

Abstract
Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1) activates NF-κB and c-Jun N-terminal kinase (JNK), which is essential for LMP1 oncogenic activity. Genetic analysis has revealed that tumor necrosis factor receptor-associated factor 6 (TRAF6) is an indispensable intermediate of LMP1 signaling leading to activation of both NF-κB and JNK. However, the mechanism by which LMP1 engages TRAF6 for activation of NF-κB and JNK is not well understood. Here we demonstrate that TAK1 MAP kinase kinase kinase and TAK1-binding protein 2 (TAB2), together with TRAF6, are recruited to LMP1 through its N-terminal transmembrane region. The C-terminal cytoplasmic region of LMP1 facilitates the assembly of this complex and enhances activation of JNK. In contrast, IκB kinase γ (IKKγ) is recruited through the C-terminal cytoplasmic region and this is essential for activation of NF-κB. Furthermore, we found that ablation of TAK1 resulted in the loss of LMP1-induced activation of JNK, but not of NF-κB. These results suggest that an LMP1-associated complex containing TRAF6, TAB2 and TAK1 plays an essential role in the activation of JNK. However, TAK1 is not an exclusive intermediate for NF-κB activation in LMP1 signaling.
Persistent latent infection with Epstein-Barr virus (EBV), a γ herpes virus that is classified as a human DNA tumor virus, is widespread in the human population, and can cause the development of malignancies such as Hodgkin’s lymphoma, Burkitt’s lymphoma and nasopharyngel carcinoma. The latent membrane protein 1 (LMP1) is an oncoprotein encoded by EBV and is critically involved in the effective immortalization and proliferation of B-cells latently infected by EBV (1-3).
LMP1 is a transmembrane protein of 386 amino acids containing a short N-terminal cytoplasmic domain of 24 amino acids, six transmembrane-spanning domains and a C-terminal cytoplasmic tail of 200 amino acids (Figure 1A). LMP1 mimics a constitutively activated tumor necrosis factor (TNF) receptor-like molecule, in the absence of ligand binding (4,5). The transmembrane domains of LMP1 mediate spontaneous autoaggregation within plasma membrane, and this aggregation is a prerequisite for LMP1 function (6,7). Previous studies have identified two domains in the LMP1 C-terminal cytoplasmic tail, called the C-terminal activator regions (CTARs) 1 (aa 194-231) and 2 (aa 332-386), that are important for the cell transformation activity of LMP1 (4,5,8). Both CTAR1 and CTAR2 participate in the activation of the transcription factor NF-κ B. CTAR1 binds to several TNF receptor-associated factors (TRAFs), TRAF1, 2, 3, and 5, through a consensus TRAF-binding motif, while CTAR2 has been shown to bind to the TNF receptor-associated death domain protein (TRADD). However, experiments using gene knockout and small interfering RNA (siRNA) treatment have determined that neither TRAF2, TRAF5 nor TRADD is essential for LMP1 signaling (9,10). Therefore, these molecules are likely to act redundantly. In contrast, two independent studies have shown that TRAF6, which was previously established as an essential mediator of interleukin 1 (IL-1) and receptor activator of NF-κB (RANK), is also a critical factor for LMP1-induced activation of NF-κB and JNK (9,10). Unlike TRAF2 and TRAF5, TRAF6 has not been extensively studied in connection with LMP1. It is still unclear which LMP1 domains are involved in its association with TRAF6. Moreover, the mechanism by which TRAF6 mediates LMP1 signaling has not been well studied.
Fig. 1.


LMP1 associates with TAK1, TAB2 and TRAF6.
(A) Schematic drawing of wild type LMP1 and mutants.
(B-D) 293 cells were transfected with expression vectors for various versions of Flag-LMP1 or Flag-Transferrin Receptor (TfR) together with HA-TAK1 (B), HA-TAB2 (C) or HA-TRAF6 (D). Cell lysates were immunoprecipitated (IP) and analyzed by immunoblotting (IB). WCE, whole cell extracts; HC, immunoglobulin heavy chain; LC, immunoglobulin light chain.
Transforming growth factor β (TGF-β) activated kinase 1 (TAK1) is a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family and is activated by various cytokines, including the family of TGF-β ligands (11). TAK1 is also involved in the IL-1 signaling pathway (12). Following exposure of cells to IL-1, endogenous TAK1 is recruited to the TRAF6 complex and activated, upon which it stimulates both the JNK and NF-κB pathways. Several lines of evidence indicate that TAK1 is an essential mediator of innate immunity signaling: (i) TAK1 deletion or siRNA targeting TAK1 abolishes IL-1-induced NF-κB activation (13,14); (ii) a selective inhibitor against TAK1 inhibits IL-1-induced activation of NF-κB and JNK (15); (iii) TAK1 deficiency in Drosophila causes an impaired immune response to bacterial infection (16). In previous studies, we have isolated TAB2 and its homologue TAB3, proteins that interact with TAK1. TAB2 and TAB3 are also intermediates in a proinflammatory signaling pathways (17,18). Knockdown of both TAB2 and TAB3 by siRNA diminishes IL-1 responses. Although TAB2 and TAB3 have overlapping functions, TAB2 is more directly involved in the TRAF6 pathway. TAB2 directly associates with TRAF6 as well as TAK1, and this interaction facilitates the assembly of a signaling complex consisting of TRAF6, TAB2, and TAK1 in response to IL-1 and RANK ligand stimulation (18-20). A recent report has demonstrated that TAK1 is activated by LMP1 and that knockdown of TAK1 expression by siRNA causes a defect in LMP1-induced JNK activation (10). This suggests that TAK1 participates in LMP1 signaling. However, the exact connections among LMP1, TRAF6, TAK1, NF-κB and JNK remain to be established.
Here we report that TAK1 and TAB2 are involved in LMP1 signaling. We found that TRAF6, TAB2, and TAK1 were assembled into a complex with LMP1. Assembly of these factors was capable of activating JNK, and was mediated through the transmembrane region of LMP1. In contrast, activation of NF-κB absolutely requires the C-terminal cytoplasmic region of LMP1 where IKK is recruited. Furthermore, we found that TAK1 was essential for LMP1-induced JNK activation but dispensable for LMP1-mediated NF-κB activation. These results suggest that the formation of the TRAF6-TAB2-TAK1 complex with LMP1 mediates activation of JNK, while IKK recruited into the C-terminal region of LMP1 can be activated through multiple pathways that may include TAK1.
MATERIALS AND METHODS
Plasmids
pSG5-Flag-LMP1 vectors were generously provided by Dr. Kieff (21). To generate pCMV-Flag-LMP1 and pCMV-HA-JNK vectors, a BamHI fragment from pSG5-Flag-LMP1 or pSRα-HA-JNK was subcloned into a pCMV mammalian expression vector. Flag-LMP1(1-186) and (25-186) mutants were generated by PCR and subcloned into pCMV vectors. The PCR products were verified by sequencing. To generate expression vector of C-terminal Flag-fused Transferrin Receptor, cDNA were excised from pcD-Transferrin Receptor (purchased from ATCC) and subcloned into pCMV-C-Flag vector. T7-TAK1 and HA-TRAF6 were also subcloned into pCMV vectors. To knockdown the expression of TAK1, siRNA targeting TAK1 were used. Origonuclotides corresponded to nucleotides 88-106 of the TAK1 coding region were subclonedinto the BS/H1 vector to produce small interference RNA (siRNA).
Cell cultures and transfection
293 cells, TAK1 +/+, and TAK1 Δ/Δ mouse embryonic fibroblasts (MEFs) (14), and HeLa S3 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum or Bovine Growth Serum (Hyclone) at 37°C in 5% CO2. For the transfection studies, 293 cells (5 x 105 cells) were plated in 10 cm-diameter dishes, transfected with a total of 10 μg of DNA containing various expression vectors by the calcium phosphate precipitation method, and incubated for 36 to 48 hr. TAK1 +/+ and TAK1 Δ/Δ MEFs and HeLa S3 cells were transfected using FuGENE6 Transfection Reagent (Roche Diagnostics) and TransFast™ Transfection Reagent (Promega).
Chemicals and antibodies
Polyclonal rabbit antibodies to TRAF6 (anti-TRAF6), TAK1 (anti-TAK1), and TAB2 (anti-TAB2) have been described previously (12,18). Anti-Flag M2 monoclonal antibody (Sigma), anti-HA (HA.11) monoclonal antibody (Babco), anti-HA (Y-11) polyclonal antibody (Santa Cruz), anti-T7 monoclonal antibody (Novagen), anti-TRAF2 (H-249) polyclonal antibody (Santa Cruz), anti-JNK1 (FL) polyclonal antibody (Santa Cruz), anti-β-actin (Sigma) and anti-p65 (A) polyclonal antibody (Santa Cruz) were used for immunoprecipitation and immunoblotting. Anti-phospho-SAPK/JNK (Thr-183/Tyr-185) rabbit polyclonal antibody (Cell Signaling) was used to detect the phosphorylated forms of JNK. The TAK1 inhibitor, 5Z-7-oxozeaenol, has been described previously (15). Recombinant human TNF-α (Roche Diagnostics) and anisomycin (Calbiochem) were used.
Generation of cell lines stably expressing T7-TAK1
To establish stable cell lines that express wild type TAK1, TAK1 Δ/Δ MEFs were transfected with pCMV-T7-TAK1 together with an expression vector for hygromycin resistance. Hygromycin resistant clones were selected in medium containing 200 μ g/ml hygromycin B (Calbiochem), and expression of T7-TAK1 was verified by immunoblotting with anti-TAK1.
Generation of cell lines stably expressing siRNA for TAK1
To establish stable cell lines that express siRNA targeting TAK1, HeLa S3 cells were transfected with pBS/H1-TAK1 siRNA vector together with an expression vector for neomycin resistance. Neomycin resistant clones were selected in medium containing 500 mg/ml G418 (Invitrogen), and expression of TAK1 was determined by immunoblotting.
Reporter gene assays
For the reporter gene assays, 293 cells (8 x 104 cells), TAK1 +/+ or TAK1 Δ/Δ MEFs (3 x 104 cells) were plated into six-well dishes (35 mm), or HeLa S3 cells (1.5 x 104 cells) were plated into 12-well dishes. At 24 hr after seeding, cells were transfected with a reporter plasmid and expression vectors as indicated. Some cells were treated with TNF (20 ng/ml) for 36 hr. An Ig-κ-firefly luciferase reporter was used to measure NF-κB-dependent gene activation. An AP-1-firefly luciferase reporter was used to measure AP-1-dependent gene activation. Plasmids encoding β-galactosidase under the control of the β -actin promoter or Renilla luciferase under the control of the EF1α promoter were used to normalize transfection efficiency.
Immunoprecipitation and immunoblotting
Cells were washed once with ice-cold phosphate-buffered saline and lysed in 0.3 ml of lysis buffer (20 mM HEPES [pH 7.4], 150 mM NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 2 mM EGTA, 10 mM NaF, 2 mM DTT, 1 mM Na3VO4, 1 mM PMSF, 20 μM aprotinin, 0.5% Triton X-100). For co-precipitation assay, cells were lysed in 0.3 ml of RIPA buffer (0.1% NP-40, 0.5% Sodium Deoxycholate, 0.1% SDS, 1 mM PMSF, 1 mM Na3Vo4, 20 μM aprotinin). Cell debris was removed by centrifugation at 15,000 x g for 5 min. Proteins from cell lysates were immunoprecipitated with 1 μ g of various antibodies and 20 μ l of protein G-Sepharose (Amersham Biosciences). The immunoprecipitates were washed three times with washing buffer (20mM HEPES, 10 mM MgCl2, 500 mM NaCl), once with rinse buffer (10 mM HEPES [pH 7.4], 5 mM MgCl2, 1 mM DTT) and resuspended in 30 μl of rinse buffer. For immunoblotting, the immunoprecipitates or whole-cell lysates were resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Hybond-P membranes (Amersham Biosciences). The membranes were immunoblotted with various antibodies, and the bound antibodies were visualized with horseradish peroxidase-conjugated antibodies against rabbit or mouse immunoglobulin G (IgG) using the Enhanced Chemiluminesense (ECL) Western Blotting System (Amersham Biosciences).
Gel retardation analysis
293 cells, TAK1 +/+ or TAK1 Δ/Δ MEFs were either left untreated or treated with TNF (20 ng/ml) for 30 min and harvested and lysed for use in gel retardation assays. 32P-labeled NF-κB oligonucleotides (Promega) were used. The binding reactions containing the radiolabeled probe, 15 μg of cell extracts, 4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris-HCl [pH 7.5], 500 ng poly (dI-dC) (Amersham) and 10 μg BSA in a final volume of 30 μl were incubated at room temperature for 1 hr and subjected to electrophoresis on a 4% (w/v) polyacrylamide gel in TBE buffer. For supershift assays, 1 μg of control rabbit IgG or anti-p65 were added to the reactions and incubated for 30 min at room temperature before probes were added. The gels were dried and exposed to X-ray film.
RESULTS
We have previously demonstrated that TAK1 and its adapter protein TAB2 play important roles in IL-1 and RANKL signaling pathways via complex formation with TRAF6 (18,19). Recently, it was shown that TRAF6 is an indispensable intermediate in LMP1 signaling (9,10). This raised the possibility that TAK1 and TAB2 also participate in LMP1 signaling. To test this possibility, we used co-precipitation assays in human embryonic kidney 293 cells to examine whether LMP1 associates with TAK1, TAB2 and TRAF6 (Figure 1B-D). 293 cells were transfected with expression vectors for Flag-tagged LMP1 (Flag-LMP1), together with HA-tagged TAK1 (HA-TAK1), HA-tagged TAB2 (HA-TAB2), or HA-tagged TRAF6 (HA-TRAF6). Flag-tagged transferrin receptor (Flag-TfR) was used as a control membrane protein. Cell extracts were immunoprecipitated with anti-Flag or anti-HA monoclonal antibody, and co-precipitated proteins were detected by immunoblotting. We found that TAK1, TAB2 and TRAF6 were capable of associating with LMP1.
The N-terminal transmembrane region of LMP1 mediates autoaggregation, which is a prerequisite for LMP1 activation (6,7), while CTAR1 and CTAR2 domains in the C-terminal cytoplasmic tail are thought to mediate LMP1 signaling. TRAF2, TRAF5, and TRADD are involved in CTAR1/2-mediated signaling (4,5,8). To determine which domain is involved in the binding of TAK1, TAB2, and TRAF6, we utilized several LMP1 mutant constructs (Figure 1A). The LMP1-AA, -ID and -DM mutants (kindly provided by Dr. Kieff) have mutations in essential amino acid residues of CTAR1, CTAR2 and both, respectively (21). In addition, we generated the mutant LMP1(1-186) and LMP1(25-186), which lack the entire C-terminal cytoplasmic tail and both the N-terminal and C-terminal cytoplasmic domains, respectively. We transfected expression vectors for these various LMP1 mutants together with HA-TAK1, HA-TAB2, or HA-TRAF6 (Figure 1B-D). We found that TAK1, TAB2 and TRAF6 were co-precipitated with all three types of LMP1 mutations: LMP1-AA, -ID and -DM. These results suggest that the CTAR1 and CTAR2 domains are not involved in the association of LMP1 with TAK1, TAB2 and TRAF6. Furthermore, even LMP1(1-186) or (25-186) could co-precipitate TAK1, TAB2 and TRAF6, although they seem to associate less efficiently with TAK1 and TRAF6. The reciprocal immunoprecipitation study (Figure 1 B-D right panels) confirmed that TAK1, TAB2 and TRAF6 could co-precipitate WT LMP1, LMP1-AA, -ID, -DM and (1-186). These results suggest that TAK1, TAB2 and TRAF6 interact with LMP1 primarily through the N-terminal transmembrane region. The C-terminalregion of LMP1 might stabilize the assembly of the complex.
To verify the physiological relevance of these interactions, we next investigated the complexes between LMP1 and endogenous molecules. We transfected 293 cells with expression vectors for various versions of Flag-LMP1 and used anti-Flag antibody to immunoprecipitate complexes. We then attempted to detect co-precipitated endogenous proteins including TRAF2, TRAF6, TAB2, and TAK1 (Figure 2A). TRAF2 is known to associate with LMP1 through the CTAR1 domain (22). Consistent with this, TRAF2 was found to associate with WT LMP1 and the LMP1-ID mutant, but not with the LMP1-AA, -DM, or (1-186) mutants (Figure 2A). In contrast, we found that TRAF6 was co-precipitated with WT LMP1, LMP1-AA. -ID, -DM, and (1-186) mutants. TAB2 and TAK1 were also co-precipitated with WT LMP1 LMP1-AA, -ID, -DM and less efficiently with (1-186). This is consistent with the results from overexpression proteins shown above. The reciprocal immunoprecipitation was performed in cell extracts from WT LMP1 and LMP1(1-186) expressing cells (Figure 2B). TAK1 could co-precipitate WT LMP1 but less effectively LMP1(1-186). These results suggest that LMP1 can recruit TRAF6, TAB2 and TAK1 through its N-terminal transmembrane region and that the C-terminal region functions to stabilize the complex.
Fig. 2.

LMP1 associates with endogenous TAK1, TAB2 and TRAF6.
(A) 293 cells were transfected with expression vectors for various versions of Flag-LMP1. Cell lysates were immunoprecipitated with anti-Flag, and co-precipitated endogenous proteins were analyzed by immunoblotting. The precipitated of Flag-LMP1 is also shown. WCE: whole cell extracts; HC, immunoglobulin heavy chain.
(B) 293 cells were transfected with expression vectors for Flag-LMP1 WT or 1-186 mutant. Cell lysates were immunoprecipitated with anti-TAK1, and co-precipitated Flag-LMP1 was analyzed by immunoblotting. The precipitated TAK1 and TAB2 are also shown. WCE: whole cell extracts. The asterisk indicates that a partially degraded Flag-LMP1 is also coprecipitated with TAK1 and TAB2.
The N-terminal transmembrane region is essential for LMP1’s action. However, the functional role of the N-terminus has not been fully addressed. We therefore examined the effects of LMP1(1-186) expression on the activation of JNK and NF-κ B (Figure 3). 293 cells were transfected with expression vectors for various Flag-LMP1 mutants. Activation of JNK was detected by immunoblotting with phospho-specific JNK antibody. The CTAR domains of LMP1 has been implicated in JNK activation (23,24). Consistently, we found that mutations at both CTAR domains and the deletion of the C-terminal region impaired JNK activation (Figure 3A). However, we found that the N-terminal transmembrane region alone had some ability to activate JNK when it was highly expressed (Figure 3A, left and right panels). This activation might be correlated with the recruitment of TAK1, TAB2 and TRAF6 into the LMP1 complex, which occur weakly with the LMP1(1-186) and most strongly with the WT LMP1.
Fig. 3.


Effect of LMP1 mutations on activation of JNK and NF-κB.
(A) 293 cells were transfected with expression vectors for various versions of Flag-LMP1. Increasing amount of Flag-LMP1 1-186 vectors were used. The activated form of JNK was detected with phospho-JNK antibody (P-JNK).
(B) 293 cells were transfected with expression vectors for various versions of Flag-LMP1. The amounts of plasmids were same as (A). Cell lysates were subjected to electrophoretic mobility shift assay (EMSA) using NF-κB DNA probe (top panel). NF-κB binding was confirmed by supershift with anti-p65. Amounts of p65 in the cell lysates and expression levels of LMP1 are also shown. WCE: whole cell extracts.
(C) 293 cells were transfected with an NF-κB-dependent luciferase reporter and pAct-β-galactosidase plasmids, together with expression vectors for various versions of Flag-LMP1 or Flag-TfR. Increasing amount of Flag-LMP1 1-186 vectors were used. Luciferase activity was determined and normalized to the levels of β-galactosidase activity. Stimulation relative to control transfection with empty vector is shown.
(D) Left panels: 293 cells were transfected with expression vectors for various versions of Flag-LMP1 or Flag-Transferrin Receptor (TfR) together with HA-IKKγ. Cell lysates were immunoprecipitated (IP) and analyzed by immunoblotting (IB). WCE: whole cell extracts. Right panels: 293 cells were transfected with expression vectors for T7-TAK1 together with HA-IKKγ in the absence or presence of Flag-LMP1 WT or 1-186 mutant. Cell lysates were immunoprecipitated (IP) and analyzed by immunoblotting (IB). WCE: whole cell extracts.
NF-κB activation was examined by employing an NF-κB electrophoretic mobility shift assay (Figure 3B) and NF-κB-dependent reporter assay (Figure 3C). In agreement with earlier studies (22,25), disruption of either CTAR1 or CTAR2 (LMP1-AA or -ID mutants) reduced NF-κB binding activity, while the double-mutation (LMP1-DM) abolished NF-κB activation (Figure 3B). The LMP1(1-186) mutant were completely unable to stimulate NF-κB binding activity. The NF-κB-dependent reporter assay further confirmed that LMP1(1-186) mutant could not activate NF-κB (Figure 3C). Taken together, these results suggest that formation of a complex containing TRAF6, TAB2 and TAK1, assembled through interactions with the LMP1 N-terminal transmembrane region, might directly participate in JNK activation, but is not sufficient to mediate NF-κB activation.
Lack of NF-κB activation by the LMP1 transmembrane region suggests that the signaling complex formed through the transmembrane region is not sufficient to activate NF-κB. We therefore hypothesized that the downstream effectors may not be recruited in the complex of LMP1 transmembrane region. We examined whether IKK is contained in the LMP1 complex. We transfected expression vectors for IKKγ together with the WT LMP1 LMP1-AA, -ID, -DM, LMP1(1-186) or LMP1(25-186) (Figure 3D left panels). We found that IKKγ was precipitated with WT LMP1, LMP1-AA, -ID, and -DM but not with the C-terminal truncated mutants. This result indicates that the LMP1 recruits IKKγ through its C-terminal region. Therefore, the transmembrane region alone is not sufficient for recruitment of the IKK complex. LMP-DM has ability to recruit IKKγ despite the fact that it does not activate NF-κB. This suggests that any of TRAF2, TRAF5 or TRADD, which is recruited through CTAR domains, is likely to be essential for NF-κB activation. We next looked at relationship between TAK1 and IKKγ in the LMP1 complex. Wetransfected expression vectors for IKKγ, TAK1 together with the WT LMP1 or LMP1(1-186) (Figure 3D right panels). TAK1 was not coprecipited with IKKγ in the absence of LMP1. Expression of WT LMP1 but not LMP1(1-186) greatly increased interaction of TAK1 with IKKγ. These results suggest that TAK1 by itself does not interact with IKKγ and that integrity of LMP1 is important for assembly of the signaling complex leading to NF-κB activation.
We next asked whether TAK1 is essential for LMP1 signaling by utilizing a selective inhibitor of TAK1, 5Z-7-oxozeaenol (Figure 4). As shown previously (15), 10-30 nM of 5Z-7-oxozeaenol can inhibit activation of JNK by TNF (Figure 4A) and IL-1 treatments (data not shown). Cells expressing LMP1 were treated with vehicle or 5Z-7-oxozeaenol every 12 hr, and JNK activity was measured. 5Z-7-oxozeaenol completely blocked LMP1-induced JNK activation, to an extent similar to its inhibition of the TNF response (Figure 4A).
Fig. 4.
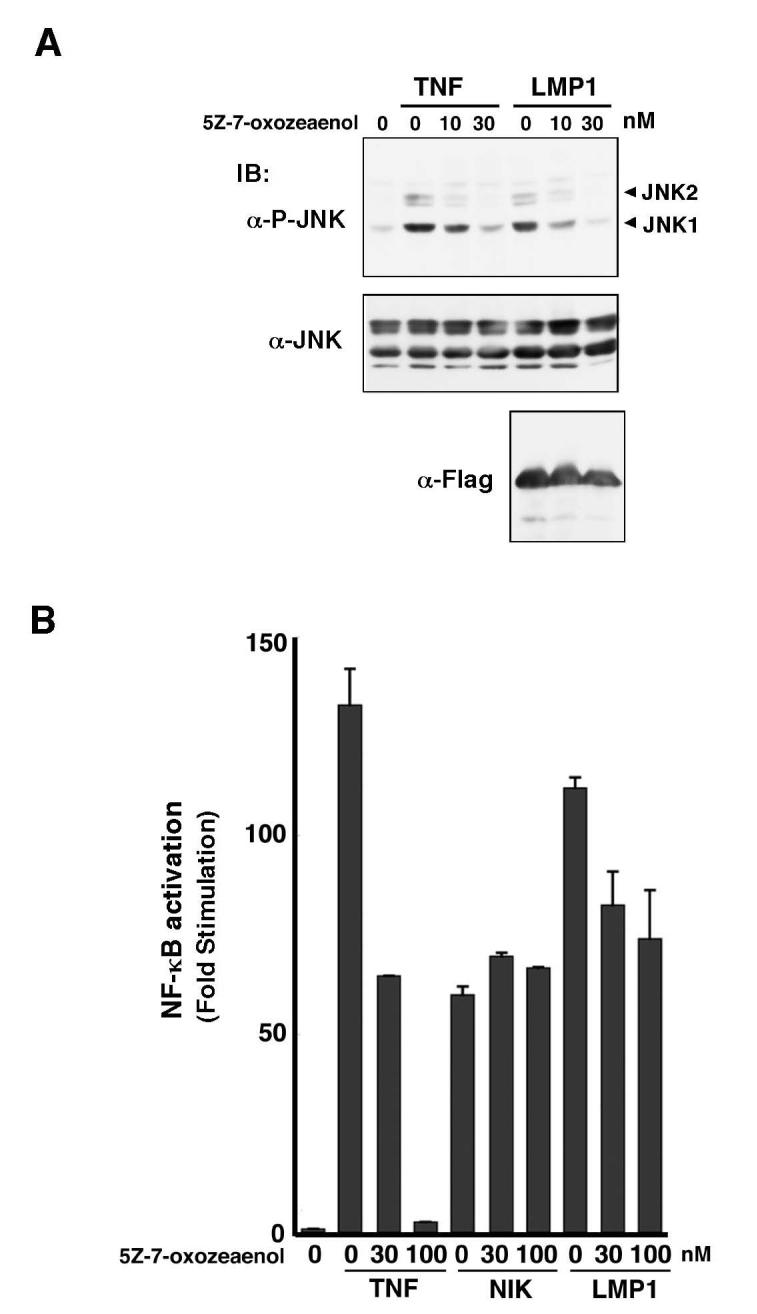
Effect of TAK1 inhibitor.
(A) 293 cells were transfected with empty vector or expression vectors for Flag-LMP1. 5Z-7-oxozeaenol was added to the culture medium every 12 hr after transfection and some cells were treated with 10 ng/ml TNF for 30 min prior to harvest. The activated form of JNK was detected with phospho-JNK antibody (P-JNK).
(B) 293 cells were transfected with NF-κB-dependent luciferase reporter and pAct-β-galactosidase plasmids, together with expression vectors for NIK or Flag-LMP1. 5Z-7-oxozeaenol was added to the culture medium at the concentrations indicated every 12 hr after transfection. Luciferase activity was determined and normalized to the levels of β-galactosidase activity. Stimulation relative to control transfection with empty vector is shown.
We next tested the effect of 5Z-7-oxozeaenol on LMP1-mediated NF-κB activation using an NF-κB-dependent reporter construct (Figure 4B). As described previously (15), 5Z-7-oxozeaenol effectively blocked TNF-induced activation of NF-κB, while overexpression of NF-κB-inducing kinase (NIK) activated NF-κB independently of TAK1. We found that 5Z-7-oxozeaenol had only marginal effect on LMP1-induced NF-κB activation. These results suggest that TAK1 is primarily involved in LMP1 signaling leading to activation of JNK, but not of NF-κB.
To further define the role of TAK1 in LMP1 signaling, we utilized mouse embryonic fibroblasts having a homozygous deletion of the ATP binding site within the TAK1 kinase domain (aa 41-77) (TAK1 Δ/Δ MEFs) (Figure 5A). This cell line was obtained by infecting MEFs containing homozygous TAK1 genes flanked by loxP sites with a Cre-expressing retroviral vector (14). The line was subsequently immortalized using the 3T3 protocol. We examined LMP1-induced JNK activation in TAK1 +/+ and TAK1 Δ/Δ MEFs transfected with HA-tagged JNK. JNK activity was measured by immunoprecipitation of HA-JNK (HA-JNK) followed by immunoblotting with anti-phospho-JNK (Figure 5B). An inhibitor of the peptidyltransferase reaction, anisomycin, can trigger a ribotoxic stress response that strongly activates JNK. We found that anisomycin activated JNK in both TAK1 +/+ and TAK1 Δ/Δ MEFs. In contrast, LMP1 activated JNK in TAK1 +/+ but not in TAK1 Δ/Δ MEFs. To verify whether the defect is due to the TAK1-deficiency, we generated TAK1 Δ/Δ MEFs stably expressing wild type T7-tagged TAK1 (T7-TAK1) (Figure 5B, right panels). Expression of TAK1 in TAK1 Δ/Δ MEFs could restore LMP1-induced JNK activation. These results confirmed that TAK1 is an essential mediator of LMP1 signaling leading to JNK activation.
Fig. 5.


TAK1 is essential for LMP1-induced JNK activation.
(A) Schematic drawing of TAK1δ
(B) TAK1 +/+, TAK1 Δ/Δ, and T7-TAK1-expressing TAK1 Δ/Δ MEFs were transfected with expression vectors for HA-JNK and Flag-LMP1. Some cells were treated with anisomycin for 30 min prior to harvest. HA-JNK was immunoprecipitated and the activated form of JNK was detected with phospho-JNK antibody (P-JNK). WCE: whole cell extracts.
(C) TAK1 +/+ and TAK1 Δ/Δ MEFs were transfected with expression vectors for Flag-LMP1. Cell lysates were subjected to electrophoretic mobility shift assay (EMSA) using an NF-κB DNA probe. NF-κB binding was confirmed by supershift with anti-p65. Amounts of p65 in the cell lysates and expression levels of LMP1 are shown. WCE: whole cell extracts.
(D) TAK1 +/+ and TAK1 Δ/Δ MEFs were transfected with NF-κB-dependent luciferase reporter and pEF1α Renilla luciferase plasmids together with expression vector for Flag-LMP1. Luciferase activity was determined and normalized to the levels of Renilla luciferase activity. Stimulation relative to control transfection with empty vector is shown.
(E) Left panels: Proteins from HeLaS3 control or TAK1 siRNA cells were immunoblotted with anti-TAK1 (upper panel) and anti-β-actin (lower panel). Right panels: HeLaS3 control or TAK1 siRNA cells were transfected with expression vectors for HA-JNK and Flag-LMP1. Some cells were treated with anisomycin for 30 min prior to harvest. HA-JNK was immunoprecipitated and the activated form of JNK was detected with phospho-JNK antibody (P-JNK). WCE: whole cell extracts.
(F) HeLaS3 control or TAK1 siRNA cells were transfected with AP-1- or NF-κB-dependent luciferase reporter and pEF1α Renilla luciferase plasmids together with expression vector for Flag-LMP1. Luciferase activity was determined and normalized to the levels of Renilla luciferase activity. Stimulation relative to control transfection with empty vector is shown.
We next tested LMP1-mediated NF-κB activation in TAK1 Δ/Δ MEFs (Figure 5C and D). An earlier study had shown that deletion of TAK1 causes impaired TNF signaling (13,14). As expected, an electrophoretic mobility shift assay showed that TNF-induced NF-κB binding activity was impaired in TAK1 Δ/Δ MEFs (Figure 5C). In contrast, we found that LMP1 was able to activate NF-κB binding activity to a similar or somewhat higher degree in TAK1 Δ/Δ MEFs compared to TAK1 +/+ MEFs. We also examined activity of an NF-κB-dependent reporter construct (Figure 5D). We found that LMP1 activated NF-κB in both TAK1 +/+ and TAK1 Δ/Δ MEFs.
TAK1 Δ/Δ expresses a truncated version of TAK1 that only lacks the ATP binding site as described above. Although this mutant TAK1 has no kinase activity, it may be possible that the truncated TAK1 still can promote activation of NF-κB in a manner of independent of kinase activity. To clarify this possibility, we used HeLa S3 cells stably expressing TAK1 siRNA in which expression of TAK1 is significantly decreased (Figure 5E, left panels). To assess whether lack of TAK1 protein alters LMP1-dependent signaling pathways, we examined LMP-1-induced JNK activation (Figure 5E, right panels), and activity of AP-1- or NF-κB-dependent reporter construct (Figure 5F). AP-1-dependent reporter construct was used to assess the downstream events of JNK. LMP1 could not activate JNK in the cells expressing TAK1 siRNA, which is consistent with the results from TAK1 deletion MEFs. Activation of AP-1 was also impaired by TAK1 depletion, while activation of NF-κB was not affected. These results confirm that TAK1 is dispensable for LMP1-induced NF-κB activation. It is likely that TAK1 redundantly functions to activate NF-κB with other kinases in LMP1 pathway.
DISCUSSION
The transmembrane region of LMP1 is essential for LMP1 signaling. Autoaggregation through the LMP1 transmembrane region is thought to trigger downstream signaling (6,7). However, the functional role of the N-terminal transmembrane region has not been closely examined. Here we demonstrate that TRAF6, TAK1 and its binding protein TAB2 can associate with LMP1 through the LMP1 N-terminal transmembrane region (Figure 1 and 2). This reveals a novel role of LMP1 transmembrane region in the facilitating assembly of a signaling complex consisting of TRAF6, TAB2 and TAK1 (Figure 6). The C-terminal region appears to enhance this assembly as shown in Figure 1 and 2.
Fig. 6.
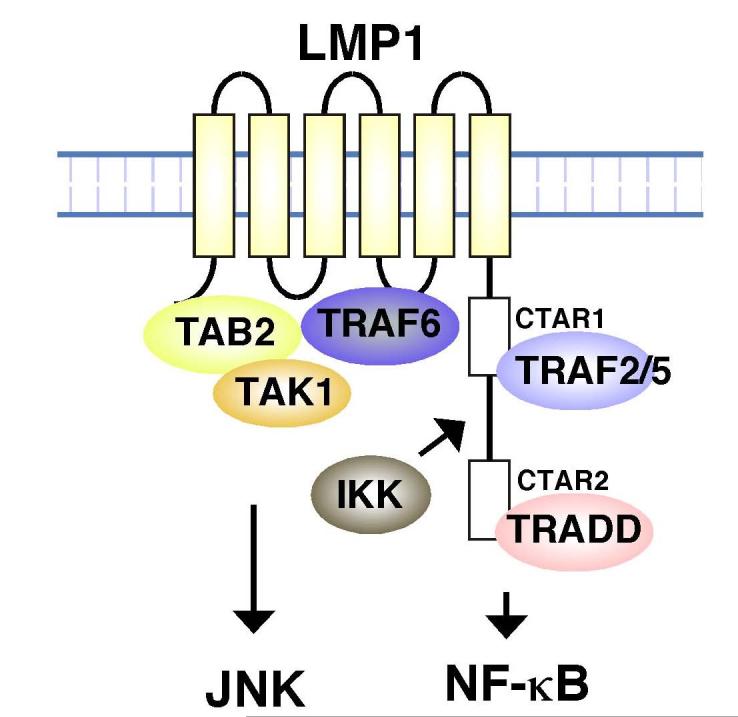
LMP1 signaling complexes.
We also demonstrated that expression of just the N-terminal transmembrane region of LMP1 has the ability to activate JNK (Figure 3A). This suggests that an LMP1 transmembrane complex containing TRAF6, TAB2 and TAK1 can mediate activation of JNK. LMP1 C-terminal region functions to stabilize the complex and enhances JNK activation. The present study also revealed that TAK1 is essential for LMP1-induced activation of JNK (Figure 4A and 5B, E, F). Taken together, assembly of a signaling complex composed of LMP1-TRAF6-TAB2-TAK1 mediates activation of JNK (Figure 6).
In agreement with earlier studies, we demonstrated that the N-terminal transmembrane region alone is completely unable to activate NF-κB (Figure 3B and C). This suggests that the LMP1-TRAF6-TAB2-TAK1 signaling complex could not mediate activation of NF-κB. In IL-1 signaling pathway, TAK1, TAB2 and TRAF6 make a signaling complex and activate IKK. However, in this process, not only forming complex of TRAF6-TAB2-TAK1 but also recruitment of IKKs into this complex is indispensable for IKK activation (26,27). We therefore examined whether LMP1 mutants can recruit IKKs. We found that IKKγ is recruited by WT LMP1 but not by LMP1 (1-186) (Figure 3D). This result suggests that LMP1 transmembrane region recruits TRAF6, TAB2 and TAK1 but not IKK complex resulting in failure of IKK activation.
TAK1 is essential for IL-1- and TNF-induced activation both of JNK and of NF-κB (14). In contrast to this, we found that TAK1 is dispensable for LMP-1-induced NF-κB activation despite the fact that TAK1 is recruited into the LMP1 complex. This raises possibility that other MAPKKK kinases such as NIK, which can activate NF-κB, may function redundantly with TAK1. NIK is indeed implicated in LMP1 signaling (27,28). LMP1 may be able to recruit not only TAK1 but also NIK and transmits the signal leading to activation of JNK and NF-κ B. It is worth noting that NIK does not have ability to activate JNK, whereas TAK1 can activate both JNK and NF-κB.
TAB2 can bind directly TRAF6 and TAK1 and thereby mediate assembly of a TRAF6-TAB2-TAK1 signaling complex (19). We show here that TRAF6, TAB2 and TAK1 are also components of the LMP1 complex. Thus, it is likely that TAB2 facilitates assembly of the LMP1 complex by binding to both TRAF6 and TAK1. This may appear inconsistent with earlier studies showing that TAB2 is dispensable for activation of both JNK and NF-κ B in LMP1 signaling (9,10). However, we previously reported that a homologue of TAB2, TAB3, plays a redundant role with TAB2 in the IL-1 and TNF signaling pathways (16). It is therefore possible that in TAB2 knockout cells, TAB3 might compensate for the function of TAB2 in LMP1 complex assembly.
This newly identified LMP1 complex containing TRAF6, TAB2 and TAK1 appears to be important for the activity of LMP1. Further studies utilizing TAK1 deletion mice may help revealing the in vivo role of this complex in B-cell transformation.
Glossary
The abbreviations used are
- EBV
Epstein-Barr virus
- LMP1
latent membrane protein 1
- JNK
c-Jun N-terminal kinase
- TRAF
tumor necrosis factor receptor-associated factor
- TNF
tumor necrosis factor
- TGF-β
Transforming growth factor β
- TAK1
TGF-β activated kinase 1
- TAB
TAK1-binding protein
- IKK
IκB kinase
- CTAR
C-terminal activator region
- TRADD
TNF receptor-associated death domain protein
- IL-1
interleukin 1
- RANK
receptor activator of NF-κB
- NIK
NF-κB-inducing kinase
- MEF
mouse embryonic fibroblast.
Footnotes
We thank E. Kieff and E. Nishida for materials. This work was supported by special grants for SORST and Advanced Research on Cancer from the Ministry of Education, Culture and Science of Japan (KM), and by NIH grants GM068812 and AR050972 (JNT).
REFERENCES
- 1.Farrell PJ. Trends. Microbiol. 1995;3:105–109. doi: 10.1016/s0966-842x(00)88891-5. [DOI] [PubMed] [Google Scholar]
- 2.Klein G. Cell. 1994;77:791–793. doi: 10.1016/0092-8674(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 3.Rickinson A, Kieff E. In: Fields Virology. Howley PM, Knipe D, editors. Lippincott; Philadelphia: 2001. pp. 2575–2627. [Google Scholar]
- 4.Cahir-McFarland ED, Izumi KM, Mosialos G. Oncogene. 1999;18:6959–6964. doi: 10.1038/sj.onc.1203217. [DOI] [PubMed] [Google Scholar]
- 5.Lam N, Sugden B. Cell. Signal. 2003;15:9–16. doi: 10.1016/s0898-6568(02)00083-9. [DOI] [PubMed] [Google Scholar]
- 6.Coffin WF, 3rd, Geiger TR, Martin JM. J. Virol. 2003;77:3749–3458. doi: 10.1128/JVI.77.6.3749-3758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yasui T, Luftig M, Soni V, Kieff E. Proc. Natl. Acad. Sci. USA. 2004;101:278–283. doi: 10.1073/pnas.2237224100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eliopoulos AG, Young LS. Semin. Cancer Biol. 2001;11:435–444. doi: 10.1006/scbi.2001.0410. [DOI] [PubMed] [Google Scholar]
- 9.Luftig M, Prinarakis E, Yasui T, Tsichritzis T, Cahir-McFarland E, Inoue J, Nakano H, Mak TW, Yeh WC, Li X, Akira S, Suzuki N, Suzuki S, Mosialos G, Kieff E. Proc. Natl. Acad. Sci. USA. 2003;100:15595–15600. doi: 10.1073/pnas.2136756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wan J, Sun L, Mendoza JW, Chui YL, Huang DP, Chen ZJ, Suzuki N, Suzuki S, Yeh WC, Akira S, Matsumoto K, Liu ZG, Wu Z. Mol. Cell. Biol. 2004;24:192–199. doi: 10.1128/MCB.24.1.192-199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 12.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 13.Takaesu G, Surabhi RM, Park KJ, Ninomiya-Tsuji J, Matsumoto K, Gaynor RB. J. Mol. Biol. 2003;326:105–115. doi: 10.1016/s0022-2836(02)01404-3. [DOI] [PubMed] [Google Scholar]
- 14.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takewuchi O, Akira S. Nat. Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 15.Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, Mihara M, Tsuchiya M, Matsumoto K. J. Biol. Chem. 2003;278:18485–18490. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- 16.Vidal S, Khush RS, Leulier F, Tzou P, Nakamura M, Lemaitre B. Genes Dev. 2001;15:1900–1912. doi: 10.1101/gad.203301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishitani T, Takaesu G, Ninomiya-Tsuji J, Shibuya H, Gaynor RB, Matsumoto K. EMBO J. 2003;22:6277–6288. doi: 10.1093/emboj/cdg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, Ninomiya-Tsuji J, Matsumoto K. Mol. Cell. 2000;5:649–658. doi: 10.1016/s1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 19.Mizukami J, Takaesu G, Akatsuka H, Sakurai H, Ninomiya-Tsuji J, Matsumoto K, Sakurai N. Mol. Cell. Biol. 2002;22:992–1000. doi: 10.1128/MCB.22.4.992-1000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takaesu G, Ninomiya-Tsuji J, Kishida S, Li X, Stark GR, Matsumoto K. Mol. Cell. Biol. 2001;21:2475–2484. doi: 10.1128/MCB.21.7.2475-2484.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higuchi M, Izumi KM, Kieff E. Proc. Natl. Acad. Sci. USA. 2001;98:4675–4680. doi: 10.1073/pnas.081075298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devergne O, Hatzivassiliou E, Izumi KM, Kaye KM, Kleijnen MF, Kieff E, Mosialos G. Mol. Cell. Biol. 1996;16:7098–7108. doi: 10.1128/mcb.16.12.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eliopoulos AG, Blake SM, Floettmann JE, Rowe M, Young LS. J. Virol. 1999;73:1023–1035. doi: 10.1128/jvi.73.2.1023-1035.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kieser A, Kaiser C, Hammerschmidt W. EMBO J. 1999;18:2511–2521. doi: 10.1093/emboj/18.9.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izumi KM, Kieff ED. Proc. Natl. Acad. Sci. USA. 1997;94:12592–12597. doi: 10.1073/pnas.94.23.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 27.Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. Mol. Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 28.Saito N, Courtois G, Chiba A, Yamamoto N, Nitta T, Hironaka N, Rowe M, Yamaoka S. J. Biol. Chem. 2003;278:46565–46575. doi: 10.1074/jbc.M302549200. [DOI] [PubMed] [Google Scholar]
- 29.Luftig M, Yasui T, Soni V, Kang MS, Jacobson N, Cahir-McFarland E, Seed B, Kieff E. Proc. Natl. Acad. Sci. USA. 2004;101:141–146. doi: 10.1073/pnas.2237183100. [DOI] [PMC free article] [PubMed] [Google Scholar]