

Abstract
Random mutations under neutral or near-neutral conditions are studied by considering plausible evolutionary trajectories on “neutral nets”—i.e., collections of sequences (genotypes) interconnected via single-point mutations encoding for the same ground-state structure (phenotype). We use simple exact lattice models for the mapping between sequence and conformational spaces. Densities of states based on model intrachain interactions are determined by exhaustive conformational enumeration. We compare results from two very different interaction schemes to ascertain robustness of the conclusions. In both models, sequences in a majority of neutral nets center around a single “prototype sequence” of maximum mutational stability, tolerating the largest number of neutral mutations. General analytical considerations show that these topologies by themselves lead to higher steady-state evolutionary populations at prototype sequences. On average, native thermodynamic stability increases toward a maximum at the prototype sequence, resulting in funnel-like arrangements of native stabilities in sequence space. These observations offer a unified perspective on sequence design, native stability, and mutational stability of proteins. These principles are generalizable from native stability to any measure of fitness provided that its variation with respect to mutations is essentially smooth.
Keywords: fitness landscapes, neutral mutations, folding funnels, thermodynamic stability, lattice protein models
The study of evolution requires an understanding of how sequences are mapped onto structures and functions. Fitness landscape is a useful conceptualization of sequence-space properties. It was originally proposed almost 70 years ago by Sewall Wright (1), who envisioned evolution as walks of populations on this landscape toward higher fitness.
Many evolutionary questions, such as those pertinent to random genetic drift as advocated by Motoo Kimura (2), entail modeling broad areas of the fitness landscape. Analytical models often assume a random mapping between genotype and phenotype, because the correlation among mutational effects proves to be mathematically complex to account for (3). However, such correlations are crucial in understanding neutral mutations and mutational stability. To address these issues, many recent theoretical efforts have been computational, focusing on constructing models of sequence-structure mapping for RNA (4–7) and proteins (8–22) that are motivated by various aspects of polymer physics. Because of the immense sizes of the systems, all these models involve significant simplifications (23–25). One of these models (8, 9) has been applied (10) to explore whether nonlethal mutations form a connected network, as envisioned by Maynard Smith (26).
Many proteins maintain their native structures while undergoing single and double mutations at many different sites. Of considerable evolutionary interest, therefore, is the number of converging sequences encoding for the same structure (9, 14, 16, 21). In a recent insightful study, Li et al. found that structures differ markedly in terms of their designability, i.e., their numbers of converging sequences, and that there are a small number of highly designable sequences (14).
Using a hydrophobic polar (HP) model with exhaustive conformational enumeration in two dimensions, one of us (16) recently obtained results consistent with that of Li et al. (17), including a Zipf-like distribution of designability. In addition, neutral nets are found to be centered around prototype sequences (16).
Another line of inquiry, beginning with the work of Bryngelson and Wolynes (27), has emphasized the importance of kinetic accessibility of the native structures as an evolutionary selection criterion. In this view, preferred structures are those that can be encoded by sequences with high native stabilities and minimal ruggedness on the folding landscape to allow for fast folding (13, 18–23). Structures preferred by the kinetic criterion are expected to be highly designable by the thermodynamic criterion of Li et al., because they observed that high native stability is likely to correlate with high designability (14, 22).
Building on these results, we now turn our attention to sequence-space topologies of neutral nets (5, 16, 18), i.e., how sequences encoding for the same structure are related to one another by mutations. We observe the following features: (i) Independent of functional fitness, topology per se can lead to concentration of evolutionary population at some sequences. This perspective may rationalize certain mutagenesis experiments. (ii) The organization of native stabilities is funnel like among certain sets of sequences encoding for the same native structure. (iii) The presence of repulsive interactions can lead to more rugged sequence-space landscape.
Thermodynamic stability of the native structure is treated here as one possible “fitness” measure. Locally optimal sequences have previously been pictured as “peaks” (3) on the fitness landscape. Here we choose to conform to conventional imageries in physics and physical chemistry; we associate higher fitness with lower altitude on sequence-space landscapes instead. In this picture, neutral nets often are basins of attraction, with prototype sequences at their bottom.
MODELS OF NEUTRAL MUTATIONS
We study two models; both admit all possible permutations of two types of monomers along chain sequences, and chain conformations are configured on two-dimensional square lattices. We first consider the HP model (28), which assigns a favorable contact energy ℰ (<0) for each contact between two H monomers (an HH contact), whereas hydrophobic-polar (HP) and polar-polar (PP) contacts are neutral (zero contact energy). For comparison, we study also the “AB” model (29), with monomer types A and B. The contact energies for AA, BB, and AB contacts are, respectively ℰ, ℰ (<0), and −ℰ (>0). The HP model is motivated by the physics of hydrophobic interactions. Native conformations in the HP model tend to have a hydrophobic core and a mostly polar surface, as in real proteins. On the other hand, the interactions in the AB model are very different; like monomers attract and unlike monomers repel. Hence the two types of monomers tend to segregate in native conformations, with mostly A monomers on one side and mostly B monomers on the other, which is not very protein like. The AB model is used here as a control, and also as a means to address effects of repulsive interactions and more disruptive mutations (29). Results below are presented for chain length n = 18.
For each of 218 possible sequences in both models, exhaustive enumeration is used to identify the ground-state (lowest-energy) conformations among all 5,808,335 possibilities. There are, respectively, 6,349 and 34,700 sequences in the HP and AB models with a unique ground-state (native) conformation. They are used as model proteins (29). All single-point mutations (8, 11) among these sequences are determined. There are 16,340 such H ↔ P mutations (12) and 121,472 such A ↔ B mutations.
A neutral net is defined as a collection of unique sequence(s) encoding for the same native structure that are interconnected by single-point mutations. There are 1,706 and 16,270 neutral nets of various sizes in the HP and AB models, respectively. Fig. 1 shows the largest neutral nets. The Hamming distance between two sequences is the total number of monomers that are different along the alignment of the two sequences (16). Sequences within a neutral net that differ by only a single-point mutation (i.e., Hamming distance of one) are called neutral neighbors.
Figure 1.

Largest neutral net in (a) the HP model (48 sequences) and (b) AB model (26 sequences). The native structures are given in their respective prototype sequences. H, P, A, and B monomers are represented by filled and open circles and filled and open squares, respectively (29). The topology of each neutral net is shown by representing each sequence by a dot. A connecting line with arrow indicates that two sequences are neutral neighbors (16). Arrows point toward the sequence with higher native stability (see Fig. 2). Larger dots represent sequences with the maximum number of neutral neighbors within the neutral net. Concentric circles in dotted lines indicate Hamming distance from the prototype sequence.
Most neutral nets center around prototype sequences (16) with the maximum number of neutral neighbors (Fig. 1). Motivated by their suggestive topologies, we first ask whether network connectivity alone can give rise to enhanced populations at the prototype sequences, based on an extremely simple model of evolutionary dynamics: for a neutral net with ω sequences, let the population of the ith sequence be Pi (i = 1, 2, … , ω), and the mutation rate μ is the same for each of the n monomers. A mutation resulting in a sequence outside the neutral net is taken as lethal, corresponding to a population loss. Neglecting population entering the neutral net from the outside,
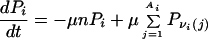 |
1 |
gives the time (t) dependence of the system, where νi(j)’s label the Ai neutral neighbors of i. We note that the overall absolute population can increase or decrease depending on whether the reproductive rate [an additional term proportional to Pi in Eq. 1] is sufficient to offset losses to lethal mutations. However, this does not affect the steady-state relative population distribution, which is determined by the expected large-t behavior, dPi/(dt) = −μλ′Pi, where λ′ is some constant to be determined. Here μ can be factored out because the overall population decay rate in Eq. 1 must be proportional to the mutational rate. It follows that the μ-independent steady-state relative population distribution is given by the eigenvector for the largest λ in the eigenvalue problem
 |
2 |
where λ ≡ (n − λ′). It is straightforward to see that this leads to enhanced population at prototype sequences. If all ω populations are equal initially, mutations within the neutral net would not at first contribute to population change, because population flux from forward and backward mutations cancel. In this case, those sequences with fewer neutral neighbors will lose population faster because of their higher probabilities for lethal mutations. It follows that population distribution would shift subsequently in favor of sequences with more neutral neighbors.
For a more quantitative illustration, consider a hypothetical neutral net with perfect symmetry, which has two circles of sequences surrounding a single prototype sequence at the center. The prototype sequence has A0 neutral neighbors. The A0 sequences in the first circle each has A1 neutral neighbors; they are also connected to a second circle of sequences, each of which has A2 neutral neighbors. It can be shown that the ratios of steady-state populations between that of the prototype sequence (P0) and one single sequence in the first (P1) and the second (P2) circles are given by (P0/P1) = 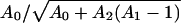 and
and 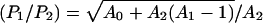 . For the special case of a symmetric neutral net with only one circle (A1 = 1, A2 = 0), these results reduce to
. For the special case of a symmetric neutral net with only one circle (A1 = 1, A2 = 0), these results reduce to 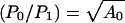 , suggesting that in general steady-state population scales roughly as the square root of a sequence’s number of neutral neighbors A.
, suggesting that in general steady-state population scales roughly as the square root of a sequence’s number of neutral neighbors A.
For neutral nets in Fig. 1, steady-state populations are determined numerically. In both cases they peak at prototype sequences, with 6.31% for the HP and 7.09% for the AB neutral nets shown. In Fig. 1, the HP prototype sequence has 10 neutral neighbors; as for the rest, the highest and lowest steady-state populations are 4.07% and 0.97% for a sequence with 7 and 3 neutral neighbors, respectively. In the same figure, the AB prototype sequence has six neutral neighbors. Two other sequences have the same number of neutral neighbors; their steady-state populations are 6.91% and 6.89%. The lowest steady-state population is 1.04% for a sequence with two neutral neighbors.
These variations among steady-state populations are modest. This is a result of their roughly ≈ 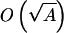 dependence. For sequences with two types of monomers, the maximum A is equal to the chain length n, whereas n is small for our highly simplified short-chain models. However, this general formulation suggests that the steady-state population distribution can be much more uneven and possibly highly peaked at prototype sequences for real proteins made up of 20 amino acid types with chain lengths n ≈ 100, because A can then be of the order 19n.
dependence. For sequences with two types of monomers, the maximum A is equal to the chain length n, whereas n is small for our highly simplified short-chain models. However, this general formulation suggests that the steady-state population distribution can be much more uneven and possibly highly peaked at prototype sequences for real proteins made up of 20 amino acid types with chain lengths n ≈ 100, because A can then be of the order 19n.
In this simple model of evolutionary dynamics, the fitness measure is effectively a step function—a constant favorable fitness for every sequence in the neutral net and lethal outside, and an effectively infinite population size is assumed. Realistically, other factors such as native stability (see below) and genetic drift because of finite population (30) are likely to skew this simple picture. The most important observation here, however, is that even with just the simple ingredients of neutral net topology and the existence of lethal mutants, uneven distributions that peak at prototype sequences can naturally arise, and this feature should be general because its derivation does not depend on any particular chain model.
FUNNELS IN SEQUENCE SPACE
In the present analysis, sequences within a neutral net are neutral only with respect to their ability to encode for the same native structure. We now consider the different thermodynamic stabilities of the native structure they encode. For each sequence, we enumerate the distribution of conformations over all possible energetic states. The density of states g(E) is the number of conformations with energy E, where E is the sum of intrachain contact energies. The free energy of folding to one of the ground-state conformations is given by (9, 11)
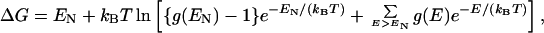 |
3 |
where kBT is Boltzmann constant times absolute temperature, EN is the ground-state energy, and g(EN) = 1 for the unique sequences considered in this section. A more negative ΔG means a more stable native structure. Here we use the “sticking” parameter −ℰ/(kBT) at the folding-denaturation midpoint (ΔG = 0) as stability measure. In some neutral nets, there is more than one sequence with the maximum number of neutral neighbors. We then define the prototype sequence to be the one that also has the highest native stability.
Fig. 2 shows examples in which the most thermodynamically stable sequence is also the prototype sequence. A striking feature, especially for the HP case, is the funnel-like arrangements of the neutral-neighbor connections, which are the “kinetic adjacencies” (12) in evolution. On average, native stability decreases for sequences further away from the prototype sequence (Fig. 2 c and d). This appears to be a general feature of sequence space, irrespective of whether native stability per se is favored by evolutionary selection. Because each sequence is itself associated with a presumably funnel-like folding landscape consisting of all conformations (23, 24, 31), sequence-space funnels represent a higher level of organization in a “cross product” of the sequence and conformational spaces, while sharing qualitatively similar features with conformational-space funnels (31). Hence we propose adding the prefix “super” to their description. We demonstrated above that prototype sequences are intrinsically favored even in the absence of any functional or reproductive advantage. If higher native stability is correlated with enhanced fitness, which may be a reasonable assumption for some biological activities, the existence of superfunnels in sequence space would imply an even higher concentration of steady-state evolutionary populations at the prototype sequences.
Figure 2.

Native stabilities of the sequences in the (a) HP and (b) AB neutral nets in Fig. 1 are represented as horizontal lines. The horizontal axis indicates Hamming distance from the prototype sequence. Neutral mutations are indicated by lines connecting horizontal levels. Heuristic views of (c) the HP and (d) AB “superfunnels” are traces through average stabilities of the sequences as a function of Hamming distance (dots). In c and d, the bottom at the center of each funnel corresponds to the prototype sequence; horizontal displacement from the center in either direction corresponds to increasing Hamming distance from the prototype sequence.
Thermodynamic statistics of all neutral nets in the two models are given in Figs. 3 and 4 as functions of neutral net size. Figs. 3 (Lower) and 4 (Lower) show that a majority of neutral nets conform to the superfunnel paradigm. There are exceptions: in some neutral nets, sequences with the maximum number of neutral neighbors do not have the highest thermodynamic stability (see Δmin traces). These cases constitute only a minority. In the HP model, this occurs in 75 neutral nets, which comprise 11.2% of the 668 neutral nets with more than two sequences. Collectively, they contain 748 sequences, which is 11.8% of all unique sequences. In the AB model, 1,348 neutral nets do not conform to the superfunnel paradigm, which is 35.5% of the 3,882 nets with more than two sequences, and they involve 6,484 sequences, 18.7% of all unique sequences. In these situations, the dominant population would be determined by two competing evolutionary effects—the selective advantage of native thermodynamic stability vs. the neutral net topology effect described above.
Figure 3.

Superfunnel geometry. (Upper) Thermodynamic stability of prototype sequences. For neutral nets of a given size, diamond shows the average stability, whereas dots show the maximum and minimum stabilities among the prototype sequences from different neutral nets. (Inset) 𝒩 (ω) is the number of neutral nets with size ω. All solid or dashed lines linking data points in Figs. 3 and 4 serve merely as visual guides. (Lower) For a given neutral net, Δ is the difference in thermodynamic stability [measured in transition-midpoint −ℰ/(kBT)] between a nonprototype sequence and the prototype sequence. The average stability gap 〈Δ〉 is the average of Δ over all nonprototype sequences in the net; thus it provides a measure of “depth” of a neutral net. The minimum stability gap Δmin is the smallest value of Δ within a neutral net. Hence Δmin < 0 implies the neutral net is not a superfunnel. Averages of 〈Δ〉 (squares) and Δmin (circles) over neutral nets of given sizes are plotted. For neutral nets that satisfy the superfunnel criterion (Δmin ≥ 0), average slopes are also computed. For every neutral mutation, δℰ is a sequence-space slope. It is equal to the transition-midpoint −ℰ/(kBT) of the sequence one Hamming step further from the prototype sequence minus that of its neutral neighbor that is one step closer. 〈δℰ〉 is the average of δℰ over all mutations within a neutral net. Because some neutral mutations lead to negative slopes (δℰ < 0), we also compute the average of their absolute values, 〈|δℰ|〉. Averages of these two quantities over neutral nets of given sizes are plotted as dots connected by solid (〈δℰ〉) and dashed (〈|δℰ|〉) lines.
Figure 4.

Same as Fig. 3 except for AB sequences.
For the majority of neutral nets that are superfunnels, there is a clear correlation between neutral net size and the stability of the prototype sequence (Figs. 3 and 4 Upper). This observation is consistent with the conclusions of Li et al. (14) and Melin et al. (22) on proteins and of Wuchty et al. (7) on RNA. This can also be seen here from the fact that, on average, the depth of superfunnels increases with size (Figs. 3 and 4 Lower, 〈Δ〉 traces). The average slope of superfunnels 〈δℰ〉, however, does not show any significant systematic increase or decrease with size.
These general trends are observed in both the HP and AB models, but details of the superfunnels depend on the intrachain interactions of the sequences. Because of the repulsive interactions, mutations in the AB model are more disruptive. As a result, the AB sequence space is more fragmented, and its neutral nets are on average smaller than that in the HP sequence space (Figs. 3 and 4 Upper). The largest HP neutral net coincides with the largest set (neutral set) of converging sequences (9, 16) encoding for the same structure. For the AB model, the structure encoded by the largest neutral net is not identical to that with the largest neutral set. The latter has 76 encoding sequences, but it is fragmented into 14 neutral nets.
HP superfunnels are also smoother. For instance, 98 of the 99 mutational connections in the largest HP neutral net in Figs. 1 and 2 have “positive slopes,” i.e., they are directed toward sequences with higher stabilities as they approach the prototype sequence. Only one mutational connection has a “negative slope.” There is no evolutionary “kinetic trap” on this HP superfunnel, because every nonprototype sequence has at least one neutral neighbor with higher thermodynamic stability [smaller −ℰ/(kBT) at ΔG = 0]. The AB neutral net in the same figures are more rugged in that 9 of the 46 mutational connections have negative slopes. The heuristic drawing in Fig. 2d also suggests that there are evolutionary “kinetic traps” in this AB neutral net. Indeed, there is one trap at Hamming distance 4, but it is very shallow. AB superfunnels are more rugged in general. This is illustrated by the much larger discrepancies between the 〈δℰ〉 and 〈|δℰ|〉 traces in Fig. 4 (Lower) vs. that in Fig. 3; 〈|δℰ|〉 − 〈δℰ〉 describes the prevalence of negative slopes and is therefore a measure of superfunnel ruggedness.
MULTIPLY-DEGENERATE SEQUENCES
So far, we have assumed that only unique sequences are viable. However, it is conceivable that a sequence with more than one ground-state conformation [i.e., degeneracy g ≡ g(EN) > 1] (29) can still possess the function performed by any one of its ground-state conformations. As a first approximation, we may assume that the activity specific to a given conformation is proportional to the fractional population p of that conformation. Its free energy of folding ΔG = −kBT ln[p/(1−p)] at a given intrachain sticking can then be used to characterize this activity. A smaller ΔG implies a higher stability for the functional form. ΔG is calculated from the sequence’s density of state by using Eq. 3. Because multiply-degenerate (g > 1) sequences can never attain more than p = 50% population for any one of its g ground-state conformations, their ΔGs are always positive.
By including sequences with as many as six ground-state conformations, an extended HP neutral net for the one in Figs. 1 and 2a is constructed (Fig. 5). Every g > 1 sequence in this net has the HP structure in Fig. 1 as one of its ground-state conformations. Fig. 5 provides these sequences’ free energies of folding to this structure, resulting in a larger superfunnel with the same general features as that in Fig. 2a. The maximum Hamming distance remains unchanged. On average, the g > 1 sequences are further away from the prototype sequence than the unique (g = 1) sequences (average Hamming distance of 2.6 vs. 2.1).
Figure 5.

Extended neutral net for the HP structure in Fig. 1, with 146 sequences. Same as Fig. 2a, except that native stabilities of the sequences are now measured by free energy of folding (Eq. 3) and that multiply-degenerate sequences (degeneracy g ≤ 6) are included. Numbers of sequences with g = 1, 2, 3, 4, 5, and 6 in this net are 48, 22, 16, 27, 14, and 19, respectively. The vertical bars and numbers on the right indicate the range of stability levels for sequences with different gs.
These observations suggest a generalization of the superfunnel concept. Basins of attraction in sequence space can be substantially enlarged to encompass more sequences if multiply-degenerate sequences are to some degree viable. In this scenario, some of the g > 1 sequences that have other encodable conformation(s) (29) in their ground states can serve as “switches” (16) to facilitate evolution to other neutral nets. In Fig. 5, 13 of the g = 2 sequences share this property.
GENERALIZATIONS AND DISCUSSION
Our model results strongly suggest that “plasticity” or mutational stability of a sequence is correlated with its thermodynamic stability. We believe that this general conclusion follows directly from a fundamental principle of sequence design—that it is important to both design in the target structure and design out nontarget structures (32). Thus native states of better designed sequences are energetically more separated from their nonnative conformations, implying that they have higher thermodynamic stabilities (14, 22). Some threshold native stability may also be needed to avoid misfolding on multimerization and aggregation (33). Insofar as stability of a given native structure varies relatively smoothly in sequence space, a superfunnel-like organization is likely. In evolutionary terms, this means that in most cases the wild-type sequence may be identified with the prototype sequence, and that most if not all single-point mutations on the wild-type sequence would be thermodynamically destabilizing. In light of these considerations, the generality of our conclusion may transcend the two models studied here, and may also be independent of questions regarding what model contact interactions and chain representations are more protein like (21, 29).
Following this logic, it appears that any fitness measure could lead to superfunnel-like organizations of that measure, provided that its variation is relatively smooth in sequence space. Several recent evolutionary studies are based on assumed selective advantages for sequences having fast folding kinetics in addition to thermodynamically stable native structures (13, 18–22). We note that mutational effects on model folding kinetics can sometimes be subtle (12, 34), and actual dynamic simulations have shown that some kinetic properties cannot always be reliably derived from the density of states alone (35) as used in some studies (18, 21). A recent experiment also shows that mutations on wild type are most likely destabilizing, but their kinetic effects are less predictable (36). Nevertheless, inasmuch as these “foldability” criteria do vary smoothly (18), superfunnel-like organization of foldability is expected. Indeed, a funnel-like variation of a “frustration” measure of kinetic accessibility along a sequence similarity parameter has recently been reported in an off-lattice study by Nelson and Onuchic (20).
Our goal here is to establish a conceptual framework. Many subtleties and complexities of biological structure and function (37) are neglected. For instance, it has been suggested that too much thermodynamic stability and conformational rigidity can be detrimental to function (38) (see also ref. 39). Real functional fitness is not expected to always correlate with native stability. The application of our results may also be less straightforward if the functional form of a protein is a multimer instead of a single-chain monomer (40). These limitations notwithstanding, the superfunnel scenario appears to be in general qualitative agreement with experiments. In an extensive mutagenesis study of 290 single-point mutations on the wild-type sequence of staphylococcal nuclease, only 33 lead to relatively small thermodynamic stabilization. All of the rest are destabilizing to various degrees (41–43). Consistent with the ruggedness consideration above, sets of mutations that are energetically more disruptive lead to higher probabilities of mutant stabilization: 2/83, 11/103, and 20/104 of the mutations on large hydrophobic (41), polar and uncharged (42) and ionizable (43) amino acid residues, respectively, lead to mutants more stable than the wild type.
A noteworthy finding here is that neutral net topology per se can be an important determining factor of evolutionary population. In some cases, mutations on wild-type sequences are found to be both stabilizing and function enhancing (44), which is puzzling from an evolutionary perspective that focuses exclusively on functional fitness (45). However, this may be rationalizable without invoking unspecified biological complexities if these mutants turn out to be mutationally unstable themselves (i.e., have few viable neutral neighbors). Our results show that it is possible for neutral net topology and functional fitness (such as native stability in our models) to have opposing evolutionary effects on population distribution. This hypothesis should be testable by experimental mapping of neutral net topologies of real proteins.
Acknowledgments
We thank José Onuchic and two anonymous referees for helpful comments on the manuscript. H. S. C. thanks the Medical Research Council of Canada (MT-15323) and the Connaught Fund for financial support.
ABBREVIATION
- HP
hydrophobic polar
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Wright S. In: Proceedings of the Sixth International Congress on Genetics, Vol. 1. Jones D F, editor. New York: Brooklyn Botanic Gardens; 1932. pp. 356–366. [Google Scholar]
- 2.Kimura M. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge Univ. Press; 1983. [Google Scholar]
- 3.Kauffman S, Levin S. J Theor Biol. 1987;128:11–45. doi: 10.1016/s0022-5193(87)80029-2. [DOI] [PubMed] [Google Scholar]
- 4.Fontana W, Schuster P. Biophys Chem. 1987;26:123–147. doi: 10.1016/0301-4622(87)80017-0. [DOI] [PubMed] [Google Scholar]
- 5.Huynen M A, Stadler P F, Fontana W. Proc Natl Acad Sci USA. 1996;93:397–401. doi: 10.1073/pnas.93.1.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fontana W, Schuster P. Science. 1998;280:1451–1455. doi: 10.1126/science.280.5368.1451. [DOI] [PubMed] [Google Scholar]
- 7.Wuchty S, Fontana W, Hofacker I L, Schuster P. Biopolymers. 1999;49:145–165. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 8.Lau K F, Dill K A. Proc Natl Acad Sci USA. 1990;87:638–642. doi: 10.1073/pnas.87.2.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan H S, Dill K A. J Chem Phys. 1991;95:3775–3787. [Google Scholar]
- 10.Lipman D J, Wilbur W J. Proc R Soc London Ser B. 1991;245:7–11. doi: 10.1098/rspb.1991.0081. [DOI] [PubMed] [Google Scholar]
- 11.Shortle D, Chan H S, Dill K A. Protein Sci. 1992;1:201–215. doi: 10.1002/pro.5560010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan H S, Dill K A. J Chem Phys. 1994;100:9238–9257. [Google Scholar]
- 13.Gutin A M, Abkevich V I, Shakhnovich E I. Proc Natl Acad Sci USA. 1995;92:1282–1286. doi: 10.1073/pnas.92.5.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Helling R, Tang C, Wingreen N. Science. 1996;273:666–669. doi: 10.1126/science.273.5275.666. [DOI] [PubMed] [Google Scholar]
- 15.Bussemaker H J, Thirumalai D, Bhattacharjee J K. Phys Rev Lett. 1997;79:3530–3533. [Google Scholar]
- 16.Bornberg-Bauer E. Biophys J. 1997;73:2393–2403. doi: 10.1016/S0006-3495(97)78268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebeling M, Nadler W. Biopolymers. 1997;41:165–180. doi: 10.1002/(SICI)1097-0282(199702)41:2<165::AID-BIP4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 18.Govindarajan S, Goldstein R A. Biopolymers. 1997;42:427–438. doi: 10.1002/(SICI)1097-0282(19971005)42:4<427::AID-BIP6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 19.Saito S, Sasai M, Yomo T. Proc Natl Acad Sci USA. 1997;94:11324–11328. doi: 10.1073/pnas.94.21.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson E D, Onuchic J N. Proc Natl Acad Sci USA. 1998;95:10682–10686. doi: 10.1073/pnas.95.18.10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buchler N E G, Goldstein R A. Proteins Struct Funct Genet. 1999;34:113–124. [PubMed] [Google Scholar]
- 22.Melin R, Li H, Wingreen N S, Tang C. J Chem Phys. 1999;110:1252–1262. [Google Scholar]
- 23.Bryngelson J D, Onuchic J N, Socci N D, Wolynes P G. Proteins Struct Funct Genet. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 24.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 25.Thirumalai D, Woodson S A. Acc Chem Res. 1996;29:433–439. [Google Scholar]
- 26.Maynard Smith J. Nature (London) 1970;225:563–564. [Google Scholar]
- 27.Bryngelson J D, Wolynes P G. Proc Natl Acad Sci USA. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dill K A, Bromberg S, Yue K, Fiebig K M, Yee D P, Thomas P D, Chan H S. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan H S, Dill K A. Proteins Struct Funct Genet. 1996;24:335–344. doi: 10.1002/(SICI)1097-0134(199603)24:3<335::AID-PROT6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 30.Futuyma D J. Evolutionary Biology. 3rd Ed. Sunderland, MA: Sinauer; 1998. , chapter 11, pp. 297–335. [Google Scholar]
- 31.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yue K, Dill K A. Proc Natl Acad Sci USA. 1992;89:4163–4167. doi: 10.1073/pnas.89.9.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrison P M, Chan H S, Prusiner S B, Cohen F E. J Mol Biol. 1999;286:593–606. doi: 10.1006/jmbi.1998.2497. [DOI] [PubMed] [Google Scholar]
- 34.Gutin A M, Abkevich V I, Shakhnovich E I. Fold Des. 1998;3:183–194. doi: 10.1016/S1359-0278(98)00026-1. [DOI] [PubMed] [Google Scholar]
- 35.Chan H S, Dill K A. Proteins Struct Funct Genet. 1998;30:2–33. doi: 10.1002/(sici)1097-0134(19980101)30:1<2::aid-prot2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 36.Kim D E, Gu H, Baker D. Proc Natl Acad Sci USA. 1998;95:4982–4986. doi: 10.1073/pnas.95.9.4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bowie J U, Reidhaar-Olson J F, Lim W A, Sauer R T. Science. 1990;247:1306–1310. doi: 10.1126/science.2315699. [DOI] [PubMed] [Google Scholar]
- 38.Rasmussen B J, Stock A M, Ringe D, Petsko G A. Nature (London) 1992;357:423–424. doi: 10.1038/357423a0. [DOI] [PubMed] [Google Scholar]
- 39.Daniel R M, Smith J C, Ferrand M, Héry S, Dunn R, Finney J L. Biophys J. 1998;75:2504–2507. doi: 10.1016/S0006-3495(98)77694-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milla M E, Brown B M, Sauer R T. Nat Struct Biol. 1994;1:518–523. doi: 10.1038/nsb0894-518. [DOI] [PubMed] [Google Scholar]
- 41.Shortle D, Stites W E, Meeker A K. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 42.Green S M, Meeker A K, Shortle D. Biochemistry. 1992;31:5717–5728. doi: 10.1021/bi00140a005. [DOI] [PubMed] [Google Scholar]
- 43.Meeker A K, Garcia-Moreno B E, Shortle D. Biochemistry. 1996;35:6443–6449. doi: 10.1021/bi960171+. [DOI] [PubMed] [Google Scholar]
- 44.Hecht M H, Hehir K M, Nelson H C M, Sturtevant J M, Sauer R T. J Cell Biochem. 1985;29:217–224. doi: 10.1002/jcb.240290306. [DOI] [PubMed] [Google Scholar]
- 45.Hecht M H, Sturtevant J M, Sauer R T. Proteins Struct Funct Genet. 1986;1:43–46. doi: 10.1002/prot.340010108. [DOI] [PubMed] [Google Scholar]