosci Lett 257: 29–32 [DOI] [PubMed] [Google Scholar]
- Kokubo Y, Kuzuhara S, Narita Y, Kikugawa K, Nakano R, Inuzuka T, Tsuji S, Watanabe M, Miyazaki T, Murayama S, Ihara Y (1999) Accumulation of neurofilaments and SOD1-immunoreactive products in a patient with familial amyotrophic lateral sclerosis with I113T SOD1 mutation. Arch Neurol 56: 1506–1508 [DOI] [PubMed] [Google Scholar]
- Lamb AL, Torres AS, O'Halloran TV, Rosenzweig AC (2001) Heterodimeric structure of superoxide dismutase in complex with its metallochaperone. Nat Struct Biol 8: 751–755 [DOI] [PubMed] [Google Scholar]
- Lindberg MJ, Bystrom R, Boknas N, Andersen PM, Oliveberg M (2005) Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc Natl Acad Sci USA 102: 9754–9759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS, Cleveland DW (2004) Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43: 5–17 [DOI] [PubMed] [Google Scholar]
- Manning M, Colon W (2004) Structural basis of protein kinetic stability: resistance to sodium dodecyl sulfate suggests a central role for rigidity and a bias toward beta-sheet structure. Biochemistry 43: 11248–11254 [DOI] [PubMed] [Google Scholar]
- Merrick WC (1990) Gene expression using cell-free systems. Curr Opin Biotechnol 1: 79–81 [DOI] [PubMed] [Google Scholar]
- Nishiyama K, Murayama S, Kwak S, Kanazawa I (1997) Expression of the copper–zinc superoxide dismutase gene in amyotrophic lateral sclerosis. Ann Neurol 41: 551–556 [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7: 710–723 [DOI] [PubMed] [Google Scholar]
- Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV (1999) Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284: 805–808 [DOI] [PubMed] [Google Scholar]
- Ratovitski T, Corson LB, Strain J, Wong P, Cleveland DW, Culotta VC, Borchelt DR (1999) Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum Mol Genet 8: 1451–1460 [DOI] [PubMed] [Google Scholar]
- Rodriguez JA, Shaw BF, Durazo A, Sohn SH, Doucette PA, Nersissian AM, Faull KF, Eggers DK, Tiwari A, Hayward LJ, Valentine JS (2005) Destabilization of apoprotein is insufficient to explain Cu, Zn-superoxide dismutase-linked ALS pathogenesis. Proc Natl Acad Sci USA 102: 10516–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JA, Valentine JS, Eggers DK, Roe JA, Tiwari A, Brown RH Jr, Hayward LJ (2002) Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J Biol Chem 277: 15932–15937 [DOI] [PubMed] [Google Scholar]
- Rosen DR (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364: 362. [DOI] [PubMed] [Google Scholar]
- Rumfeldt JA, Stathopulos PB, Chakrabarrty A, Lepock JR, Meiering EM (2006) Mechanism and thermodynamics of guanidinium chloride-induced denaturation of ALS-associated mutant Cu, Zn superoxide dismutases. J Mol Biol 355: 106–123 [DOI] [PubMed] [Google Scholar]
- Shibata N, Hirano A, Kobayashi M, Dal Canto MC, Gurney ME, Komori T, Umahara T, Asayama K (1998) Presence of Cu/Zn superoxide dismutase (SOD) immunoreactivity in neuronal hyaline inclusions in spinal cords from mice carrying a transgene for Gly93Ala mutant human Cu/Zn SOD. Acta Neuropathol (Berl) 95: 136–142 [DOI] [PubMed] [Google Scholar]
- Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY, Kato T, Asayama K (1996) Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol 55: 481–490 [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Rumfeldt JA, Scholz GA, Irani RA, Frey HE, Hallewell RA, Lepock JR, Meiering EM (2003) Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad Sci USA 100: 7021–7026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart ML, Grollman AP, Huang MT (1971) Aurintricarboxylic acid: inhibitor of initiation of protein synthesis. Proc Natl Acad Sci USA 68: 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine JS, Doucette PA, Potter SZ (2005) Copper–zinc superoxide dismutase and amyotrophic lateral sclerosis. Ann Rev Biochem 74: 563–593 [DOI] [PubMed] [Google Scholar]
- Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR (2003) Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet 12: 2753–2764 [DOI] [PubMed] [Google Scholar]
- Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD (2001) Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 8: 933–941 [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14: 1105–1116 [DOI] [PubMed] [Google Scholar]