

Abstract
We previously demonstrated that infection of cultured cells with murine coronavirus mouse hepatitis virus (MHV) resulted in activation of the mitogen-activated protein kinase (Raf/MEK/ERK) signal transduction pathway (Y. Cai et al., Virology 355:152-163, 2006). Here we show that inhibition of the Raf/MEK/ERK signaling pathway by the MEK inhibitor UO126 significantly impaired MHV progeny production (a reduction of 95 to 99% in virus titer), which correlated with the phosphorylation status of ERK1/2. Moreover, knockdown of MEK1/2 and ERK1/2 by small interfering RNAs suppressed MHV replication. The inhibitory effect of UO126 on MHV production appeared to be a general phenomenon since the effect was consistently observed in all six different MHV strains and in three different cell types tested; it was likely exerted at the postentry steps of the virus life cycle because the virus titers were similarly inhibited from infected cells treated at 1 h prior to, during, or after infection. Furthermore, the treatment did not affect the virus entry, as revealed by the virus internalization assay. Metabolic labeling and reporter gene assays demonstrated that translation of cellular and viral mRNAs appeared unaffected by UO126 treatment. However, synthesis of viral genomic and subgenomic RNAs was severely suppressed by UO126 treatment, as demonstrated by a reduced incorporation of [3H]uridine and a decrease in chloramphenicol acetyltransferase (CAT) activity in a defective-interfering RNA-CAT reporter assay. These findings indicate that the Raf/MEK/ERK signaling pathway is involved in MHV RNA synthesis.
Murine coronavirus mouse hepatitis virus (MHV) is an enveloped, positive-strand RNA virus. MHV contains four or five structural proteins, depending on the viral strain. The spike (S) protein is the virion surface glycoprotein that facilitates virus binding to the host cell receptor and entry into cells. Thus, it is responsible for virus infectivity (24). The hemagglutinin-esterase protein is present only in some MHV strains such as JHM (45, 54); its role in virus infection is not clear. The membrane (M) and small envelope (E) proteins are important for virion assembly and morphogenesis (4, 51). The nucleocapsid (N) protein is associated with the viral RNA genome to form the nucleocapsid (49); it may also play a role in viral replication and translation (9, 50).
Infection of host cells by MHV is initiated by binding of the viral S protein to the receptor on the cell surface. This interaction induces conformational change of the S protein and facilitates fusion of viral envelope with plasma membranes or with endosomal membranes after endocytosis (17, 36). Upon entry, the viral nucleocapsid is released into cytoplasm. It has been shown that dephosphorylation of the N protein occurs immediately after entry, and it is thus suggested that dephosphorylation and dissociation of the N protein from the incoming genomic RNA are a prerequisite for viral replication (19, 20). As a positive-strand RNA virus, the MHV genome functions as an mRNA for the synthesis of the viral RNA-dependent RNA polymerase (RdRp) polyprotein via the cap-dependent translation mechanism, which is subsequently processed into 16 nonstructural peptides (nsps 1 to 16) (2, 3, 5, 12, 13, 21, 30, 31, 37, 42, 44, 47). The polymerase complex in turn initiates viral RNA synthesis, including genomic RNA replication and subgenomic mRNA synthesis. In addition to the RdRp, a number of host cell proteins such as heterogeneous nuclear ribonucleoprotein A1, polypyrimidine tract binding protein, poly(A)-binding protein, mitochondrial anicotase, and the synaptotagmin binding cytoplamic RNA-interacting protein (SYNCRIP) have been implicated in modulation of MHV RNA synthesis (7, 16, 27, 28, 35, 48, 57). However, the mechanisms by which these cellular proteins regulate MHV RNA synthesis are largely unknown.
The Raf/MEK/ERK signaling pathway is one of the three mitogen-activated protein kinase (MAPK) cascades that play important roles in the regulation of cell growth and differentiation (41). Activation of the extracellular signal-regulated kinase (ERK) signaling pathway follows three signal cascades. Upon stimulation, Raf kinase is activated, which then activates the MAPK MEK1/2 by phosphorylation of its serine residues (25). Subsequently, the activated MEK1/2 in turn activates ERK1/2 by phosphorylation of its tyrosine and threonine residues (8). Finally, the activated ERK1/2 translocates from cytoplasm to nucleus and phosphorylates a large number of downstream substrates such as transcription factors c-myc, Ets, Elk-1, and Egr-1, which ultimately regulate gene expression (10). Therefore, the ERK signaling pathway involves a wide range of cellular functions including cell proliferation, differentiation, and survival (10, 43). These functions may play a role in virus propagation as well, since viruses are obligate intracellular parasites. Indeed, several viruses including human cytomegalovirus (HCMV), human immunodeficiency virus type 1, influenza virus, respiratory syncytial virus, Borna disease virus, and coxsackievirus B3 have been shown to manipulate the host ERK signaling pathway to regulate viral replication and gene expression (18, 22, 32, 38, 39, 52, 53). However, the role of the MEK/ERK signaling pathway in coronavirus replication is not known.
In a previous study we found that the Raf/MEK/ERK signaling pathway was activated in cultured cells by MHV infection (6). In the present study we examined the role of the Raf/MEK/ERK signaling pathway in MHV replication by using the MEK inhibitors (14), with the goal of identifying particular steps of the virus life cycle being regulated by this signaling pathway. We found that MHV production was severely inhibited by the MEK inhibitor UO126 and siRNAs to MEK1/2 and ERK1/2. Further experiments revealed that UO126 had an inhibitory effect on the synthesis of viral genomic and subgenomic RNAs but did not affect viral entry and viral protein translation. These data demonstrate that the Raf/MEK/ERK signaling pathway plays a role in coronavirus RNA synthesis.
MATERIALS AND METHODS
Viruses, cells, and reagents.
MHV strain JHM (33) was used throughout the present study. For some experiments, MHV strain A59 (26), MHV type 1 (MHV-1), MHV-2, and MHV-3 (kindly provided by Michael Lai, University of Southern California) and Penn98-1 (kindly provided by Ehud Levi, University of Pennsylvania) were also used. Penn98-1 is a recombinant virus containing the MHV A59 genome backbone with the replacement of the spike gene from MHV-2. The mouse astrocytoma cell line (DBT) (23) was used for virus propagation, plaque assay, and all other experiments involving cell culture throughout the present study. DBT cells were cultured in 1× minimum essential medium (MEM) containing 10% tryptose phosphate broth and 7.5% newborn calf serum (Gibco). All MHV strains were propagated in DBT cells in MEM without serum. Mouse fibroblast 17Cl-1 (kindly provided by Susan Baker, Loyola University of Chicago), NIH 3T3, and baby hamster kidney (BHK) cells were used in some experiments. Antibodies specific to the phosphorylated and unphosphorylated forms of ERK1/2 and to MEK1/2 were purchased from Cell Signaling, Inc.
The MEK1/2 inhibitors UO126 and PD98059 (purchased from Cell Signaling, Inc.) were dissolved in dimethyl sulfoxide (DMSO). Both do not compete for ATP-binding or ERK-binding to MEK and, therefore, they are noncompetitive inhibitors (1, 15). The inhibitory effect of the two compounds is very selective. UO126 has very little, if any, effect on the kinase activities of ERK, MEKK, protein kinase C, Ab1, Raf, JNK, Cdk2, Cdk4, MKK-3, MKK-4, and MKK-6 (15). It has been shown that PD98059 blocks the activation (phosphorylation) of MEK1 by its upstream kinase Raf-1 (1), whereas UO126 is not only capable of blocking the activation of MEK by upstream kinases but also inhibiting the MEK1/2 activity (11). UO126 is a more potent MEK1/2 inhibitor than PD98059 (15 ). siRNAs specific to MEK1/2 and ERK1/2 were purchased from Santa Cruz Biochem., Inc., and the EGFP-siRNA was obtained from Invitrogen.
Western blot analysis.
DBT cells were grown to a monolayer in 60-mm petri dishes and were infected with MHV or mock infected. Cells were harvested and lysed in 50 μl of strong lysis buffer (25 mM HEPES [pH 7.5], 0.5% sodium deoxycholate, 1% Triton X-100, 0.2% sodium dodecyl sulfate [SDS], 5 mM EDTA, 1 mM Na3VO4, 20 mM glycerophosphate, 50 mM NaF) containing protease inhibitor cocktail tablets (Roche) and phosphatase inhibitor (Sigma). The lysates were sonicated three times for 20 s each time on ice. The protein concentration was measured by using Bio-Rad protein assay kit (Bio-Rad). Equal amounts of cell lysates were boiled for 5 min, chilled on ice, and pelleted by centrifugation before being loaded onto gels. Proteins were separated by SDS-10% polyacrylamide gel electrophoresis (PAGE). The proteins were then transferred to nitrocellulose membrane (MSI) for 7 h at 40 V in a transfer buffer (25 mM Tris, 200 mM glycine, 20% methanol, 0.02% SDS). After being blocked with 5% skim milk in Tris-buffered saline (TBS) for 1 h at room temperature, the membrane was washed three times in TBS containing 0.5% Tween 20 and then immunoblotted with a primary antibody (1 μg/ml) overnight at 4°C, followed by a secondary antibody coupled to horseradish peroxidase (1:1,000 dilution; Sigma) for 1 h at room temperature. The presence of the immunolabeled protein was detected by enhanced chemiluminescence (ECL) using peracid as a substrate (Amersham), followed by autoradiography.
Virus internalization assay.
DBT cells were incubated with MHV at a multiplicity of infection (MOI) of 2 at 4°C for 1 h. Unbound viruses were then removed by three washes with phosphate-buffered saline (PBS). The cells were either maintained at 4°C or incubated at 37°C for another hour to allow virus internalization, in the presence of UO126 (50 μM) or DMSO, followed by washing with cold PBS and treatment with protease K (0.5 mg/ml) at 4°C for 45 min to remove the bound but uninternalized virus particles. Internalized viruses were titrated by infectious center assay. Briefly, the infected cells were then serially diluted in MEM and added onto uninfected DBT cell monolayers. After incubation for 5 h, detached cells and medium were removed and 0.8% agarose was overlaid onto the monolayer as in the plaque assay. Two days later, plaques were counted after neutral red staining.
[3H]uridine labeling of viral RNA.
DBT cells were infected with MHV or mock infected. The actinomycin D (10 μg/ml) with UO126 (50 μM) or with control DMSO were added to the medium at 1 h postinfection (p.i.). The infected cells were labeled with [3H]uridine (50 μCi/ml; Amersham Biosciences) at 2 h p.i. At various time points p.i., intracellular total RNAs were isolated and purified with Trizol reagent according to the manufacturer's instruction (Invitrogen). RNA samples with an equal volume were precipitated with 10% trichloroacetic acid (TCA). The radioactivities were determined by liquid scintillation counting (Beckman 2500T). The radioactivity reflects the amount of [3H]uridine that was incorporated into viral RNAs during synthesis.
Metabolic labeling of viral and cellular proteins.
DBT cells were infected with MHV or mock infected. After starvation for 30 min in methionine-free medium (Sigma), cells were labeled with 50 μCi of [35S]methionine/ml for 30 min at various time points p.i., washed extensively with PBS, and lysed in the strong lysis buffer as described above for Western blot analysis. Proteins in the cell lysates were separated by SDS-10% PAGE, and the gel was developed by phosphorimaging (Molecular Dynamics).
β-Gal.
DBT cells were washed twice with PBS and harvested by adding 1 ml of PBS and scraping into a tube. The cells were centrifuged at 12,000 × g for 15 s, and the supernatant was removed. Then, 125 μl of 0.25 M Tris (pH 8.0) was added into the tube, and the cells were mixed by vortexing. The cells were lysed by freezing and thawing three times, and the lysates were clarified from debris by centrifugation at 12,000 × g for 5 min. For β-galactosidase (β-Gal) assay, 3 μl of a 100× Mg solution (0.1 M MgCl2, 4.5 M β-mercaptoethanol), 66 μl of 1× ONPG (o-nitrophenyl-β-d-galactopyranoside), 30 μl of cell extract, and 201 μl of 0.1 M sodium phosphate (pH 7.5) were mixed together. The reaction was incubated at 37°C for 30 min until a faint yellow color has developed. Then, 500 μl of 1 M Na2CO3 was added to stop the reaction. The optical density of the samples was read at a wavelength of 420 nm.
Plasmid construction.
The plasmid pUTR-CAT was made in three steps. In the first PCR, a 200-nucleotide (nt) fragment corresponding to the 5′ untranslated region (5′UTR) of MHV-JHM was amplified from p25CAT (29) by using the primer pair 5′L9 (5′-TGA TTG GCG TCC GTA CGT ACC-3′, corresponding to the sequence at nt 9 to 29) and 3′UTR-CAT (5′-TTT TCT CCA TTA TGC AAC CTA TG-3′, complementary to the sequence at nt 202 to 214 with a 12-nt sequence overhang at the 5′ end of the CAT gene). In the second PCR, a 560-nt fragment of the CAT gene was synthesized from 25CAT plasmid using the primer pair 5′UTR-CAT (5′-GTT GCA TAA TGG AGA AAA AAA TC-3′, corresponding to CAT gene sequence at nt 1 to 15 with an 8-nt overhang at nt 207 to 214 of DI genome sequence) and 3′CAT 542 (5′-TTA CGC CCC GCC CTG CCA-3′, complementary to the CAT sequence at nt 542 to 560). The PCR products from these two sets of reactions were separated by agarose gel electrophoresis; the corresponding DNA fragments were excised from the gels and purified with a gel extraction kit (QIAGEN). The two PCR fragments were then mixed and used as a template for a third PCR. The third PCR was carried out with the primer pair 5′L9 and 3′CAT542. The PCR products from the third PCR were digested with SnaBI and BspEI and directionally cloned into the SnaBI and BspEI sites of p25CAT, resulting in the generation of pUTR-CAT. In this plasmid, the CAT gene is directly fused with the 5′ UTR of MHV-JHM and also contains the 3′ UTR of MHV-JHM.
In vitro transcription.
Plasmid DNAs of p25CAT, pUTR-CAT, or pUTRdelta86 were digested with XbaI. The linearized DNA fragments were purified through gel purification. The T7 in vitro transcription reaction was set up according to the manufacturer's protocol (mMessage mMachine kit; Ambion). The reaction was assembled in the RNase-free microtube containing 1 μg of linearized template DNAs, 2 μl of enzyme mixture, 10 μl of 2× NTP/Cap, and 2 μl of 10× reaction buffer. The reaction was incubated at 37°C for 4 h.
DNA and RNA transfection.
For DNA plasmid transfection, the Lipofectamine reagent was used according to the manufacturer's protocol (Invitrogen). Briefly, DBT cells were grown in six-well plates to 60% confluence. A total of 500 ng of pSV-β-Gal plasmid (Promega) and 3 μl of the Lipofectamine reagent were diluted separately in 100 μl of serum-free OPTI medium (Gibco) without antibiotics and then mixed. The DNA-Lipofectamine mixture was incubated at room temperature for 30 min. DBT cells were washed with PBS, and 300 μl of OPTI medium was added to each well of the cell culture. The DNA-Lipofectamine mixture (200 μl) was then added to each well, and the cell culture plates were incubated at 37°C for various time periods as indicated. For viral genomic RNA and in vitro-transcribed DI-RNA transfection, DOTAP was used according to the manufacturer's instructions (Roche). Briefly, 5 μg of RNA was mixed with 30 μl of DOTAP in 70 μl of 20 mM HEPES (pH 7.4). The mixture was then added to the cells.
CAT assay.
The chloramphenicol acetyltransferase (CAT) assay was carried out as described previously (58). Briefly, cell lysates were prepared as described above for β-Gal assay, except that the lysates were incubated at 60°C for 10 min before centrifugation. The CAT assay reaction containing 5 μl of n-butyl coenzyme A (5 mM), 50 μl of cell lysate, 66 μl of 0.25 M Tris-HCl (pH 8.0), and 4 μl of [14C]chloramphenicol (0.05mCi/ml) was incubated at 37°C for 16 h and was terminated by the addition of 300 μl of mixed xylenes (Sigma Aldrich). The mixture was vortexed for 30 s and spun at 12,000 × g for 3 min. The upper phase (xylenes) was transferred to a new tube, and 100 μl of fresh 0.25 M Tris-HCl (pH 8.0) was added. Finally, 200 μl of upper xylene phase was transferred to a scintillation vial containing scintillation fluid. The radioactivity of the butyrylated chloramphenicol products was measured in a liquid scintillation counter, which represents the CAT activity.
RESULTS
MHV propagation was impaired by treatment with inhibitors of the Raf/MEK/ERK signaling pathway.
We previously showed that infections of DBT and 17Cl-1 cells with MHV activated the Raf/MEK/ERK signaling pathway, as evidenced by an increased phosphorylation level of ERK1/2 (6, and unpublished data). To assess the biological relevance of the ERK1/2 activation, we used specific inhibitors of the Raf/MEK/ERK signaling pathway. DBT cells were pretreated 1 h prior to infection with UO126 at a concentration of 50 μM or with DMSO as a negative control. Cells were then infected with MHV strain JHM at an MOI of 2. UO126 or DMSO was present in the medium throughout the entire period of infection. At various time points p.i., virus titers were determined by plaque assay. As shown in Fig. 1A, the progeny virus produced in UO126-treated DBT cells was ca. 95 to 99% less than that in the DMSO-treated control cells from 6 to 14 h p.i. The inhibitory effect of UO126 on virus propagation was readily detectable at 6 h p.i. but not at 4 h p.i. This result demonstrates that treating the cells with the MEK inhibitor UO126 suppressed MHV propagation in DBT cells, suggesting that the Raf/MEK/ERK signaling pathway may play a role in MHV propagation.
FIG. 1.

MHV propagation was impaired by treatment with inhibitors of the Raf/MEK/ERK signaling pathway. (A) DBT cells were pretreated with UO126 (50 μM) or DMSO for 1 h prior to infection and were infected with MHV-JHM at an MOI of 2. UO126 or DMSO was present in the medium throughout the infection. At the indicated time points p.i., the virus titer was determined by plaque assay. The data are the average of three independent experiments, and error bars denote the standard deviation. (B) The experiments were performed as in panel A except that the inhibitor treatment was omitted in one set of samples during the first hour of infection (UO126-II). UO126-I denotes the presence of UO126 throughout the infection. Virus titers were determined by plaque assay at 8 and 12 h p.i. (C and D) Effects of inhibitors UO126 and PD98059 on ERK1/2 phosphorylation (C) and virus production (D). DBT cells were infected with MHV-JHM at an MOI of 2 or mock infected. After 1 h of incubation, the inocula were removed, and cells were cultured in fresh medium containing UO126 or PD98059 at various concentrations as indicated or DMSO. At 8 h p.i., phosphorylated or total ERK1/2 was detected by Western blot analysis (C), and virus titers were determined by plaque assay (D). Values are expressed as means of three independent experiments. Error bars represent the standard deviations.
Because this experiment was carried out in the presence of UO126 before, during, and after virus infection, to exclude the possibility that the UO126 has a direct effect on virion infectivity, we removed the UO126 compound during virus infection for 1 h to avoid direct contact of the UO126 with the virus and added it back to the cells after 1 h of infection. The virus titers were determined at 8 and 12 h p.i. and were compared to those in the presence of UO126 throughout the infection. As shown in Fig. 1B, the virus titers decreased to a similar extent regardless of whether UO126 was present during the 1 h infection. This result thus confirms that the UO126 indeed has an inhibitory effect on MHV propagation. Furthermore, the result also indicates that the inhibitory effect of UO126 is likely exerted through cellular mechanisms rather than directly on virion infectivity.
To further provide a biological correlate, we determined the phosphorylation status of the downstream components ERK1/2 after treatment with two MEK inhibitors. We found that both inhibitors inhibited ERK1/2 phosphorylation at 8 h p.i. (Fig. 1C). However, the inhibitory effect of the two inhibitors was markedly different, with a significant inhibition observed for UO126 at 25 μM. In contrast, ERK1/2 phosphorylation was only slightly reduced by treatment with the PD98059 compound even at 100 μM. This finding suggests that the PD98059 might not be as effective as UO126 in inhibiting MHV propagation. To address this hypothesis, we determined the virus titers at 8 h p.i. after the treatment with UO126 and PD98059. Indeed, the inhibitory effect on virus titers was dose dependent for both inhibitors, and it correlated with the ability of the compounds to inhibit ERK1/2 phosphorylation (Fig. 1D). PD98059 was significantly less effective in suppressing virus growth (a 50% reduction in virus titer at a concentration of 50 μM) than UO126 (a 99% reduction in virus titer at the same concentration) (Fig. 1D). Such a correlation suggests that the Raf/MEK/ERK signaling pathway may play a role in MHV propagation. Since UO126 is a much more potent inhibitor than PD98059, we used UO126 in all subsequent experiments.
MHV propagation was suppressed by siRNAs specific to MEK1/2 and ERK1/2.
To provide direct evidence that the Raf/MEK/ERK signaling pathway is involved in regulation of MHV replication, DBT cells were transfected with small interfering RNA (siRNAs) specific to MEK1/2 and ERK1/2 or a nonspecific EGFP-siRNA as a negative control. At 48 h posttransfection, cells were infected with MHV at an MOI of 2. At 12 h p.i., the level of MEK1/2 and ERK1/2 proteins was determined by Western blot analysis, and virus titers were determined by plaque assay. As shown in Fig. 2A and B, the expression levels for MEK1/2 and ERK1/2 proteins were reduced by ca. 50% after siRNA treatment in both mock- and virus-infected cells, compared to those treated with the nonspecific EGFP-siRNA, indicating specific knockdown of MEK1/2 and ERK1/2 by siRNAs. Moreover, treatment with siRNAs specific to MEK1/2 and ERK1/2 decreased virus titers by ca. 1.5 log10 (Fig. 2C). These results indicate that knockdown of MEK1/2 or ERK1/2 has a detrimental effect on MHV propagation, thus demonstrating a role for the raf/MEK/ERK signaling pathway in MHV replication.
FIG. 2.

MHV propagation was suppressed by siRNAs specific to MEK1/2 and ERK1/2. (A) DBT cells were transfected with siRNAs to MEK1/2 or EGFP-siRNA as a negative control. At 48 h posttransfection, cells were infected with MHV at an MOI of 2. At 12 h p.i., MEK1/2 proteins were detected by Western blotting with monoclonal antibodies specific to MEK1/2. Beta-actin was used as an internal control. (B) The experiments were carried out exactly as in panel A, except that the ERK1/2 siRNAs were used for transfection and ERK1/2 antibodies were used for Western blot analysis. (C) Virus titers were determined at 12 h p.i. by plaque assay. The data are the averages of three experiments, and error bars denote the standard deviations.
The inhibitory effect of UO126 on MHV production was neither virus strain specific nor cell type specific.
To determine whether UO126 has a general inhibitory effect on coronavirus propagation, we used various MHV strains. These strains have diverse biological and pathogenic properties. For example, JHM is the most neurovirulent and causes acute encephalitis and demyelination, as well as persistent infection in the central nervous system, whereas MHV-3 is primarily hepatotropic, causing fulminant hepatitis in animals. DBT cells were infected with JHM, A59, MHV-1, MHV-2, MHV-3, or Penn-98-1 at an MOI of 2 and treated with UO126 (50 μM) or DMSO. Virus titers were measured by plaque assay at 8 h p.i. As shown in Fig. 3A, virus titers for all six strains were significantly inhibited by treatment with UO126 (the reduction in virus titers ranged from 2 to 3 log10).
FIG. 3.

The inhibitory effect of UO126 on MHV production was neither virus strain specific nor cell type specific. (A) DBT cells were infected with MHV-JHM, -A59, Penn98-1, MHV-1, MHV-2, or MHV-3 at an MOI of 2. After 1 h of incubation, the inocula were replaced with medium containing either UO126 (50 μM) or DMSO. Virus titers were determined by plaque assay at 8 h p.i. (B) Various types of cells, including DBT, 17 Cl-1, and NIH 3T3 cells, were infected with MHV-JHM at an MOI of 2. At 1 h p.i., UO126 (50 μM) or DMSO was added to the medium, and virus titers were determined at 8 h p.i. The data represent the means of three independent experiments. Error bars denote the standard deviations.
Next, we determined whether the inhibitory effect of UO126 was cell type specific. We chose three types of cells that are susceptible to MHV infection. These cells (DBT, 17Cl-1, and NIH 3T3) were infected with the JHM strain at an MOI of 2 and treated with 50 μM UO126 or DMSO at 1 h p.i. Virus titers were inhibited by UO126 in all three types of cells tested, although there was some variation among the cell types (Fig. 3B). Taken together, these results demonstrate that the inhibitory effect of UO126 on MHV production was neither virus strain specific nor cell type specific.
UO126 inhibited MHV propagation at early steps of the virus life cycle but did not block virus internalization.
To identify which step(s) of the virus life cycle was inhibited by UO126 treatment, we started with experiments to assess the earliest step, i.e., virus entry. We performed the internalization assay to address this question. DBT cells were incubated with MHV-JHM at an MOI of 2 at 4°C for 1 h to allow virus binding. Virus-bound cells were washed with cold PBS and were either maintained at 4°C or moved to 37°C for another hour to allow virus internalization in the presence of UO126 (50 μM) or DMSO. Cells were then treated with protease K to remove the viruses remaining on the cell surface. The cells were then serially diluted, and virus titers were determined by infectious center assay. As shown in Fig. 4A, the virus titer was virtually the same in cells treated with UO126 as those with DMSO at 37°C. The low titers seen in cells remained at 4°C suggested an inefficient removal of the bound virus by protease K. Nevertheless, these data indicate that UO126 has no significant effect on virus entry.
FIG. 4.
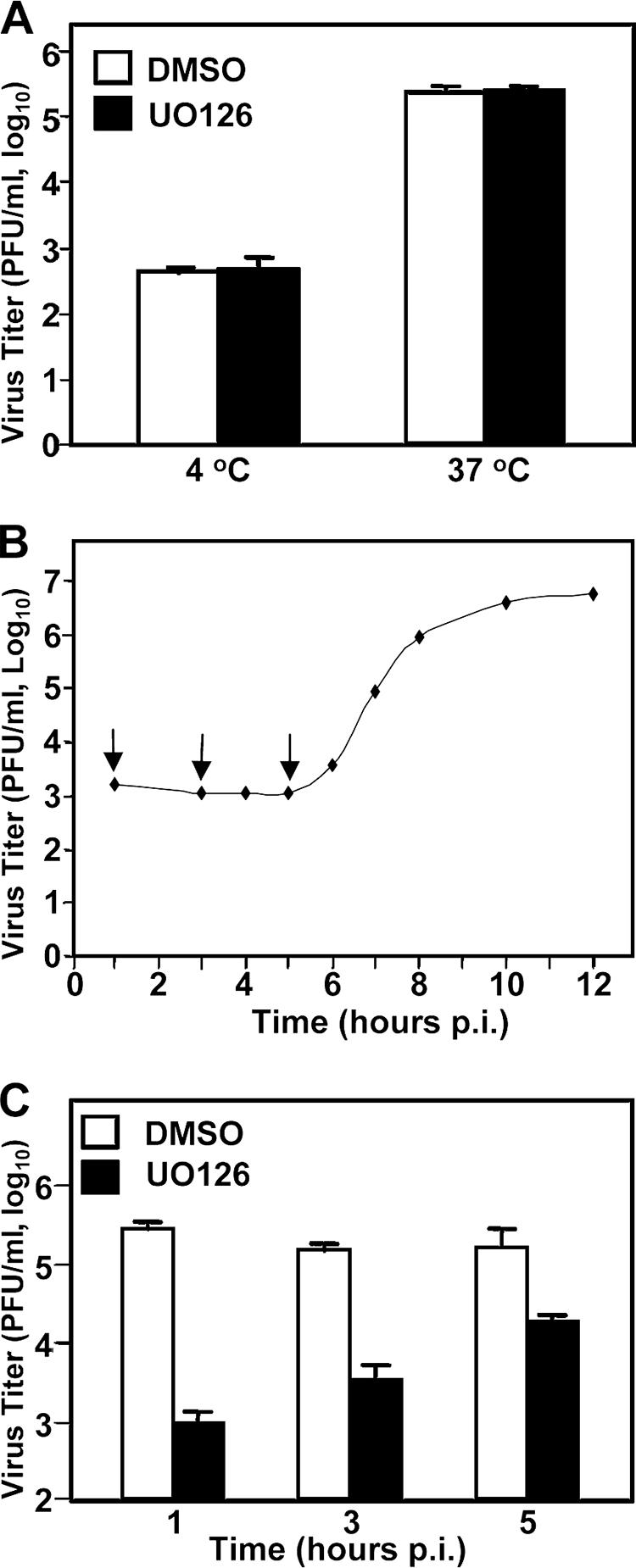
UO126 inhibited MHV propagation at early steps of the virus life cycle but did not block virus internalization. (A) Virus internalization assay. DBT cells were infected with MHV-JHM at an MOI of 2 at 4°C to allow virus binding. After 1 h of incubation, the infected cells were washed with cold PBS. In the presence of UO126 (50 μM) or DMSO, infected cells were either maintained at 4°C or moved to 37°C for an additional hour to allow the virus to internalize into cells. At the 1 h posttemperature shift, bound but uninternalized viruses were removed by treatment with protease K. The treated cells were then serially diluted and plated onto a DBT monolayer for 5 h. The medium was then removed, and agarose was overlaid on the monolayer as in the plaque assay. Values represent the means of three independent experiments. Error bars denote the standard deviation. (B) One-step growth curve. DBT cells were infected with MHV-JHM at an MOI of 2. At each time point p.i. as indicated, the virus titer was determined by plaque assay. Arrows indicate the time point for the addition of the drug as described in panel C. (C) DBT cells were infected with MHV-JHM at an MOI of 2. At various time points p.i. as indicated (1, 3, or 5 h p.i.), the cells were treated with UO126 (50 μM) or DMSO. Virus titers were determined at 8 h p.i. The values are the means of three independent experiments. The error bars denote the standard deviations.
Next, we investigated the postentry steps. Since we consistently used the virus at an MOI of 2 throughout the present study, we wanted to first establish a precise growth kinetic with this MOI to better understand the drug action. As shown in Fig. 4B, the one-step growth curve indicates that the eclipse period appeared to be 5 h p.i.; progeny viruses began to be detectable at 6 h p.i. Accordingly, we added UO126 to the culture at various time points (i.e., 1, 3, and 5 h) p.i. prior to the production of progeny virus (Fig. 4B) to examine the potential early steps that UO126 inhibits. When UO126 was added at 1 h p.i., the virus titer was decreased ∼2 log10 (Fig. 4C). The virus titer was decreased ∼1.5 log10 when UO126 was added at 3 h p.i. When UO126 was added at 5 h p.i., the reduction in virus titer was less than 1 log10 (Fig. 4C). These results indicated that the inhibitory effect of UO126 on MHV propagation was likely exerted largely on the early postentry steps of the virus life cycle, including protein translation and RNA synthesis, although these experiments could not completely exclude the possibility of the effect on late steps of the virus life cycle, such as assembly and budding.
UO126 did not inhibit cellular and viral protein translation.
For positive-strand RNA viruses, initiation of viral RNA replication requires de novo synthesis of the viral RdRp and its associated proteins. The polymerase polyprotein of coronavirus is translated from the viral genome via the same cellular cap-dependent mechanism. Thus, if the cellular or viral translation machinery is inhibited by UO126, it is conceivable that all subsequent steps in the virus life cycle would be affected. For this reason, we first determined whether UO126 has an inhibitory effect on cellular translation. DBT cells were treated with UO126 (50 μM) or DMSO alone for 11.5 h, and protein synthesis was monitored by labeling with [35S]methionine. At 12.5 h posttreatment, cells were lysed, and proteins were separated by SDS-PAGE. Overall, there was no obvious difference in the amount and species of the proteins between UO126-treated and DMSO-treated cells (Fig. 5A). We then used an alternative approach to quantitatively determine the potential effect of UO126 on cellular translation. DBT cells were transfected with a β-Gal reporter plasmid, whose gene expression is under the control of simian virus 40 early promoter and enhancer (Fig. 5B). Consistent with the results from radiolabeling, the β-Gal activity in UO126-treated cells was similar to that in DMSO-treated cells (Fig. 5B). Taken together, these results demonstrate that UO126 at the concentration of 50 μM had no inhibitory effect on cellular translation.
FIG. 5.
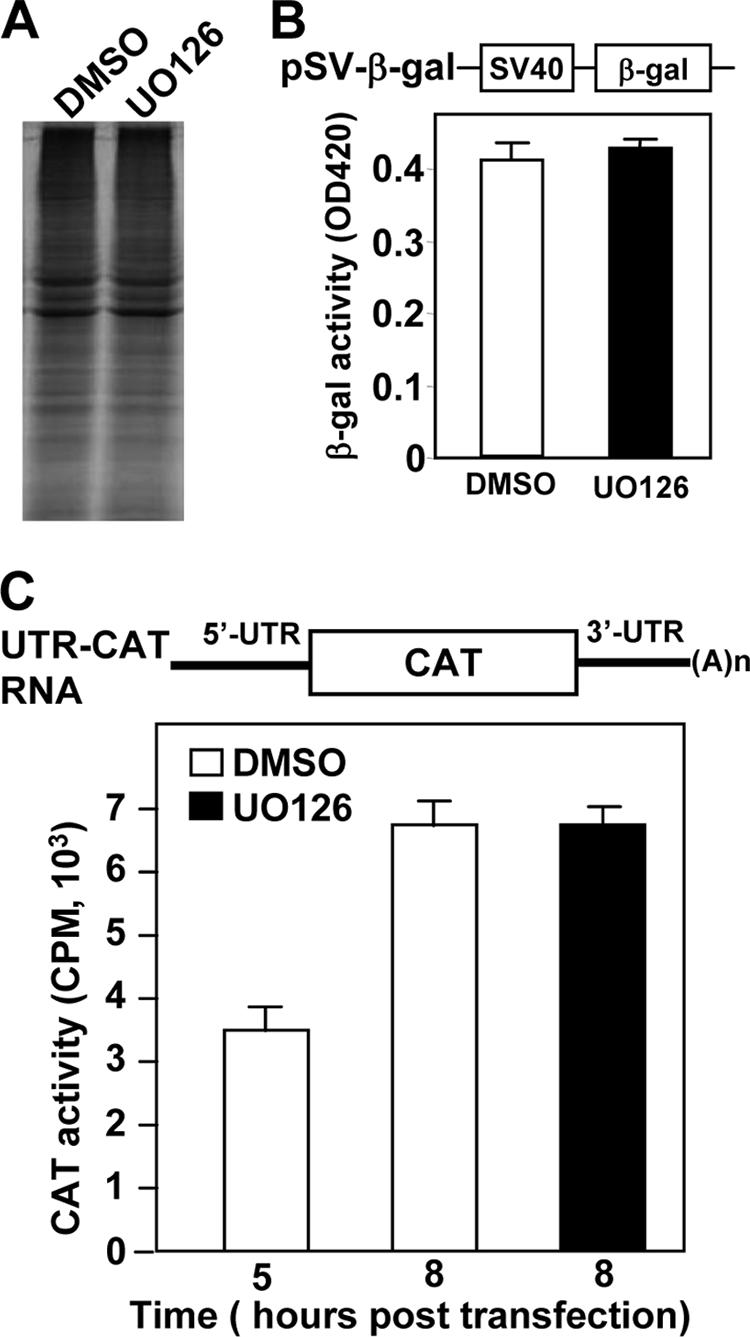
Translation of cellular and viral mRNAs was not inhibited by UO126. (A) Uninfected DBT cells were treated with UO126 (50 μM) or DMSO for 11 h and labeled with [35S]methionine (50 μCi/ml) for 30 min. One eighth of the cell lysates for each sample was analyzed by SDS-10% PAGE, and the gel was autoradiographed. (B) The top panel shows a simplified diagram of the structure of pSV-β-Gal. The transcription of β-Gal gene is under the control of simian virus 40 early promoter and enhancer. For the bottom panel, DBT cells were transfected with plasmid pSV-β-Gal and treated with either UO126 (50 μM) or DMSO at 5 h posttransfection. The β-Gal activity was measured at 24 h posttransfection. The data represent the means of six independent experiments. Error bars denote the standard deviation. (C) The top panel shows a diagram of the reporter mRNA (UTR-CAT RNA). 5′UTR and 3′UTR are derived from MHV-JHM sequences. For the bottom panel, DBT cells were transfected with in vitro-transcribed UTR-CAT RNA and treated with UO126 (50 μM) or DMSO. The CAT activity was measured at 5 or 8 h posttransfection. Values are means of six independent experiments. Error bars denote the standard deviations.
These results also imply that UO126 does not have an inhibitory effect on viral translation since the cell and the virus use the same translation machinery. However, there is a possibility that virus infection may alter the translation machinery in favor of viral translation (50), and thus viral translation may have a different sensitivity to UO126 treatment. To address this issue, we designed a minigenomic RNA, in which the viral coding genes were replaced with the CAT reporter gene, whereas the 5′UTR and 3′UTR are identical to the viral genome (Fig. 5C, top panel). Thus, the CAT activity expressed from the minigenome would most likely quantitatively reflect the efficiency of viral genome translation. Indeed, we found that the CAT activity was detectable at 5 h posttransfection of the in vitro-transcribed minigenomic RNAs and continued to increase. Significantly, the CAT activities in UO126-treated and DMSO-treated cells were virtually identical (Fig. 5C, bottom panel) at 8 h posttransfection. These results establish that UO126 did not have any inhibitory effect on cellular and viral protein translation at this concentration.
UO126 inhibited viral gene expression.
We then examined the effect of UO126 on viral RNA synthesis. DBT cells were infected with MHV-JHM at an MOI of 2. At 1 h p.i., cells were treated with UO126 (50 μM) or DMSO in the presence of actinomycin D (10 μg/ml). At 2 h p.i., newly synthesized viral RNAs were labeled with [3H]uridine (50 μCi/ml) and were isolated at different time points p.i. Labeled viral RNAs were then quantified after TCA precipitation. As shown in Fig. 6A, the amounts of radiolabeled, TCA-precipitable RNAs were approximately 10- to 100-fold lower in the cells treated with UO126 than in those treated with DMSO alone from 4 to 12 h p.i. The reduced incorporation of [3H]uridine indicated that viral RNA synthesis was inhibited by UO126.
FIG. 6.
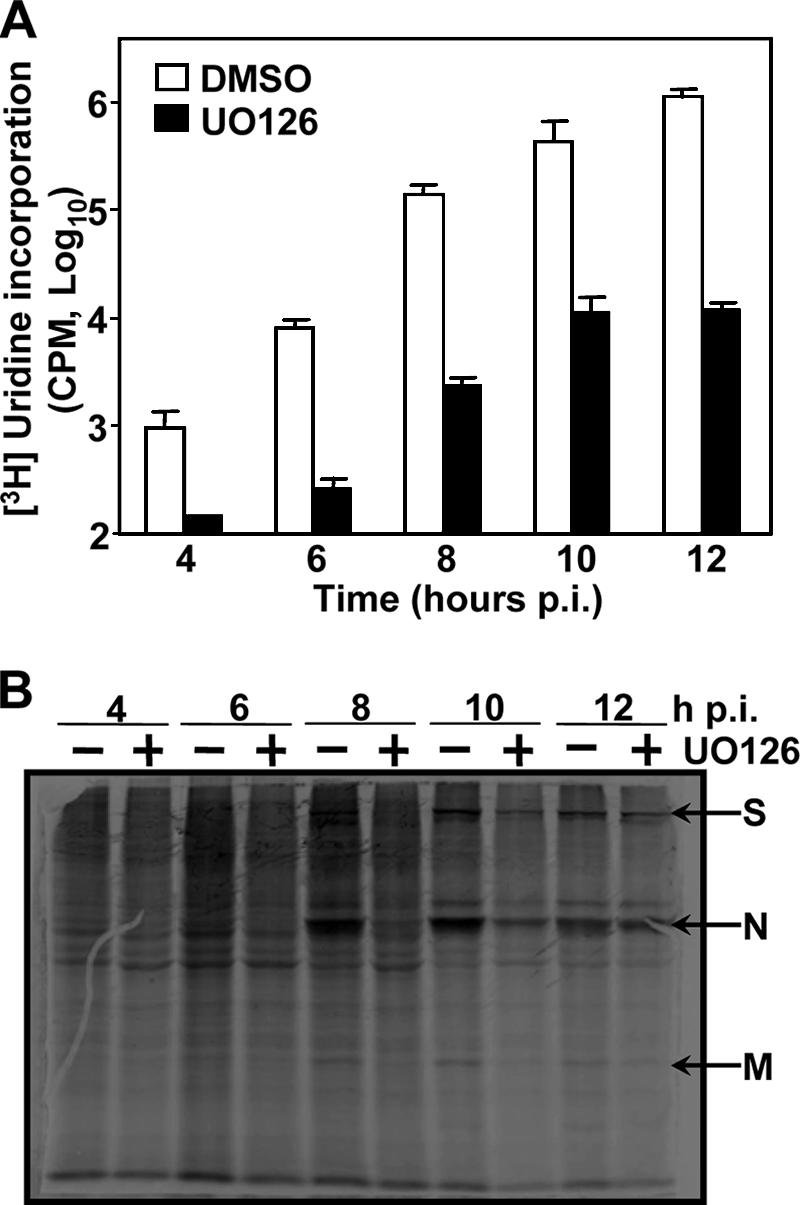
Inhibition of MHV gene expression by UO126. (A) Inhibition of viral RNA synthesis by UO126. DBT cells were infected with MHV-JHM at an MOI of 2 and treated with UO126 or DMSO in the presence of actinomycin D (10 μg/ml). At 2 h p.i., [3H]uridine was added to the medium at a concentration of 50 μCi/ml. The viral RNAs were isolated at various time points p.i. as indicated and were precipitated by TCA. The radioactivity levels of the TCA precipitates were determined by a scintillation counter and are expressed as counts per minute. The data indicate the means of three separate experiments. Error bars denote the standard deviations. (B) Inhibition of viral protein synthesis by UO126. DBT cells were infected with MHV-JHM at an MOI of 2, treated with UO126 [UO126 (+)] or DMSO [UO126(−)] at 1 h p.i., and labeled with [35S]methionine (50 μCi/ml) at various time points for 30 min. Cell extracts were prepared at the end of the 30 min labeling. One-eighth of each sample with equivalent cell numbers was analyzed by SDS-10% PAGE, and the gel was autoradiographed. Arrows indicate the position of three predominant MHV structural proteins (S, N, and M).
We next examined the effect of UO126 on viral protein synthesis. MHV-infected DBT cells were labeled with [35S]methionine for 30 min in the presence or absence of UO126. At various time points p.i., cells were lysed, and proteins were separated by PAGE. As shown in Fig. 6B, the amounts of viral structural proteins were clearly reduced in UO126-treated cells compared to those in DMSO-treated cells, demonstrating that UO126 also inhibited the synthesis of MHV proteins. However, this inhibition of viral structural proteins was likely a secondary effect from inhibition of viral mRNA synthesis (see next section).
UO126 inhibited viral genomic and subgenomic mRNA synthesis.
To further establish whether UO126 has a specific effect on viral RNA synthesis, we used the DI-CAT reporter system to monitor viral RNA synthesis. The transfection of a DI-CAT RNA into helper virus-infected cells results in the expression of CAT activity (29, 58). The CAT activity thus quantitatively reflects on the efficiency of subgenomic mRNA transcription and translation, as well as the efficiency of genomic RNA replication. DBT cells were transfected with in vitro-transcribed DI-RNA. At 5 h posttransfection, the cells were infected with MHV-JHM at an MOI of 2. UO126 (50 μM) or DMSO was added to the medium at 1 h p.i. The CAT activities were determined by CAT assay at 8 h p.i. As shown in Fig. 7, the CAT activity decreased ∼100-fold in the presence of UO126 compared to that in the presence of DMSO alone.
FIG. 7.
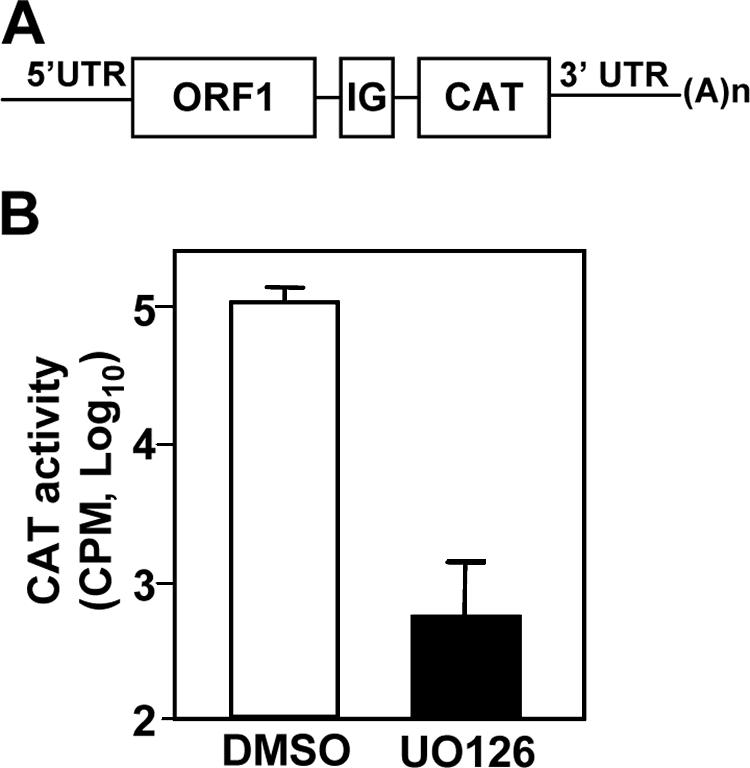
UO126 inhibited viral RNA synthesis. In the top panel is a schematic diagram depicting the bicistronic structure of the reporter 25CAT DI RNA (see Materials and Methods). ORF1, the first ORF of the DI RNA; IG, MHV intergenic sequence essential for CAT gene transcription; the second ORF is the CAT gene. For the lower panel, DBT cells were transfected with in vitro-transcribed 25CAT DI RNA. At 5 h posttransfection, cells were infected with MHV-JHM at an MOI of 2 and treated with UO126 (50 μM) or DMSO at 1 h p.i. CAT activities were determined at 8 h p.i. The data indicate the means of three independent experiments. Error bars represent the standard deviations.
Virus particles were used to establish infections in all of the experiments described above. If UO126 inhibits the uncoating step after entry but before polymerase translation, as did the proteasome inhibitor MG132 (55), then virus replication would be delayed or reduced in UO126-treated cells. To address this possibility, we isolated viral genomic RNAs from sucrose-gradient purified virus particles and then cotransfected them with the DI-CAT reporter RNA (Fig. 8A) into BHK cells. At 5 h posttransfection, the transfection mixture was removed, and fresh medium containing UO126 (50 μM) or DMSO was added. Cells were then harvested at 12 and 24 h posttransfection. Intracellular RNAs were extracted, and reverse transcription-PCR (RT-PCR) was performed to determine the level of DI genomic RNA replication. The results showed that the genomic RNA replication was significantly inhibited by UO126 treatment, while in the absence of UO126, DI genomic RNA continued to replicate from 12 to 24 h posttransfection (Fig. 8B). Similarly, the CAT activity was ≥10-fold lower in the UO126-treated cells than in the control DMSO-treated cells at 12 and 24 h posttransfection (Fig. 8C). These data further establish that UO126 specifically inhibited viral RNA synthesis, including genomic RNA replication.
FIG. 8.
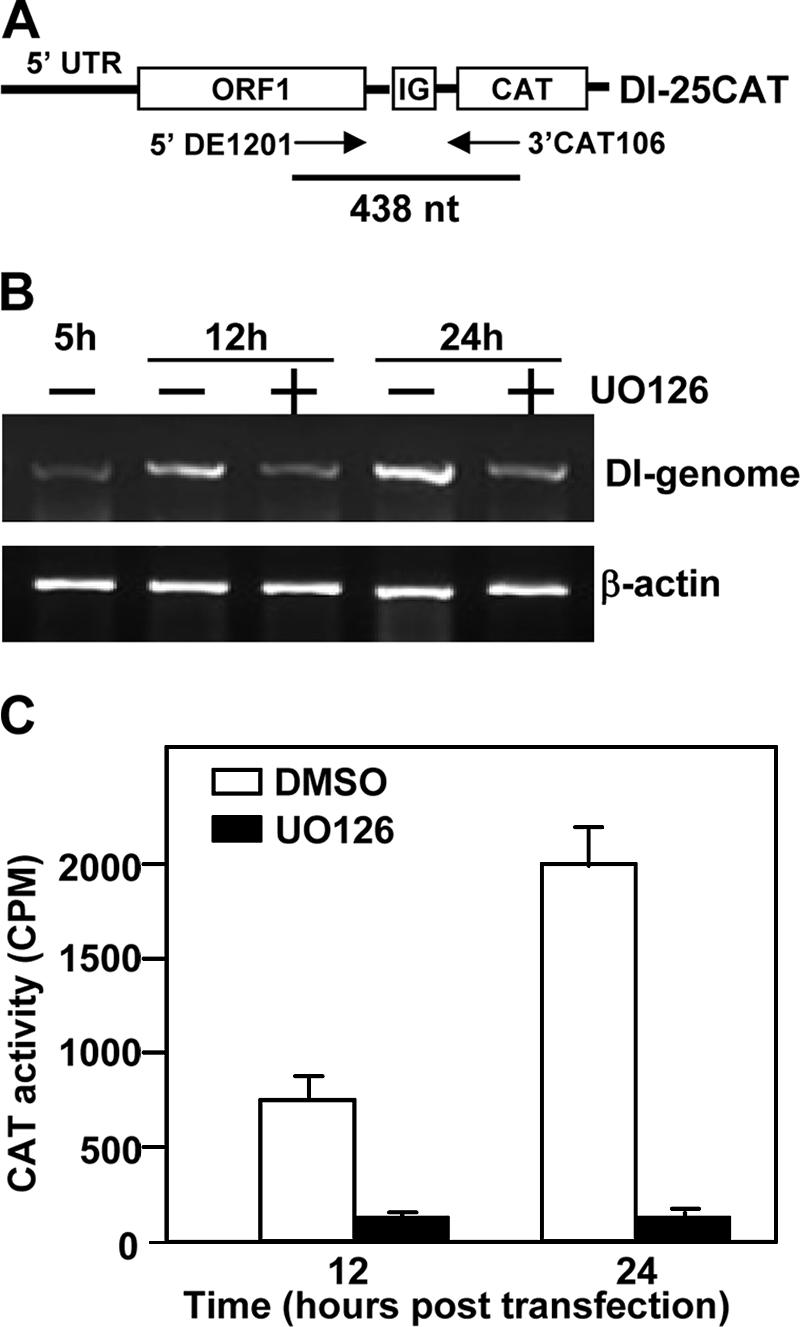
UO126 specifically inhibited viral genomic RNA replication. (A) Schematic diagram of the DI RNA-CAT reporter construct. Two primers used for RT-PCR are indicated with arrows, and the size of the expected RT-PCR product is also shown. (B) BHK cells were cotransfected with MHV-JHM viral genomic RNA and in vitro-transcribed 25CAT DI RNA. Cells were treated with UO126 (50 μM) or DMSO at 5 h posttransfection and then harvested at 12 and 24 h posttransfection. Intracellular RNAs were extracted and RT-PCR was performed to measure the genomic DI-RNA replication. β-Actin was used as an internal control. (C) CAT activity. The experiments were done as in panel B, except that the cell lysates were used for the CAT assay. Values are means of three independent experiments. Error bars represent the standard deviations.
Since the CAT activity measured in the experiment described above represented both the replication of viral genomic RNA and the synthesis of subgenomic mRNAs, we wanted to determine whether UO126 has an inhibitory effect on subgenomic mRNA synthesis independent of its effect on genomic RNA replication. Another DI construct UTR-86 was used in this experiment, which contains a deletion from nt 60 to nt 85 in the 5′UTR of DI-25CAT (Fig. 9A) and is defective in replication (56). The inability of UTR-86 to replicate was confirmed by RT-PCR in an experiment in which UTR86 RNA was transfected into helper MHV-infected DBT cells (Fig. 9B). To determine the CAT activity, BHK cells were cotransfected with MHV-JHM genomic RNA and in vitro-transcribed UTR-86 DI RNA. At 5 h posttransfection, the transfection mixture was removed, and fresh medium containing UO126 (50 μM) or DMSO was added. The CAT activity was determined at 12 and 24 h posttransfection. Because UTR-86 is replication defective (56; Fig. 9B), the CAT activity would solely reflect the efficiency of subgenomic mRNA synthesis. As shown in Fig. 9C, the CAT activity was sixfold lower in the UO126-treated cells than in the control DMSO-treated cells at 24 h posttransfection. These data further demonstrate that UO126 also specifically inhibited MHV subgenomic mRNA synthesis. We thus conclude that the MEK inhibitor UO126 specifically inhibited the viral genomic and subgenomic mRNA synthesis.
FIG. 9.
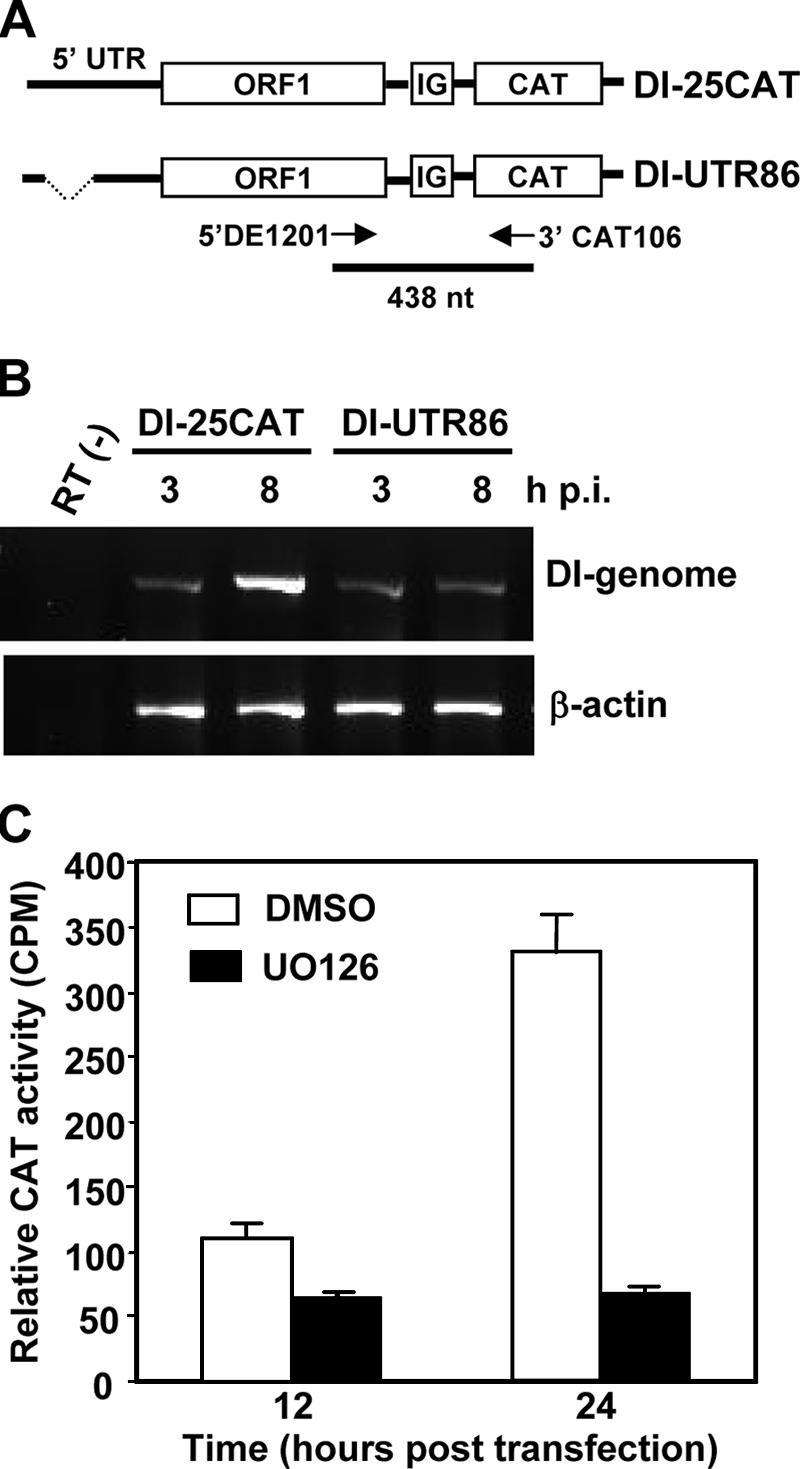
UO126 specifically inhibited the synthesis of viral subgenomic mRNAs. (A) Schematic diagram of DI-25CAT and DI-UTR86 reporter constructs. Two primers used for RT-PCR are indicated with arrows, and the size of the expected RT-PCR product is also shown. (B) DBT cells were infected with MHV-JHM at an MOI of 5. At 1 h p.i., cells were transfected with 25CAT or UTR-86 DI RNA. The RNA-transfection mixtures were removed from the culture at 3 h p.i. RNAs were isolated either at 3 or 8 h posttransfection. RT-PCR was performed to determine the DI-genomic RNA replication. (C) The CAT activity expressed from UTR86 DI RNA. BHK cells were cotransfected with viral genomic RNA and in vitro-transcribed UTR-86 DI RNA. Cells were treated with UO126 (50 μM) or DMSO at 5 h posttransfection. The CAT activity was measured at 12 and 24 h posttransfection. Values are the means of three independent experiments. Error bars represent the standard deviations.
DISCUSSION
We have shown previously that the Raf/MEK/ERK signaling pathway was activated upon MHV infection in cultured cells (6). In the present study, we show that two specific inhibitors of MEK1/2 (UO126 and PD98059) suppressed MHV propagation. Furthermore, the inhibitory effects of the MEK1/2 inhibitors on virus propagation positively correlated with their inhibitory effects on phosphorylation of the substrate ERK1/2 (Fig. 1C and D), suggesting a role for the Raf/MEK/ERK pathway in MHV propagation. In support of this conclusion is our finding that knockdown of MEK1/2 and ERK1/2 with specific siRNAs significantly suppressed MHV replication (Fig. 2). In addition, we recently showed that MHV infection induced transcription factor Egr-1 expression in DBT cells (6). Since Egr-1 is a downstream substrate of the Raf/MEK/ERK signaling pathway, its induction by MHV was blocked by treating the cells with the MEK inhibitor UO126. Importantly, knockdown of Egr-1 by siRNA inhibited MHV replication by more than 2 log10. These findings strongly support the notion that the Raf/MEK/ERK signaling pathway plays a beneficial role in MHV production, possibly through the activation of its downstream components such as Egr-1 (6).
The inhibitory effect of UO126 on virus propagation was examined in three different cell types and with six different MHV strains. The results demonstrated that UO126 has a general inhibitory effect on MHV propagation, independent of cell type and virus strain (Fig. 3). Moreover, UO126 at the concentration of 50 μM used throughout the present study did not affect cell viability (as determined by trypan blue staining) (data not shown) and cellular transcription and translation (Fig. 5), suggesting that the inhibition of MHV replication is not an indirect effect due to the toxicity of the drug to the host cell. It has been shown that activated ERK1/2 enhanced the infectivity of human immunodeficiency virus (HIV), whereas treatment of cells with the MEK1/2 inhibitor PD98059 significantly inhibited HIV infectivity (52, 53). Treating cells with UO126 also significantly inhibited the propagation of influenza A virus, Borna disease virus, Coxsackievirus B3, and HCMV (18, 32, 38, 39). Thus, it appears that the MEK1/2 inhibitors have a broad effect on propagations of viruses from various families (positive-strand and negative-strand RNA viruses, retroviruses, and DNA viruses). Alternatively, the Raf/MEK/ERK signaling pathway may be a common pathway that regulates not only the growth of host cells but also the growth of the infecting viruses. In this regard, our current findings add a new member to the growing list of viruses whose growth is modulated by the Raf/MEK/ERK signaling pathway. These findings also identify UO126 as a potential antiviral candidate for treatment of coronavirus-caused diseases.
The inhibitory effect of UO126 on the MHV life cycle was further investigated in the present study. Our data showed that treatment of cells with UO126 suppressed virus propagation at the step of viral RNA synthesis, but not viral entry and protein translation. This result suggests that the antiviral action of UO126 may be different from the proteasome inhibitor MG132 that has been shown to inhibit MHV uncoating from endosomes or lysosomes after virus internalization (55) or the inhibitors (E-64, CA-074, and Z-FY-DMK) of the endosomal cysteine proteases cathepsins B and L that inhibited severe acute respiratory syndrome coronavirus and MHV-2 infections by inhibiting the cleavage of the viral spike protein (34, 40, 46). Although our results cannot completely exclude the possibility that UO126 may also have an inhibitory effect on virus entry and uncoating, experiments with direct transfection of genomic viral and DI RNAs, which bypass the entry and uncoating steps prior to viral replication, establish that the inhibition involves postentry steps of the virus life cycle. The finding that the reporter DI RNA translation (Fig. 5C) was not affected by UO126 treatment further supports the notion that the translation of the polymerase polyprotein from the incoming viral genomic RNA is not inhibited by treatment with UO126. Thus, the reduction of radiolabeled viral RNAs in UO126-treated cells at early times postinfection must have resulted from inhibition of viral RNA synthesis. These results suggest that the antiviral action of UO126 on MHV is also different from those on other viruses. For example, UO126 inhibits influenza A virus propagation by interfering with the RNA import and export functions of NEP/NS2 proteins, resulting in blockage of the import of incoming ribonucleoproteins into the nucleus or nuclear retention of newly assembled ribonucleoproteins, and consequently in inhibition of virion formation. However, UO126 does not affect the RNA synthetic process of influenza A virus (39). For the HCMV, the phosphorylation of the viral immediate-early genes IE1-72 and IE2-86 was significantly inhibited by UO126 treatment, resulting in inhibition of the viral DNA synthesis and viral progeny production (18). However, a detailed mechanistic understanding of the inhibition of UO126 on MHV RNA synthesis requires the identification of particular cellular factors that are the components of the Raf/MEK/ERK signaling pathway or the targets of UO126. In this pursuit, the transcription factor Egr-1 may provide an opportunity as a candidate cellular factor to address this important issue in the future.
Acknowledgments
We thank Kelli Halcom (Zhang laboratory) for proofreading the manuscript.
This study was supported in part by grants from the National Institutes of Health (AI47188, AI61204, and NS47499).
Footnotes
Published ahead of print on 1 November 2006.
REFERENCES
- 1.Alessi, D. R., A. Cuenda, P. Cohen, D. T. Dudley, and A. R. Saltiel. 1995. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 270:27489-27494. [DOI] [PubMed] [Google Scholar]
- 2.Bonilla, P. J., S. A. Hughes, J. D. Pinon, and S. R. Weiss. 1995. Characterization of the leader papain-like proteinase of MHV-A59: identification of a new in vitro cleavage site. Virology 209:489-497. [DOI] [PubMed] [Google Scholar]
- 3.Bonilla, P. J., S. A. Hughes, and S. R. Weiss. 1997. Characterization of a second cleavage site and demonstration of activity in trans by the papain-like proteinase of the murine coronavirus mouse hepatitis virus strain A59. J. Virol. 71:900-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bos, E. C., W. Luytjes, H. V. Van der Meulen, H. K. Koerten, and W. J. Spaan. 1996. The production of recombinant infectious DI-particles of a murine coronavirus in the absence of helper virus. Virology 218:52-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bost, A. G., R. H. Carnahan, X. T. Lu, and M. R. Denison. 2000. Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly. J. Virol. 74:3379-3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai, Y., Y. Liu, and X. Zhang. 2006. Induction of transcription factor Egr-1 gene expression in astrocytoma cells by murine coronavirus infection. Virology 355:152-163. [DOI] [PMC free article] [PubMed]
- 7.Choi, K. S., A. Mizutani, and M. M. Lai. 2004. SYNCRIP, a member of the heterogeneous nuclear ribonucleoprotein family, is involved in mouse hepatitis virus RNA synthesis. J. Virol. 78:13153-13162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crews, C. M., and R. L. Erikson. 1992. Purification of a murine protein-tyrosine/threonine kinase that phosphorylates and activates the Erk-1 gene product: relationship to the fission yeast byr1 gene product. Proc. Natl. Acad. Sci. USA 89:8205-8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Compton, S. R., D. B. Rogers, K. V. Holmes, D. Fertsch, J. Remenick, and J. J. McGowan. 1987. In vitro replication of mouse hepatitis virus strain A59. J. Virol. 61:1814-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis, R. J. 1995. Transcriptional regulation by MAP kinases. Mol. Reprod. Dev. 42:459-467. [DOI] [PubMed] [Google Scholar]
- 11.Davies, S. P., H. Reddy, M. Caivano, and P. Cohen. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351:95-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denison, M. R., S. A. Hughes, and S. R. Weiss. 1995. Identification and characterization of a 65-kDa protein processed from the gene 1 polyprotein of the murine coronavirus MHV-A59. Virology 207:316-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denison, M. R., W. J. Spaan, Y. van der Meer, C. A. Gibson, A. C. Sims, E. Prentice, and X. T. Lu. 1999. The putative helicase of the coronavirus mouse hepatitis virus is processed from the replicase gene polyprotein and localizes in complexes that are active in viral RNA synthesis. J. Virol. 73:6862-6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dudley, D. T., L. Pang, S. J. Decker, A. J. Bridges, and A. R. Saltiel. 1995. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 92:7686-7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Favata, M. F., K. Y. Horiuchi, E. J. Manos, A. J. Daulerio, D. A. Stradley, W. S. Feeser, D. E. Van Dyk, W. J. Pitts, R. A. Earl, F. Hobbs, R. A. Copeland, R. L. Magolda, P. A. Scherle, and J. M. Trzaskos. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273:18623-18632. [DOI] [PubMed] [Google Scholar]
- 16.Furuya, T., and M. M. Lai. 1993. Three different cellular proteins bind to complementary sites on the 5′-end-positive and 3′-end-negative strands of mouse hepatitis virus RNA. J. Virol. 67:7215-7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallagher, T. M., C. Escarmis, and M. J. Buchmeier. 1991. Alteration of the pH dependence of coronavirus-induced cell fusion: effect of mutations in the spike glycoprotein. J. Virol. 65:1916-1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson, R. A., X. L. Ma, A. D. Yurochko, and E. S. Huang. 2001. The role of MKK1/2 kinase activity in human cytomegalovirus infection. J. Gen. Virol. 82:493-497. [DOI] [PubMed] [Google Scholar]
- 19.Kalicharran, K., and S. Dales. 1995. Dephosphorylation of the nucleocapsid protein of inoculum JHMV may be essential for initiating replication. Adv. Exp. Med. Biol. 380:485-489. [DOI] [PubMed] [Google Scholar]
- 20.Kalicharran, K., D. Mohandas, G. Wilson, and S. Dales. 1996. Regulation of the initiation of coronavirus JHM infection in primary oligodendrocytes and L-2 fibroblasts. Virology 225:33-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanjanahaluethai, A., and S. C. Baker. 2000. Identification of mouse hepatitis virus papain-like proteinase 2 activity. J. Virol. 74:7911-7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kong, X., H. San Juan, A. Behera, M. E. Peeples, J. Wu, R. F. Lockey, and S. S. Mohapatra. 2004. ERK-1/2 activity is required for efficient RSV infection. FEBS Lett. 559:33-38. [DOI] [PubMed] [Google Scholar]
- 23.Hirano, N., K. Fujiwara, S. Hino, and M. Matumoto. 1974. Replication and plaque formation of mouse hepatitis virus (MHV-2) in mouse cell line DBT culture. Arch. Gesamte Virusforsch. 44:298-302. [DOI] [PubMed] [Google Scholar]
- 24.Lai, M. M. 1990. Coronaviruses: organization, replication and expression of genome. Annu. Rev. Microbiol. 44:303-333. [DOI] [PubMed] [Google Scholar]
- 25.Lange-Carter, C. A., C. M. Pleiman, A. M. Gardner, K. J. Blumer, and G. L. Johnson. 1993. A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science 260:315-319. [DOI] [PubMed] [Google Scholar]
- 26.Leibowitz, J. L., K. C. Wilhelmsen, and C. W. Bond. 1981. The virus-specific intracellular RNA species of two murine coronaviruses: MHV-a59 and MHV-JHM. Virology 114:39-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, H. P., X. Zhang, R. Duncan, L. Comai, and M. M. Lai. 1997. Heterogeneous nuclear ribonucleoprotein A1 binds to the transcription-regulatory region of mouse hepatitis virus RNA. Proc. Natl. Acad. Sci. USA 94:9544-9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, H. P., P. Huang, S. Park, and M. M. Lai. 1999. Polypyrimidine tract-binding protein binds to the leader RNA of mouse hepatitis virus and serves as a regulator of viral transcription. J. Virol. 73:772-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao, C. L., and M. M. Lai. 1994. Requirement of the 5′-end genomic sequence as an upstream cis-acting element for coronavirus subgenomic mRNA transcription. J. Virol. 68:4727-4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu, X., Y. Lu, and M. R. Denison. 1996. Intracellular and in vitro-translated 27-kDa proteins contain the 3C-like proteinase activity of the coronavirus MHV-A59. Virology 222:375-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu, X. T., A. C. Sims, and M. R. Denison. 1998. Mouse hepatitis virus 3C-like protease cleaves a 22-kilodalton protein from the open reading frame 1a polyprotein in virus-infected cells and in vitro. J. Virol. 72:2265-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo, H., B. Yanagawa, J. Zhang, Z. Luo, M. Zhang, M. Esfandiarei, C. Carthy, J. E. Wilson, D. Yang, and B. M. McManus. 2002. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J. Virol. 76:3365-3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makino, S., and M. M. Lai. 1989. Evolution of the 5′-end of genomic RNA of murine coronaviruses during passages in vitro. Virology 169:227-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuyama, S., M. Ujike, S. Morikawa, M. Tashiro, and F. Taguchi. 2005. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA 102:12543-12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nanda, S. K., and J. L. Leibowitz. 2001. Mitochondrial aconitase binds to the 3′ untranslated region of the mouse hepatitis virus genome. J. Virol. 75:3352-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nash, T. C., and M. J. Buchmeier. 1997. Entry of mouse hepatitis virus into cells by endosomal and nonendosomal pathways. Virology 233:1-8. [DOI] [PubMed] [Google Scholar]
- 37.Pinon, J. D., H. Teng, and S. R. Weiss. 1999. Further requirements for cleavage by the murine coronavirus 3C-like proteinase: identification of a cleavage site within ORF1b. Virology 263:471-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Planz, O., S. Pleschka, and S. Ludwig. 2001. MEK-specific inhibitor U0126 blocks spread of Borna disease virus in cultured cells. J. Virol. 75:4871-4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pleschka, S., T. Wolff, C. Ehrhardt, G. Hobom, O. Planz, U. R. Rapp, and S. Ludwig. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 3:301-305. [DOI] [PubMed] [Google Scholar]
- 40.Qiu, Z., S. T. Hingley, G. Simmons, C. Yu, J. Das Sarma, P. Bates, and S. R. Weiss. 2006. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 80:5768-5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson, M. J., and M. H. Cobb. 1997. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 9:180-186. [DOI] [PubMed] [Google Scholar]
- 42.Schiller, J. J., A. Kanjanahaluethai, and S. C. Baker. 1998. Processing of the coronavirus MHV-JHM polymerase polyprotein: identification of precursors and proteolytic products spanning 400 kilodaltons of ORF1a. Virology 242:288-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sebolt-Leopold, J. S., D. T. Dudley, R. Herrera, K. Van Becelaere, A. Wiland, R. C. Gowan, H. Tecle, S. D. Barrett, A. Bridges, S. Przybranowski, W. R. Leopold, and A. R. Saltiel. 1999. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 5:810-816. [DOI] [PubMed] [Google Scholar]
- 44.Shi, S. T., J. J. Schiller, A. Kanjanahaluethai, S. C. Baker, J. W. Oh, and M. M. Lai. 1999. Colocalization and membrane association of murine hepatitis virus gene 1 products and de novo-synthesized viral RNA in infected cells. J. Virol. 73:5957-5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shieh, C.-K, H.-J. Lee, K. Yokomori, N. La Monica, S. Makino, and M. M. C. Lai. 1989. Identification of a new transcriptional initiation site and the corresponding functional gene 2b in the murine coronavirus RNA genome. J. Virol. 63:3729-3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simmons, G., D. N. Gosalia, A. J. Rennekamp, J. D. Reeves, S. L. Diamond, and P. Bates. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 102:11876-11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snijder, E. J., P. J. Bredenbeek, J. C. Dobbe, V. Thiel, J. Ziebuhr, L. L. M. Poon, Y. Guan, M. Rozanov, W. J. M. Spaan, and A. E. Gorbalenya. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331:991-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spagnolo, J. F., and B. G. Hogue. 2000. Host protein interactions with the 3′ end of bovine coronavirus RNA and the requirement of the poly (A) tail for coronavirus defective genome replication. J. Virol. 74:5053-5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stohlman, S. A., and M. M. Lai. 1979. Phosphoproteins of murine hepatitis viruses. J. Virol. 32:672-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tahara, S. M., T. A. Dietlin, G. W. Nelson, S. A. Stohlman, and D. J. Manno. 1998. Mouse hepatitis virus nucleocapsid protein as a translational effector of viral mRNAs. Adv. Exp. Med. Biol. 440:313-318. [DOI] [PubMed] [Google Scholar]
- 51.Vennema, H., G. J. Godeke, J. W. Rossen, W. F. Voorhout, M. C. Horzinek, D. J. Opstelten, and P. J. Rottier. 1996. Nucleocapsid-independent assembly of coronavirus-like particles by coexpression of viral envelope protein genes. EMBO J. 15:2020-2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang, X., and D. Gabuzda. 1998. Mitogen-activated protein kinase phosphorylates and regulates the HIV-1 Vif protein. J. Biol. Chem. 273:29879-29887. [DOI] [PubMed] [Google Scholar]
- 53.Yang, X., and D. Gabuzda. 1999. Regulation of human immunodeficiency virus type 1 infectivity by the ERK mitogen-activated protein kinase signaling pathway. J. Virol. 73:3460-3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yokomori, K., N. La Monica, S. Makino, C. K. Shieh, and M. M. C. Lai. 1989. Biosynthesis, structure, and biological activities of envelope protein gp65 of murine coronavirus. Virology 173:683-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu, G. Y., and M. M. Lai. 2005. The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J. Virol. 79:644-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang, Y., and X. Zhang. 2000. The leader RNA of coronavirus mouse hepatitis virus contains an enhancer-like element for subgenomic mRNA transcription. J. Virol. 74:10571-10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang, X., and M. M. Lai. 1995. Interactions between the cytoplasmic proteins and the intergenic (promoter) sequence of mouse hepatitis virus RNA: correlation with the amounts of subgenomic mRNA transcribed. J. Virol. 69:1637-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang, X., C. L. Liao, and M. M. Lai. 1994. Coronavirus leader RNA regulates and initiates subgenomic mRNA transcription both in trans and in cis. J. Virol. 68:4738-4746. [DOI] [PMC free article] [PubMed] [Google Scholar]