

Abstract
In Saccharomyces cerevisiae, the highly conserved Rho-type GTPase Cdc42 is essential for cell division and controls cellular development during mating and invasive growth. The role of Cdc42 in mating has been controversial, but a number of previous studies suggest that the GTPase controls the mitogen-activated protein (MAP) kinase cascade by activating the p21-activated protein kinase (PAK) Ste20. To further explore the role of Cdc42 in pheromone-stimulated signaling, we isolated novel alleles of CDC42 that confer resistance to pheromone. We find that in CDC42(V36A) and CDC42(V36A, I182T) mutant strains, the inability to undergo pheromone-induced cell cycle arrest correlates with reduced phosphorylation of the mating MAP kinases Fus3 and Kss1 and with a decrease in mating efficiency. Furthermore, Cdc42(V36A) and Cdc42(V36A, I182T) proteins show reduced interaction with the PAK Cla4 but not with Ste20. We also show that deletion of CLA4 in a CDC42(V36A, I182T) mutant strain suppresses pheromone resistance and that overexpression of CLA4 interferes with pheromone-induced cell cycle arrest and MAP kinase phosphorylation in CDC42 wild-type strains. Our data indicate that Cla4 has the potential to act as a negative regulator of the mating pathway and that this function of the PAK might be under control of Cdc42. In conclusion, our study suggests that control of pheromone signaling by Cdc42 not only depends on Ste20 but also involves interaction of the GTPase with Cla4.
The Rho-type GTPase Cdc42 is a member of the Ras superfamily of small GTP-binding proteins that play an essential role in regulating proliferation and differentiation in all eukaryotes. In the budding yeast Saccharomyces cerevisiae, Cdc42 is required for generation and maintenance of cell polarity and controls cell division, invasive and filamentous growth development, and mating of haploid cells (13, 24, 36). The exact role of Cdc42 in pheromone-induced mating, however, has been controversial. A major question concerning the function of Cdc42 in the mating process is the role of this small GTPase in the signal transduction pathway that confers cell cycle arrest, the formation of mating projections, expression of mating-specific genes, and fusion of partner cells of opposite mating types in response to peptide pheromone signals. In the mating pathway (3), signaling is initiated by binding of pheromones to the G-protein-coupled receptors at the membrane, causing the release of the Gβγ subunits from the heterotrimeric G protein and activation of a mitogen-activated protein kinase (MAP kinase) cascade. At least two proteins have been found to mediate activation of the MAP kinase cascade after binding to the Gβγ subunits, the PAK (p21-activated protein kinase) Ste20 and the scaffold protein Ste5 (15, 30, 46). Activation of the MAP kinase cascade consisting of Ste11, Ste7, Kss1, and Fus3 in turn promotes cell cycle arrest in G1 and stimulates expression of mating-specific genes, e.g., FUS1 (3).
Because PAKs are well-documented effector proteins of the Cdc42/Rac protein family (1, 12), a number of studies have addressed the function of Cdc42 and its guanine nucleotide exchange factor (GEF) Cdc24 in pheromone-stimulated signaling in S. cerevisiae. Initial studies suggested a direct role of Cdc42 by showing that temperature-sensitive cdc42 and cdc24 mutant strains are defective for pheromone-induced expression of FUS1 and arrest of cells in G1 at the restrictive temperature, and that the GTP-bound form of Cdc42 is able to stimulate Ste20 kinase activity in vitro (42, 47). Unfortunately, cells of cdc42 and cdc24 temperature-sensitive mutant strains shifted to the restrictive temperature become unresponsive to mating pheromone due to accumulation of high levels of Cln1/2-Cdc28 kinase, indicating that Cdc42 might not directly control activity of the mating signaling pathway (34, 35). In addition, further studies demonstrated that in vivo the interaction of Cdc42 and Ste20 is not required for pheromone-stimulated G1 arrest, FUS1 transcription, and mating, suggesting that functions of Cdc42 during mating might be restricted to actin cytoskeleton rearrangements during shmoo formation and control of cell fusion by localizing Ste20 to the shmoo tips (14, 29, 37).
More recent data suggest that Cdc42 might yet have a more direct role in pheromone-stimulated signal transduction, because CDC42 alleles could be isolated that confer polarization functions required for viability but cause resistance to pheromone treatment, reduction of FUS1 transcription, and mislocalization of Ste20 (32). Moreover, Ste20 kinase activity was demonstrated to be stimulated by GTP-bound Cdc42 in vivo (28). These data led to the current view that Cdc42 is able to control activity of the mating response pathway by binding to Ste20 and antagonizing the autoinhibitory interaction between the domains of Ste20 conferring interaction with Cdc42 (CRIB domain) and kinase activity. However, although the Cdc42-Ste20 interaction is critical for pheromone-stimulated signaling, it seems to operate independently of receptor activation and does not appear to require regulatory input from pheromone or the Gβγ subunits.
To further explore the role of Cdc42 in pheromone-stimulated signal transduction, we have isolated novel alleles of CDC42 that confer resistance to mating pheromone without affecting functions of the GTPase required for cell division. In contrast to previous studies, we have measured the functions of these variants of Cdc42 during mating when expressed at endogenous levels and in the complete absence of the wild-type protein. Our study indicates that control of pheromone signaling by Cdc42 not only involves Ste20 but might also depend on proper interaction of the GTPase with its well-known effector Cla4, a PAK that we here identified as a negative regulator of pheromone-induced G1 arrest and MAP kinase phosphorylation.
MATERIALS AND METHODS
Yeast strains and media.
Yeast strains used in this study are described in Table 1 and are congenic to the Σ1278b background with the exception of EGY48-p1840. CDC42 control and mutant strains were obtained by integration of desired CDC42 alleles at the LEU2 locus of strain YHUM1168 (cdc42Δ) and subsequent elimination of plasmid pME1534 carrying CDC42. Only strains with single integrations of CDC42 mutant alleles were selected after Southern blot analysis (40). Strains carrying bar1Δ::kanR, ste20Δ::HIS3, ste7Δ::loxP, cla4Δ::natR, and skm1Δ::natR deletion alleles were obtained by transformation using respective deletion cassettes, verification by Southern blot analysis, and genetic crosses following standard yeast genetic methods. Yeast culture medium was prepared as described previously (18). Invasive growth tests were described earlier (6).
TABLE 1.
Strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| YHUM70 | MATα his1 | H.-U. Mösch, laboratory collection |
| YHUM1168 | MATa bar1Δ::kanR ura3-52 leu2::hisG his3::hisG cdc42Δ::HIS3 [pME1534] | This study |
| YHUM1064 | MATabar1Δ::kanR ura3-52 his3::hisG cdc42Δ::HIS3 CDC42::LEU2 | This study |
| YHUM1060 | As YHUM1064 with CDC42(T58M)::LEU2 | This study |
| YHUM1061 | As YHUM1064 with CDC42(V36A, I182T)::LEU2 | This study |
| YHUM1062 | As YHUM1064 with CDC42(V36A)::LEU2 | This study |
| YHUM1063 | As YHUM1064 with CDC42(E62D, S71L, K128R, T161R)::LEU2 | This study |
| YHUM1066 | As YHUM1064 with CDC42(I46M)::LEU2 | This study |
| YHUM1067 | As YHUM1064 with CDC42(S71P)::LEU2 | This study |
| YHUM1068 | As YHUM1064 with CDC42(E100G)::LEU2 | This study |
| YHUM1069 | As YHUM1064 with CDC42(S158T)::LEU2 | This study |
| YHUM1071 | MATa bar1Δ::kanR ura3-52 leu2::hisG trp1::hisG his3::hisG kss1Δ::hisG | This study |
| YHUM1072 | MATa bar1Δ::kanR ura3-52 leu2::hisG trp1::hisG his3::hisG fus3Δ::TRP1 | This study |
| YHUM1073 | MATa bar1Δ::kanR ura3-52 leu2::hisG trp1::hisG his3::hisG fus3Δ::TRP1 kss1Δ::hisG | This study |
| YHUM1074 | MATa bar1Δ::kanR ura3-52 leu2::hisG, his3::hisG ste7Δ::loxP | This study |
| YHUM1006 | MATa bar1Δ::kanR ura3-52 leu2::hisG trp1::hisG his3::hisG ste20Δ::HIS3 | This study |
| YHUM1165 | As YHUM1064 with cla4Δ::natR | This study |
| YHUM1296 | As YHUM1064 with skm1Δ::natR | This study |
| YHUM1299 | As YHUM1064 with CDC42(V36A, I182T)::LEU2 cla4Δ::natR | This study |
| YHUM1327 | As YHUM1064 with CDC42(V36A, I182T)::LEU2 skm1Δ::natR | This study |
| YHUM1294 | As YHUM1064 with CDC42(E62D, S71L, K128R, T161R)::LEU2 cla4Δ::natR | This study |
| YHUM1302 | As YHUM1064 with CDC42(E62D, S71L, K128R, T161R)::LEU2 skm1Δ::natR | This study |
| YHUM1297 | As YHUM1074 with cla4Δ::natR | This study |
| YHUM1337 | As YHUM1074 with skm1Δ::natR | This study |
| EGY48-p1840 | MATα ura3 his3 trp1 leu2 lexAop-lacZ-URA3 | 19 |
Pheromone sensitivity and mating assays.
For tests of α-factor sensitivity, strains were grown in yeast-peptone-dextrose (YPD) to logarithmic phase and equal amounts of cells were spotted on YPD solid medium containing α-factor at 0.025 μM (bar1Δ strains) after serial dilutions of cultures. Growth was analyzed after incubation at 30°C for 2 to 3 days. Alternatively, strains from precultures were streaked on solid YPD media containing α-factor and incubated at 30°C for 3 days. Pheromone-induced cell cycle arrest was assayed by growing strains into logarithmic phase and determination of percentage of budded cells (n = 300 cells) before and after addition of α-factor at the desired concentration to the cultures. Quantitative mating assays were performed as previously described (20).
Plasmids.
Plasmids used in this study are listed in Table 2. Integrative plasmids BHUM316, BHUM322, BHUM328, BHUM330, and BHUM1081 to BHUM1085 were obtained by cloning the different CDC42 alleles isolated from the CDC42 library (see below) as 1.7-kb BamHI-HindIII fragments from pRS315 to pRS305. CDC42 alleles tested in two-hybrid assays were subjected to site-directed mutagenesis in order to introduce the C188S mutation, avoiding membrane localization of the proteins. CDC42 alleles were amplified by PCR using appropriate mutagenic primers and subjected to sequence analysis before introduction as 576-bp EcoRI-BamHI fragments into vector pEG202 (19) to obtain plasmids BHUM1044 to BHUM1047.
TABLE 2.
Plasmids used in this study
| Plasmid | Genotype | Reference or source |
|---|---|---|
| pME1534 | CDC42 in pTF27 | 31 |
| pRS305 | LEU2-marked integrative vector | 41 |
| BHUM1085 | CDC42 in pRS305 | This study |
| BHUM316 | CDC42(I46M) in pRS305 | This study |
| BHUM322 | CDC42(S71P) in pRS305 | This study |
| BHUM328 | CDC42(E100G) in pRS305 | This study |
| BHUM330 | CDC42(S158T) in pRS305 | This study |
| BHUM1081 | CDC42(T58M) in pRS305 | This study |
| BHUM1082 | CDC42(V36A, I182T) in pRS305 | This study |
| BHUM1083 | CDC42(V36A) in pRS305 | This study |
| BHUM1084 | CDC42(E62D, S71L, K128R, T161R) in pRS305 | This study |
| pEG202 | Vector for construction of lexA fusion proteins | 19 |
| pME1913 | LexA-CDC42(C188S) in pEG202 | 31 |
| pME1916 | LexA-CDC42(I46M, C188S) in pEG202 | 31 |
| pME1919 | LexA-CDC42(S71P, C188S) in pEG202 | 31 |
| BHUM1044 | LexA-CDC42(T58M, C188S) in pEG202 | This study |
| BHUM1045 | LexA-CDC42(V36A, C188S) in pEG202 | This study |
| BHUM1046 | LexA-CDC42(V36A, I182T, C188S) in pEG202 | This study |
| BHUM1047 | LexA-CDC42(E62D, S71L, K128R, T161R, C188S) in pEG202 | This study |
| pJG4-5 | Vector for construction of B42 fusion proteins | 19 |
| pJG4-5(STE20) | B42AD-STE20 in pJG4-5 | 31 |
| pJG4-5(CLA4) | B42AD-CLA4 in pJG4-5 | 11 |
| pJG4-5(SKM1) | B42AD-SKM1 in pJG4-5 | 39 |
| pJG4-5(GIC1) | B42AD-GIC1 in pJG4-5 | 8 |
| pJG4-5(GIC2) | B42AD-GIC2 in pJG4-5 | 8 |
| pJG4-5(BNI1) | B42AD-BNI1 in pJG4-5 | 14 |
| B1497 | FUS1-lacZ URA3 CEN | G. R. Fink, lab collection |
| pME2280 | myc3-TEC1 URA3, 2 μm | 26 |
| pME2287 | myc3-TEC1(T273M) URA3, 2 μm | 26 |
| YEplac195 | URA3-based 2 μm vector | 16 |
| pDLB722 | CLA4 URA3, 2 μm | Erfei Bi |
Screen for CDC42 alleles conferring pheromone resistance.
CDC42 mutant alleles conferring increased pheromone resistance were isolated from a library of randomly mutagenized CDC42 mutant genes in the LEU2-based vector pRS315 (31). A pool of approximately 25,000 CDC42 mutants was created by transformation of yeast strain YHUM1168 (cdc42Δ deletion strain carrying CDC42 on the URA3-based centromeric plasmid pME1534) with the CDC42 mutant library, selection on synthetic complete (SC)-Leu medium, and subsequent growth on plates containing 0.1% 5-fluoroorotic acid to remove plasmid pME1534 by counterselection. The resulting pool of strains was plated on YPD medium containing α-factor, and pheromone-resistant colonies were picked after 3 days of growth. CDC42-containing plasmids were isolated, and mutations were determined by sequence analysis of both strands of CDC42 alleles using the ABI Prism Big Dye terminator sequencing kit and an ABI 310 Genetic Analyzer (Perkin-Elmer Applied Biosystems GmbH, Weiterstadt, Germany). To confirm phenotypes, CDC42 alleles were integrated as single copies into the genome of the parental strain YHUM1168 by plasmid shuffling using integrative plasmids (Table 2).
Cell extracts and Western blot analysis.
Preparation of total cell extracts and subsequent Western blot analysis were performed as described previously (6). After transfer to a nitrocellulose membrane, proteins were detected using the enhanced chemiluminescence system and incubation with primary antibodies against Cdc42 or Fus3 (Santa Cruz Biotechnology, Santa Cruz, California), Cdc28 (kind gift from S. Irniger), or the myc epitope followed by incubation with a peroxidase-coupled secondary antibody (Santa Cruz Biotechnology, Santa Cruz, California). Phosphorylation of Fus3 and Kss1 was detected using a Phospho-p44/42 MAP kinase (Thr202/Tyr204) antibody from Cell Signaling Technology, Inc. (Danvers, Massachusetts). For quantification of signals, a scanner and the Quantity One software (Bio-Rad, Munich, Germany) were used.
Northern blot analysis.
Total RNA was prepared according to Cross and Tinkelenberg (10). RNA was separated on a 1.4% agarose gel containing 3% formaldehyde and transferred onto nylon membrane as described previously (6). CDC42 and ACT1 transcripts were detected using gene-specific 32P-labeled DNA probes.
β-Galactosidase assays.
Strains carrying FUS1-lacZ reporter constructs were grown to the exponential growth phase, and extracts were prepared and assayed for β-galactosidase activity as described previously (6). β-Galactosidase activity was normalized to the total protein in each extract with the following formula: (optical density at 420 nm × 1.7)/(0.0045 × protein concentration × extract volume × time). Assays were performed in triplicate on at least three transformants, and the mean values ± standard deviations were calculated.
Two-hybrid protein interactions.
Two-hybrid analysis was performed as described previously (19). The reporter strain EGY48-p1840 was cotransformed pairwise with various pEG202(CDC42) and pJG4-5 constructs (Table 2), and transformants were selected on SC-His-Trp medium. Transformants were grown in SC-His-Trp containing 2% galactose and 2% raffinose to an optical density at 600 nm of between 1 and 2 before β-galactosidase assays were performed. All assays were performed in triplicate on at least four independent transformants for each combination of plasmids. Specific β-galactosidase activities were determined as described above and were normalized to the mean values obtained for Cdc42(C188S) (wild-type control) and the different effectors, which corresponded to 41 U for Ste20, 69 U for Cla4, 23 U for Skm1, 175 U for Bni1, 396 U for Gic1, and 283 U for Gic2. Standard deviation did not exceed 25%.
RESULTS
Isolation of CDC42 alleles conferring resistance to mating pheromone.
We have previously shown that single-amino-acid substitutions in Cdc42 are sufficient to uncouple functions of this essential Rho-type GTPase required for cell division from those regulating filamentous and invasive growth (31). Other studies have addressed the role of Cdc42 in mating, but in some cases this led to controversial conclusions (32, 35, 42). To further investigate the role of this Rho-type GTPase during mating, we attempted to obtain novel variants of Cdc42 that interfere with the pheromone response of S. cerevisiae. For this purpose, a library of PCR-mutagenized CDC42 genes on centromeric plasmids was introduced into a haploid cdc42Δ deletion mutant strain by plasmid shuffling and was screened for CDC42 alleles that permit growth in the presence of otherwise inhibiting concentrations of mating pheromone. This scheme allowed isolation of the previously identified CDC42(V36A) allele (32) and three novel alleles, CDC42(V36A, I182T), CDC42(T58M), and CDC42(E62D, S71L, K128R, T161R) [named CDC42(S71L*) for ease of reference]. To prevent general perturbations often observed when CDC42 is expressed at nonendogenous levels (24), we created strains carrying single copies of the isolated CDC42 alleles in the genome instead of the endogenous CDC42 and verified resistance of strains to mating pheromone (Fig. 1A). Expression of these alleles (from the natural CDC42 promoter) resulted in CDC42 transcripts and Cdc42 proteins at endogenous levels (Fig. 1B), indicating that altered protein structure and not protein levels caused growth resistance.
FIG. 1.

Isolation of pheromone-resistant CDC42 mutant strains. (A) Growth arrest assay of strains expressing wild-type CDC42 (YHUM1064) or alleles CDC42(T58M) (YHUM1060), CDC42(V36A,I182T) (YHUM1061), CDC42(V36A) (YHUM1062), and CDC42(S71L*) (YHUM1063) conferring resistance to pheromone. Growth at 30°C is shown after 3 days on solid YPD medium without (−pheromone) or with (+pheromone) 0.025 μM α-factor. (B) Expression of different CDC42 alleles. CDC42 transcript and Cdc42 protein levels were measured in strains described for panel A by Northern (CDC42) and immunoblot analysis (Cdc42), respectively. Expression of the ACT1 gene was used as an internal standard for CDC42 transcript analysis, and protein levels of Cdc28 served as internal controls for immunoblot analysis of Cdc42.
Pheromone tolerance of CDC42 mutant strains was further analyzed by observing growth on solid medium containing α-factor (Fig. 2A). As controls we included isogenic ste7Δ and kss1Δ fus3Δ mutant strains that are completely sterile, as well as four CDC42 mutants [CDC42(I46M), CDC42(S71P), CDC42(E100G) and CDC42(S158T)] that we previously isolated as strains that are suppressed for invasive growth. We found that growth of the CDC42(V36A), CDC42(V36A, I182T), and CDC42(S71L*) mutants was comparable to that of the sterile mutants, whereas pheromone resistance of CDC42(T58M) mutant was less pronounced. In contrast, the CDC42 control strain and the CDC42(I46M), CDC42(E100G), and CDC42(S158T) mutants were unable to grow at any dilution. Interestingly, the CDC42(S71P) allele also conferred pheromone tolerance that was comparable to that of the CDC42(S71L*) allele, indicating that the residue Ser71 might be crucial. In summary, our data show that residue Val36, which has previously been reported to be involved in pheromone resistance (32), and residues Thr58 and Ser71 of Cdc42 are involved in rendering haploid yeast strains sensitive to mating pheromone.
FIG. 2.

Pheromone sensitivity of different CDC42 mutants. (A) Strains YHUM1064 (control), YHUM 1073 (kss1Δ fus3Δ), YHUM1074 (ste7Δ), YHUM1060 [CDC42(T58M)], YHUM1063 [CDC42(S71L*)], YHUM1061 [CDC42(V36A, I182T)], YHUM1062 [CDC42(V36A)], YHUM1066 [CDC42(I46M)], YHUM1067 [CDC42(S71P)], YHUM1068 [CDC42(E100G)], and YHUM1069 [CDC42(S158T)] were grown to logarithmic phase, and serial fivefold dilutions of the cultures were spotted onto YPD plates supplemented without or with 0.025 μM of α-factor before incubation for 2 days at 30°C. (B) Pheromone-induced cell cycle arrest. Strains of the indicated genotype (described for panel A) were grown in YPD to logarithmic phase, and the percentage of budded cells in each culture was determined before (time, 0 h) and 1, 3, and 5 h, respectively, after addition of α-factor to 0.15 μM. Values indicate means of three independent assays with a standard deviation not exceeding 15%.
Pheromone-induced cell cycle arrest and mating efficiency of CDC42 mutants.
In haploid strains, the pheromone response includes a growth arrest in the G1 phase of the cell cycle, activation of FUS1 gene expression, and fusion with mating partner cells to generate diploid zygotes. To determine how mutations in CDC42 identified above affect these processes, we measured the growth arrest of CDC42 mutant strains in response to pheromone, expression of a FUS1-lacZ reporter gene, and the ability of mutants to fuse to partner cells of opposite mating type (Fig. 2B, 3, and 4). Pheromone-induced cell cycle arrest was measured by monitoring the percentage of budded cells before and after pheromone addition of 0.15 μM α-factor to exponentially growing cultures (Fig. 2B). In the CDC42 control strain, the fraction of budded cells starts to decrease immediately after addition of pheromone; after 3 h, less than 10% of cells have formed a bud, and after 5 h more than 99% of cells in the population have arrested in G1. In contrast, no significant change in the proportion of budded cells was observed in the case of the CDC42(V36A) and CDC42(V36A, I182T) mutant strains, which behaved similar to the ste7Δ and kss1Δ fus3Δ mutants. In the case of the CDC42(T58M), CDC42(S71P), and CDC42(S71L*) strains, the number of budded cells started to decline after 1 h to reach 35 to 40% after 3 h and 10 to 15% after 5 h. In both cases, the percentage of budded cells did not significantly decrease further (data not shown). These results indicate that Val36 is crucial for pheromone-induced cell cycle arrest and that residues Thr58 and Ser71 also contribute to this process, albeit to a lesser extent.
FIG. 3.
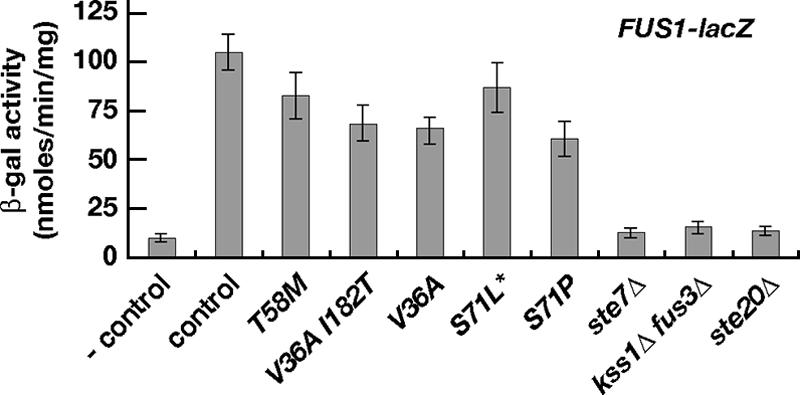
Pheromone-induced expression of FUS1-lacZ in CDC42 mutants. Strains of the indicated genotype (described in the legend to Fig. 2 and in Table 1) were transformed with plasmid B1497 carrying the FUS1-lacZ reporter gene and were grown to exponential phase before addition of 0.15 μM α-factor. Specific β-galactosidase (β-gal) activities were determined after 2 h. The bars depict mean values ± standard deviations of three transformants, each assayed in triplicate. Activity measured in the control strain before addition of α-factor (−control) is shown in the first bar.
FIG. 4.

Mating of CDC42 mutants. (A) Qualitative mating assay. Strains of the indicated genotype (described in the legend to Fig. 2A) were grown overnight as patches on YPD medium, replica plated to fresh YPD medium together with a lawn of a MATα tester strain (YHUM 70), and incubated for 5 h at 30°C for mating. Formation of diploids was monitored by replica plating of mating reactions on selective yeast nitrogen base (YNB) medium and incubation for 2 days. (B) Quantitative mating assay. Liquid cultures of strains of the indicated genotype and of the tester strain YHUM70 were mixed and subjected to growth selection for diploids after 5 h of mating. Mating frequencies shown correspond to the number of diploids formed after 3 days, normalized to the number of cells used in the mating reactions and to the mean value obtained with the control strain YHUM1064. The bars indicate the means ± standard deviations of three independent assays.
When measuring expression of FUS1-lacZ in response to pheromone, we found that gene expression in all CDC42 mutants reached between 58% and 80% of the control strain. In contrast, FUS1-lacZ could not be significantly activated above basal levels in strains lacking STE20, STE7, or FUS3 and KSS1 (Fig. 3).
Mating efficiency of CDC42 mutant strains was measured by qualitative patch assays and by quantitative mating in liquid cultures using a tester strain of opposite mating type (Fig. 4). The strongest effect was observed in the case of CDC42(V36A) and CDC42(V36A, I182T), where mating efficiency dropped more than 90%, although suppression of mating was not as pronounced as that observed in the ste7 mutant strain. In the case of the CDC42 alleles with changed codons for Thr58 and Ser71, mating frequency was still between 40% and 60% of that of the CDC42 control strain. In conclusion, the ability of the different CDC42 mutant strains to undergo pheromone-induced cell cycle arrest appears to correlate with mating efficiency and to a lesser extent with mating-specific gene expression.
Phosphorylation of MAP kinases Fus3 and Kss1 in CDC42 mutants.
Because we found that certain mutations in CDC42 efficiently block cell cycle arrest, we measured the phosphorylation status of the mating MAP kinases Fus3 and Kss1 under a number of different conditions, including the presence of different CDC42 alleles, the presence and absence of Ste7 (and Ste20), and dependence on the pheromone concentration and time after pheromone administration (Fig. 5). We found that Fus3 and Kss1 were efficiently phosphorylated in CDC42(T58M) and CDC42(S71L*) mutants, whereas phosphorylation was slightly reduced in CDC42(S71P) strains. In strains expressing CDC42(V36A) and CDC42(V36A, I182T), however, phosphorylation of the MAP kinases was clearly reduced compared to that of unaltered CDC42 (Fig. 5D). Phosphorylation is likely to stem from Ste7, because phosphorylated forms of both MAP kinases are completely absent in strains lacking STE7 (Fig. 5A, C, and D). In addition, dependence of MAP kinase phosphorylation on exposure time and pheromone concentration was not significantly altered in the different CDC42 mutant strains (Fig. 5B and C). Thus, a clear correlation between MAP kinase phosphorylation, G1 arrest, and mating is obvious only in the case of the CDC42(V36A) and CDC42(V36A, I182T) variants but not in the case of the CDC42 alleles that cause milder mating defects.
FIG. 5.

Activation of MAP kinases Fus3 and Kss1 by pheromone. (A) Pheromone-induced phosphorylation of Fus3 and Kss1 was determined in protein extracts prepared from strains of the indicated genotype before (−) and 20 min after (+) addition of 0.15 μM α-factor by immunoblot analysis using phosphospecific antibodies against human p44 and p42 kinases (Erk1 and Erk2) phosphorylated at Thr202 and Tyr204. As an internal control, total levels of Fus3 were detected by anti-Fus3 polyclonal antibodies. (B) Time-dependent phosphorylation of MAP kinases. Phosphorylation of Fus3 and Kss1 was assayed as described for panel A in extracts of strains of the indicated genotype exposed for 0, 20, and 120 min to 0.15 μM α-factor. Measurement of expression levels of Cdc28 served as a loading control. (C) Dose-dependent phosphorylation of Fus3 and Kss1 was measured by exposure of cultures of indicated strains to α-factor at concentrations of 0, 0.025, and 0.15 μM, respectively, for 20 min. (D) Quantification of phosphorylated forms of Kss1 and Fus3 after 20 min in 0.15 μM α-factor. Relative Kss1-P (black bars) and Fus3-P (gray bars) amounts are shown in arbitrary units and were obtained by normalizing Kss1-P and Fus3-P signals to Cdc28 signals and to the values obtained for the control strain YHUM1064. Bars indicate means ± standard deviations of three independent measurements.
We further measured pheromone-induced degradation of the transcription factor Tec1 under identical conditions as MAP kinase phosphorylation and cell cycle arrest, because it was previously shown that Tec1 is phosphorylated by activated Fus3, leading to degradation of the transcription factor (6). We found that Tec1 is normally degraded in the different CDC42 mutant strains, whereas deletion of FUS3 or expression of the pheromone-resistant TEC1(T273M) allele was sufficient to suppress degradation of Tec1 (data not shown). Thus, the amount of phosphorylated Fus3 observed in the different CDC42 mutant strains appears to be sufficient to trigger degradation of Tec1.
Interaction of Cdc42 mutant proteins with PAK family protein kinases Cla4, Ste20, and Skm1.
To further characterize the Cdc42 mutant proteins, we measured their capacity to interact with the PAK kinases Ste20, Cla4, and Skm1 in the yeast two-hybrid system, which in the case of Ste20 and Cla4 has been shown to accurately report in vitro interactions between Cdc42 and these effector proteins (11, 17, 27, 31, 38, 39, 43). In the case of Ste20, each of the variants except Cdc42(S71P) showed the same or enhanced interaction compared to wild-type Cdc42 (Fig. 6A). In contrast, interaction with Cla4 and Skm1 was significantly reduced in the case of Cdc42(V36A) and Cdc42(V36A, I182T). This finding is in agreement with a previous study demonstrating that Cdc42(V36A, Q61L) is significantly reduced for Cla4 binding in vitro (17). In the case of Cdc42(S71L*), only interaction with Skm1 was clearly affected, whereas interaction with all three PAKs was reduced in the case of Cdc42(S71P). No significant effect on interaction with Cla4 and Skm1 could be detected with Cdc42(T58M). These data demonstrate that pheromone-resistant variants of Cdc42, with the exception of Cdc42(S71P), are not blocked for interaction with Ste20 and indicate that pheromone-induced cell cycle arrest in yeast might depend on interaction of Cdc42 with Cla4 and/or Skm1.
FIG. 6.

Two-hybrid protein interactions between Cdc42 proteins and PAK kinases Cla4, Ste20, and Skm1 (A) as well as Cdc42 proteins and effector proteins Bni1, Gic1, and Gic2 (B). Wild-type Cdc42 (control), Cdc42 mutants, and effector proteins are indicated. All Cdc42 proteins carry the C188S mutation to prevent plasma membrane localization. The bars indicate the means ± standard deviations of four transformants, each measured in triplicate and expressed as percent mean specific β-galactosidase (β-gal) activity in the control strain (41 U for Ste20, 69 U for Cla4, 23 U for Skm1, 175 U for Bni1, 396 U for Gic1, and 283 U for Gic2).
We reasoned that if the above hypothesis was true, either Cla4 or Skm1 or both might affect pheromone responses. Therefore, we created cla4Δ and skm1Δ deletion mutations in CDC42(V36A, I182T) and CDC42(S71L*) mutant strains and, as controls, in CDC42 wild-type and ste7Δ strains to measure effects of such mutations on pheromone sensitivity and phosphorylation of the MAP kinases Fus3 and Kss1. We chose Cdc42(V36A, I182T) due to its reduced interaction with both Cla4 and Skm1 and Cdc42(S71L*) as a variant showing reduced interaction with Skm1 but not Cla4. We were not able to introduce cla4Δ mutations into strains carrying the CDC42(V36A) allele, a finding that is in agreement with the previously reported synthetic lethality caused by these two mutations (32). We expected that if pheromone-induced and Cdc42-dependent G1 arrest was mediated by Cla4 and/or Skm1, cla4Δ and/or skm1Δ mutations might cause pheromone resistance in a CDC42 control strain and induce synthetic effects on G1 arrest in CDC42(V36A, I182T) and CDC42(S71L*) mutant strains. No such effects could be observed in the case of skm1Δ mutations (Fig. 7A), indicating that Skm1 does not affect pheromone signaling. Similarly, cla4Δ mutations did not alter pheromone sensitivity in the CDC42 control strain and the ste7Δ or CDC42(S71L*) mutant strains. However, we found that deletion of CLA4 affected pheromone sensitivity of a CDC42(V36A, I182T) strain, albeit not as expected by further aiding to the pheromone tolerance of such a strain, but in contrast by suppressing its enhanced pheromone resistance phenotype (Fig. 7A). We also found that in the CDC42(V36A, I182T) genetic background, a cla4Δ mutation was sufficient to restore phosphorylation of Fus3 and Kss1 to levels observed in the CDC42 control strain (Fig. 7B and C). These data indicate that Cla4 might negatively act on the pheromone signaling pathway, an effect that is not observed in strains expressing wild-type CDC42 and that seems to be unmasked by introduction of the CDC42(V36A, I182T) mutation.
FIG. 7.

Genetic interactions between mutations in CDC42, CLA4, and SKM1. (A) Pheromone sensitivity. Strains YHUM1064 (control), YHUM1165 (cla4Δ), YHUM1061 [CDC42(V36A, I182T)], YHUM 1299 [CDC42(V36A, I182T) cla4Δ], YHUM1074 (ste7Δ), YHUM1297 (ste7Δ cla4Δ), YHUM1063 [CDC42(S71L*)], YHUM1294 [CDC42(S71L*) cla4Δ], YHUM1296 (skm1Δ), YHUM1327 [CDC42(V36A, I182T) skm1Δ], YHUM1337 (ste7Δ skm1Δ), and YHUM1302 [CDC42(S71L*) skm1Δ] were grown to logarithmic phase, and serial fivefold dilutions of the cultures were spotted onto YPD plates supplemented without or with 0.025 μM of α-factor before incubation for 2 days at 30°C. (B and C) Phosphorylation of Kss1 (black bars) and Fus3 (gray bars) in strains of the indicated genotype was measured before (−) and 20 min after (+) addition of 0.15 μM α-factor and quantified (C) as described in the legend to Fig. 5.
We also measured two-hybrid interactions between the different Cdc42 variants and the known effector proteins Bni1, Gic1, and Gic2 (Fig. 6B). These data did not reveal any obvious correlation between effector binding patterns and G1 arrest behavior or MAP kinase phosphorylation. However, they uncover a significant difference between the binding of Cdc42(V36A) and Cdc42(V36A, I182T) to the effector proteins Gic1 and Gic2, which might account for our finding that the cla4Δ mutation could be introduced in the CDC42(V36A, I182T) but not in the CDC42(V36A) genetic background.
Effects of CLA4 overexpression on pheromone sensitivity and MAP kinase phosphorylation.
To further explore a possible negative function of Cla4 in pheromone signaling, we overexpressed CLA4 in different CDC42 strains. We found that overexpression of CLA4 is sufficient to induce pheromone resistance in a CDC42 wild-type strain to a degree observed in a CDC42(V36A, I182T) mutant strain that does not overexpress CLA4 (Fig. 8A) and to completely block G1 arrest (Fig. 8B). Similarly, pheromone-induced phosphorylation of Fus3 and Kss1 in the Cla4-overexpressing CDC42 wild-type strain was reduced to levels comparable to the CDC42(V36A, I182T) strain carrying a control vector (Fig. 8C and D). Overexpression of CLA4 in the CDC42(V36A, I182T) mutant strain did not significantly enhance pheromone resistance or alter G1 arrest but caused a further decrease in MAP kinase phosphorylation. Effects of CLA4 overexpression on pheromone resistance, G1 arrest, and MAP kinase phosphorylation in a CDC42(S71L*) mutant strain were comparable to the effects observed in the CDC42 control strain. Finally, overexpression of CLA4 was sufficient to fully block G1 arrest in CDC42(T58M) and CDC42(S71P) mutant strains and did not significantly alter G1 arrest behavior in the ste7Δ mutant (data not shown). Thus, Cla4 appears to be able to negatively act on pheromone signaling, a function of the PAK that has not been reported yet. Cla4 might exert this negative function when not associated with Cdc42, because overexpression could lead to an increase in the GTPase-free form of the PAK. This conclusion is in agreement with our finding that the negative effects on pheromone signaling exerted by Cdc42(V36A, I182T), which binds Cla4 with low efficiency, depends on the presence of Cla4.
FIG. 8.

Effects of CLA4 overexpression on pheromone sensitivity and MAP kinase phosphorylation. (A) Pheromone sensitivity. Strains YHUM1064 (control), YHUM1063 [CDC42(S71L*)], and YHUM1061 [CDC42(V36A, I182T)] carrying either plasmid YEplac195 (vector) or plasmid pDLB722(CLA4) were grown to logarithmic phase, and serial fivefold dilutions of the cultures were spotted onto SC-Ura plates supplemented without or with 0.025 μM of α-factor before incubation for 2 days at 30°C. (B) Pheromone-induced cell cycle arrest. Strains of the indicated genotype (described for panel A) were grown in SC-Ura to logarithmic phase, and the percentage of budded cells in each culture was determined before (time, 0 h) and 1, 2, 3, and 5 h, respectively, after addition of α-factor to 0.15 μM. Values indicate means of three independent assays with a standard deviation not exceeding 15%. (C and D) Phosphorylation of Kss1 (black bars) and Fus3 (gray bars) in strains described for panel A was measured before (−) and 20 min after (+) addition of 0.15 μM α-factor (C) and quantified (D).
DISCUSSION
The current view of how Cdc42 controls pheromone-stimulated signaling is by activating Ste20 through interaction with the CRIB domain of this PAK family protein kinase (3, 13). This model is based on several studies, which suggest that the Cdc42-Ste20 interaction is not under direct control of pheromone but is required for efficient pheromone-induced activation of the mating MAP kinase cascade (17, 28, 32). A recent study has identified a CDC42 allele, CDC42(V36M), that specifically interferes with cell fusion but not with initial pheromone-induced signaling, indicating that Cdc42 has a second function during mating at the step of cell fusion (2). Here, we have aimed at isolating additional CDC42 mutations that confer enhanced pheromone resistance and interfere with efficient G1 arrest. Although this type of genetic screen has been performed before and our independent approach has uncovered the previously identified CDC42(V36A) variant (32), we have uncovered novel CDC42 variants that aid in further assessing the role of Cdc42 in pheromone signaling. Importantly, we have analyzed the different CDC42 alleles when expressed at endogenous levels in the complete absence of endogenous Cdc42 and by investigating phenotypes that previous studies have not addressed, foremost MAP kinase phosphorylation (for a summary, see Table 3).
TABLE 3.
Summary of phenotypes of different CDC42 variantsa
| CDC42 variant | Pheromone resistanceb
|
G1 arrestc
|
Mating | FUS1-lacZ | MAP kinase phosphorylation | PAK binding
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CLA4 | cla4Δ | hc CLA4d | CLA4 | hc CLA4d | Cla4 | Ste20 | Skm1 | ||||
| Wild type | − | − | ++ | ++ | − | ++ | ++ | ++ | ++ | ++ | ++ |
| V36A | ++ | NDe | ++ | − | − | +/− | + | + | +/− | ++ | +/− |
| V36A, I182T | ++ | − | ++ | − | − | +/− | + | + | +/− | ++ | +/− |
| T58M | + | ND | ND | + | − | + | ++ | ++ | ++ | ++ | ++ |
| S71L* | + | + | ++ | + | − | + | ++ | ++ | ++ | ++ | + |
| S71P | + | ND | ND | + | − | + | + | ++ | +/− | + | +/− |
| ste7Δ | ++ | ++ | ++ | − | − | − | +/− | − | NAf | NA | NA |
Quantifiable phenotypes were evaluated as >70% of wild type (++), 30 to 70% of wild type (+), <30% of wild type (+/−), or not detectable (−).
−, no growth on pheromone plates in serial dilutions; +, robust growth at the minimal dilution; ++, growth comparable to that of an ste7Δ mutant.
Reduction of percentage of budded cells after 5 h in pheromone in the wild type was set to 100 and in the ste7Δ mutant to 0.
CLA4 was overexpressed from a high-copy (hc) plasmid.
ND, not determined.
NA, not applicable.
The most surprising outcome of our study is that the Cdc42 effector protein Cla4 appears to be able to act as a negative regulator of pheromone-induced signaling. This previously undescribed function of Cla4 is revealed by the newly isolated CDC42(V36A, I182T) allele, which in contrast to the CDC42(V36A) allele can be combined with a cla4Δ mutation and whose ability to confer pheromone resistance depends on CLA4. Although we do not know the reason for the differential viability of CDC42(V36A, I182T) cla4Δ and CDC42(V36A) cla4Δ double mutant strains, one might speculate that the significantly higher affinity of Cdc42(V36A, I182T) to Gic1 and Gic2 compared to Cdc42(V36A) might be of crucial importance, because Gic1 and Gic2 are known to fulfill essential functions during bud initiation and cytokinesis (5, 21, 23). Importantly, our study has independently revealed the negative effects of Cla4 on pheromone responses by overexpression of this PAK in strains that express endogenous levels of different variants of Cdc42. Our findings that expression of Cdc42 variants, which inefficiently bind to Cla4 and whose effects depend on Cla4 as well as overexpression of Cla4, block pheromone responses could be explained by a model in which increased amounts of the PAK that are not bound to Cdc42 interfere with pheromone signaling. This interpretation of our data could explain why overexpression of CLA4 was not found to enhance pheromone resistance in a previous study (32). In contrast to our study, these experiments were performed in strains expressing CDC42 from plasmids and carrying an additional genomic copy of GAL1-driven CDC42, which might have caused increased levels of Cdc42 that prevented the negative effects of Cla4 on pheromone signaling observed in our study.
It is puzzling that in the case of the Cdc42(V36A, I182T) variant a decreased affinity of Cdc42 to Cla4 correlates with an increase of a Cla4-dependent function, because efficient Cla4 functionality would be expected to depend on binding of the PAK to Cdc42. However, previous studies have demonstrated a significant basal Cdc42-independent Cla4 kinase activity both in vivo in cdc42-1 and cdc24-1 strains (4) and in vitro using bacterially purified Cla4 protein (44). Moreover, a chemical genetic analysis suggested that Cla4 could have kinase-independent functions that might not depend on stimulation by Cdc42 (45). Hence, proper Cdc42-Cla4 interaction might not only ensure stimulation of PAK functions at the desired sites of action but also prevent Cla4 from acting on undesired targets. It has been shown that Cla4 and Cdc24 colocalize to the incipient bud site and tips of small buds of dividing cells and to polarized cell surface projections in pheromone-treated cells (22, 33). Thus, failure to establish proper Cdc42-Cla4 interactions at these sites might not only cause growth defects but also permit undesired interactions of Cla4 with, e.g., components of the pheromone response pathway. In this context it is interesting to note that expression of a Cla4 variant carrying a single-amino-acid substitution, Cla4(D772T), is able to partially confer Ste20-specific functions during mating by changing the substrate specificity of Cla4, thus demonstrating that Cla4 has the potential to interact with the pheromone response pathway (25). However, because it is unclear whether kinase activity of Cla4 is necessary for the observed negative regulation, future experiments must resolve the exact mechanism by which this PAK is able to control pheromone-induced cell cycle arrest and MAP kinase phosphorylation. It will be interesting to see whether Cla4, for instance, has a negative effect on the Ste20 activity. The previous finding that expression of Ste20ΔCRIB is sufficient to restore pheromone sensitivity in a CDC42(V36A) mutant (32) indicates that Cla4 might negatively act on the pheromone pathway at the level or upstream of Ste20.
Our study has uncovered a positive correlation between the degree of pheromone sensitivity, G1 arrest, mating, and phosphorylation of the MAP kinases Fus3 and Kss1 in the case of CDC42(V36A) and CDC42(V36A, I182T) mutant strains. However, MAP kinase phosphorylation is only partially blocked in these strains compared to bona fide ste mutants. Thus, either a reduction of MAP kinase phosphorylation by a factor of roughly two is sufficient to prevent an efficient G1 arrest or Cdc42 and Cla4 are able to control pheromone-induced cell cycle arrest by further mechanisms that act independently of MAP kinase activation. Our data do not allow discrimination between these two possibilities. However, if the former possibility was correct, our results would point towards a threshold level of phosphorylated MAP kinases for entry into G1 arrest that appears to be higher than one might have expected. We are aware of the fact that considerable differences exist in the capacity of individual cells to transmit signals through the mating pathway (9). However, our G1 arrest measurements reflect the budding behavior of a large number of randomly selected individual cells that should be representative for the whole population, and quantification of MAP kinase phosphorylation represents the average values of all cells in the population. In addition, we have used bar1Δ strains and saturating pheromone concentrations to ensure maximal stimulatory effects. Similarly, FUS1-lacZ measurements and Tec1 degradation were assayed under comparable conditions. Hence, correlation of these phenotypes appears justified to a certain extent. It then would appear that induction of G1 arrest requires a higher MAP kinase phosphorylation status than does the induction of FUS1-lacZ expression or the degradation of the transcription factor Tec1. Finally, our analysis of MAP kinase phosphorylation in the CDC42(V36A, I182T) mutant strain that overexpresses CLA4 revealed that these mutations cause additive effects. This result is in agreement with the idea that Cla4, when not associated with Cdc42, might negatively act on the pheromone pathway, because reducing the Cla4-binding affinity of Cdc42 in combination with overexpression of Cla4 would be expected to additively contribute to the GTPase-free form of the PAK.
Our study has uncovered CDC42(T58M), CDC42(S71L*), and CDC42(S71P) as previously unknown alleles that confer pheromone resistance. In contrast to the CDC42(V36A, I182T) allele, effector binding studies and analysis of MAP kinase phosphorylation was less revealing in the case of these variants of CDC42. All three variants, Cdc42(T58M), Cdc42(S71L*), and Cdc42(S71P), interact less efficiently with Skm1, but Skm1 does not appear to mediate inputs into the pheromone response pathway. In addition, the T58M and S71L* mutations do not affect interaction of Cdc42 with Cla4. The Cdc42(S71P) mutant shows reduced Cla4 binding, but MAP kinase phosphorylation is not lowered to the same extent as that observed in the case of Cdc42(V36A) and Cdc42(V36A, I182T). Thus, while we do not know the exact molecular reasons for the pheromone-resistant phenotype induced by these variants of Cdc42, they further demonstrate the importance of this GTPase in the mating process. Whether some of the pheromone-resistant Cdc42 variants isolated here are affected in GDP-GTP cycling as has been found in the case of Cdc42(V36M) (2) remains to be determined.
In summary, our study expands the growing number of functions attributed to Cdc42 in the control of yeast growth and development. As found in the case of the septin assembly pathway (7) and the invasive growth signaling pathway (43), our study indicates that Cdc42 might also control the pheromone response pathway by targeting more than one effector protein. It will be exciting to further uncover the mechanisms by which this GTPase fulfills its diverse functions.
Acknowledgments
We thank Diana Kruhl and Maria Meyer for excellent technical assistance in this work and Gerhard Braus for helpful comments on the manuscript. We are grateful to Robert Arkowitz, Erfei Bi, Charlie Boone, and Douglas Johnson for generous gifts of reagents.
This work was supported by grants from the Deutsche Forschungsgemeinschaft.
Footnotes
Published ahead of print on 22 December 2006.
REFERENCES
- 1.Bagrodia, S., and R. A. Cerione. 1999. Pak to the future. Trends Cell Biol. 9:350-355. [DOI] [PubMed] [Google Scholar]
- 2.Barale, S., D. McCusker, and R. A. Arkowitz. 2006. Cdc42p GDP/GTP cycling is necessary for efficient cell fusion during yeast mating. Mol. Biol. Cell 17:2824-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardwell, L. 2005. A walk-through of the yeast mating pheromone response pathway. Peptides 26:339-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benton, B. K., A. Tinkelenberg, I. Gonzalez, and F. R. Cross. 1997. Cla4p, a Saccharomyces cerevisiae Cdc42p-activated kinase involved in cytokinesis, is activated at mitosis. Mol. Cell. Biol. 17:5067-5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown, J. L., M. Jaquenoud, M. P. Gulli, J. Chant, and M. Peter. 1997. Novel Cdc42-binding proteins Gic1 and Gic2 control cell polarity in yeast. Genes Dev. 11:2972-2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brückner, S., T. Köhler, G. H. Braus, B. Heise, M. Bolte, and H.-U. Mösch. 2004. Differential regulation of Tec1 by Fus3 and Kss1 confers signaling specificity in yeast development. Curr. Genet. 46:331-342. [DOI] [PubMed] [Google Scholar]
- 7.Caviston, J. P., M. Longtine, J. R. Pringle, and E. Bi. 2003. The role of Cdc42p GTPase-activating proteins in assembly of the septin ring in yeast. Mol. Biol. Cell 14:4051-4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, G. C., Y. J. Kim, and C. S. Chan. 1997. The Cdc42 GTPase-associated proteins Gic1 and Gic2 are required for polarized cell growth in Saccharomyces cerevisiae. Genes Dev. 11:2958-2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colman-Lerner, A., A. Gordon, E. Serra, T. Chin, O. Resnekov, D. Endy, C. G. Pesce, and R. Brent. 2005. Regulated cell-to-cell variation in a cell-fate decision system. Nature 437:699-706. [DOI] [PubMed] [Google Scholar]
- 10.Cross, F. R., and A. H. Tinkelenberg. 1991. A potential positive feedback loop controlling CLN1 and CLN2 gene expression at the start of the yeast cell cycle. Cell 65:875-883. [DOI] [PubMed] [Google Scholar]
- 11.Cvrckova, F., C. De Virgilio, E. Manser, J. R. Pringle, and K. Nasmyth. 1995. Ste20-like protein kinases are required for normal localization of cell growth and for cytokinesis in budding yeast. Genes Dev. 9:1817-1830. [DOI] [PubMed] [Google Scholar]
- 12.Dan, I., N. M. Watanabe, and A. Kusumi. 2001. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 11:220-230. [DOI] [PubMed] [Google Scholar]
- 13.Etienne-Manneville, S. 2004. Cdc42-the centre of polarity. J. Cell Sci. 117:1291-1300. [DOI] [PubMed] [Google Scholar]
- 14.Evangelista, M., K. Blundell, M. S. Longtine, C. J. Chow, N. Adames, J. R. Pringle, M. Peter, and C. Boone. 1997. Bni1p, a yeast formin linking Cdc42p and the actin cytoskeleton during polarized morphogenesis. Science 276:118-122. [DOI] [PubMed] [Google Scholar]
- 15.Feng, Y., L. Y. Song, E. Kincaid, S. K. Mahanty, and E. A. Elion. 1998. Functional binding between Gbeta and the LIM domain of Ste5 is required to activate the MEKK Ste11. Curr. Biol. 8:267-278. [DOI] [PubMed] [Google Scholar]
- 16.Gietz, R. D., and A. Sugino. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527-534. [DOI] [PubMed] [Google Scholar]
- 17.Gladfelter, A. S., J. J. Moskow, T. R. Zyla, and D. J. Lew. 2001. Isolation and characterization of effector-loop mutants of CDC42 in yeast. Mol. Biol. Cell 12:1239-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guthrie, C., and G. R. Fink. 1991. Guide to yeast genetics and molecular biology, vol. 194. Academic Press, San Diego, CA.
- 19.Gyuris, J., E. Golemis, H. Chertkov, and R. Brent. 1993. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell 75:791-803. [DOI] [PubMed] [Google Scholar]
- 20.Hartwell, L. H. 1980. Mutants of Saccharomyces cerevisiae unresponsive to cell division control by polypeptide mating hormone. J. Cell Biol. 85:811-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Höfken, T., and E. Schiebel. 2004. Novel regulation of mitotic exit by the Cdc42 effectors Gic1 and Gic2. J. Cell Biol. 164:219-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holly, S. P., and K. J. Blumer. 1999. PAK-family kinases regulate cell and actin polarization throughout the cell cycle of Saccharomyces cerevisiae. J. Cell Biol. 147:845-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwase, M., J. Luo, S. Nagaraj, M. Longtine, H. B. Kim, B. K. Haarer, C. Caruso, Z. Tong, J. R. Pringle, and E. Bi. 2006. Role of a Cdc42p effector pathway in recruitment of the yeast septins to the presumptive bud site. Mol. Biol. Cell. 17:1110-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson, D. I. 1999. Cdc42: an essential Rho-type GTPase controlling eukaryotic cell polarity. Microbiol. Mol. Biol. Rev. 63:54-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keniry, M. E., and G. F. Sprague, Jr. 2003. Identification of p21-activated kinase specificity determinants in budding yeast: a single amino acid substitution imparts Ste20 specificity to Cla4. Mol. Cell. Biol. 23:1569-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Köhler, T., S. Wesche, N. Taheri, G. H. Braus, and H. U. Mösch. 2002. Dual role of the Saccharomyces cerevisiae TEA/ATTS family transcription factor Tec1p in regulation of gene expression and cellular development. Eukaryot. Cell 1:673-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozminski, K. G., A. J. Chen, A. A. Rodal, and D. G. Drubin. 2000. Functions and functional domains of the GTPase Cdc42p. Mol. Biol. Cell 11:339-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamson, R. E., M. J. Winters, and P. M. Pryciak. 2002. Cdc42 regulation of kinase activity and signaling by the yeast p21-activated kinase Ste20. Mol. Cell. Biol. 22:2939-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leberer, E., C. Wu, T. Leeuw, A. Fourest-Lieuvin, J. E. Segall, and D. Y. Thomas. 1997. Functional characterization of the Cdc42p binding domain of yeast Ste20p protein kinase. EMBO J. 16:83-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leeuw, T., C. Wu, J. D. Schrag, M. Whiteway, D. Y. Thomas, and E. Leberer. 1998. Interaction of a G-protein beta-subunit with a conserved sequence in Ste20/PAK family protein kinases. Nature 391:191-195. [DOI] [PubMed] [Google Scholar]
- 31.Mösch, H.-U., T. Köhler, and G. H. Braus. 2001. Different domains of the essential GTPase Cdc42p required for growth and development of Saccharomyces cerevisiae. Mol. Cell. Biol. 21:235-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moskow, J. J., A. S. Gladfelter, R. E. Lamson, P. M. Pryciak, and D. J. Lew. 2000. Role of Cdc42p in pheromone-stimulated signal transduction in Saccharomyces cerevisiae. Mol. Cell. Biol. 20:7559-7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nern, A., and R. A. Arkowitz. 1999. A Cdc24p-Far1p-Gβγ protein complex required for yeast orientation during mating. J. Cell Biol. 144:1187-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oehlen, L. J., and F. R. Cross. 1994. G1 cyclins CLN1 and CLN2 repress the mating factor response pathway at Start in the yeast cell cycle. Genes Dev. 8:1058-1070. [DOI] [PubMed] [Google Scholar]
- 35.Oehlen, L. J., and F. R. Cross. 1998. The role of Cdc42 in signal transduction and mating of the budding yeast Saccharomyces cerevisiae. J. Biol. Chem. 273:8556-8559. [DOI] [PubMed] [Google Scholar]
- 36.Pan, X., T. Harashima, and J. Heitman. 2000. Signal transduction cascades regulating pseudohyphal differentiation of Saccharomyces cerevisiae. Curr. Opin. Microbiol. 3:567-572. [DOI] [PubMed] [Google Scholar]
- 37.Peter, M., A. M. Neiman, H. O. Park, H. van Lohuizen, and I. Herskowitz. 1996. Functional analysis of the interaction between the small GTP binding protein Cdc42 and the Ste20 protein kinase in yeast. EMBO J. 15:7046-7059. [PMC free article] [PubMed] [Google Scholar]
- 38.Richman, T. J., and D. I. Johnson. 2000. Saccharomyces cerevisiae Cdc42p GTPase is involved in preventing the recurrence of bud emergence during the cell cycle. Mol. Cell. Biol. 20:8548-8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richman, T. J., M. M. Sawyer, and D. I. Johnson. 1999. The Cdc42p GTPase is involved in a G2/M morphogenetic checkpoint regulating the apical-isotropic switch and nuclear division in yeast. J. Biol. Chem. 274:16861-16870. [DOI] [PubMed] [Google Scholar]
- 40.Sambrook, J., E. F. Fritch, and T. Maniatias. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 41.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simon, M. N., C. De Virgilio, B. Souza, J. R. Pringle, A. Abo, and S. I. Reed. 1995. Role for the Rho-family GTPase Cdc42 in yeast mating-pheromone signal pathway. Nature 376:702-705. [DOI] [PubMed] [Google Scholar]
- 43.Truckses, D. M., J. E. Bloomekatz, and J. Thorner. 2006. The RA domain of Ste50 adaptor protein is required for delivery of Ste11 to the plasma membrane in the filamentous growth signaling pathway of the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 26:912-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Versele, M., and J. Thorner. 2004. Septin collar formation in budding yeast requires GTP binding and direct phosphorylation by the PAK, Cla4. J. Cell Biol. 164:701-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weiss, E. L., A. C. Bishop, K. M. Shokat, and D. G. Drubin. 2000. Chemical genetic analysis of the budding-yeast p21-activated kinase Cla4p. Nat. Cell Biol. 2:677-685. [DOI] [PubMed] [Google Scholar]
- 46.Whiteway, M. S., C. Wu, T. Leeuw, K. Clark, A. Fourest-Lieuvin, D. Y. Thomas, and E. Leberer. 1995. Association of the yeast pheromone response G protein beta gamma subunits with the MAP kinase scaffold Ste5p. Science 269:1572-1575. [DOI] [PubMed] [Google Scholar]
- 47.Zhao, Z., T. Leung, E. Manser, and L. Lim. 1995. Pheromone signalling in Saccharomyces cerevisiae requires the small GTP-binding protein Cdc42p and its activator CDC24. Mol. Cell. Biol. 15:5246-5257. [DOI] [PMC free article] [PubMed] [Google Scholar]