

Abstract
Solvent plays a significant role in determining the electrostatic potential energy of proteins, most notably through its favorable interactions with charged residues and its screening of electrostatic interactions. These energetic contributions are frequently ignored in computational protein design and protein modeling methodologies because they are difficult to evaluate rapidly and accurately. To address this deficiency, we report a revised form of the original Tanford–Kirkwood continuum electrostatic model [Tanford, C. & Kirkwood, J. G. (1957) J. Am. Chem. Soc. 79, 5333–5339], which accounts for the effects of solvent polarization on charged atoms in proteins. The Tanford–Kirkwood model was modified to increase its speed and to improve its sensitivity to the details of protein structure. For the 37 electrostatic self-energies of the polar side-chains in bovine pancreatic trypsin inhibitor, and their 666 interaction energies, the modified Tanford–Kirkwood potential of mean force differs from a computationally intensive numerical potential (DelPhi) by root-mean-square errors of 0.6 kcal/mol and 0.08 kcal/mol, respectively. The Tanford–Kirkwood approach makes possible a realistic treatment of electrostatics in computationally demanding protein modeling calculations. For example, pH titration calculations for ovomucoid third domain that model polar side-chain relaxation (including >2 × 1023 rotamer conformations of the protein) provide pKa values of unprecedented accuracy.
Substantial advances in protein design (1–3) and homology modeling (4–7) have resulted from the introduction of multicopy sampling algorithms (8), which simultaneously evaluate the fitness of multiple amino acids and side-chain rotamers at each position in a protein structure. Electrostatic energies in current studies have either been disregarded (1, 4–7, 9) or have been approximated by a highly damped Coulomb potential and empirical terms proportional to atomic solvent accessible surface areas (10). These methods ignore the substantial interactions of buried charges in proteins with solvent and incorrectly represent the screening of charge–charge interactions. Electrostatic energies in proteins are not small (11), and inaccurate treatments of the electrostatic potential would be expected to introduce large energetic errors into modeling calculations.
One reason for the omission of electrostatic energies is the lack of methods for their evaluation that are both rapid and accurate. Free-energy perturbation calculations with explicit solvent molecules (12), as well as finite-difference and boundary-element methods based on continuum solvent models (13), are thought to well approximate electrostatic free energies, but the extensive computation times they require prohibit their use in many protein modeling calculations. Fast analytical expressions for the electrostatic potential based on the Coulomb field approximation and the generalized Born equation (14) provide remarkably accurate small-molecule solvation energies but give rise to large errors when applied to bulky solutes such as proteins.
To address these problems, we have revisited the classic Tanford–Kirkwood (TK) electrostatic model (15). The basis for the TK model is conceptually simple (Fig. 1). Regions of space occupied by solvent and solute are treated as dielectric continua. The solvent-excluded volume of the protein is equated to a sphere of low-dielectric material, and the solvent is equated to a surrounding high-dielectric material. Single charges and charge pairs are mapped from the protein into the sphere, and their interactions are evaluated in the spherical geometry by using Kirkwood’s analytical series solution to the Poisson–Boltzmann equation (16).
Figure 1.

The TK model. The TK model equates the solvent-excluded volume of a solute (yellow) with a sphere of low dielectric material. Atoms in the solute (black and gray circles) are mapped to corresponding locations in the sphere. Electrostatic energies can be calculated rapidly in the simplified spherical geometry. Fractional solvent-accessible surface areas are determined for a series of concentric probe shells (dashed lines) centered on each charged atom of the solute (black circles). Charged atoms are mapped into the sphere such that a weighted average of the probe-shell accessible surface area is identical in the solute and sphere geometries.
We have made three alterations to the TK model to facilitate its use in protein modeling. First, we replace the original TK point-charge representation by shell charges, so that the electrostatic self energies of buried atoms in proteins can be calculated in a well defined manner (11). Second, we map charges between the protein and sphere geometries on the basis of a Coulomb field integral (17, 18), making the TK model sensitive to the structural details of the protein in the vicinity of a charge. Finally, we substitute Kirkwood’s equations with a complete and precise image-charge solution to the potential of a charge in a spherical dielectric cavity, which allows fast evaluation of electrostatic self energies and interaction energies (19–21). The modified Tanford-Kirkwood (MTK) model matches the speed of analytical small-molecule electrostatic methods but accurately reproduces in proteins the electrostatic free energies of computationally intensive numerical methods.
MATERIALS AND METHODS
Definitions.
The symbols R, ɛs, and ɛp denote, respectively, the radius of the low-dielectric sphere (LDS) used in the TK model, the dielectric constant assigned to the solvent, and the dielectric constant assigned to the protein interior. The symbols qi, bi, and di denote the charge, Born radius, and depth (distance to the surface of the LDS; negative when an atom is outside the sphere) of atom i. The symbol rij denotes the distance between atoms i and j. The symbols Sk and dSk denote the geometric-mean radius and thickness of a probe shell indexed k. The symbols ℱki, pro and ℱki, sph denote the fractional surface area of probe shell k concentric with atom i that falls inside the volume of low dielectric material in the protein and sphere geometries respectively. D⃗(x⃗) and Φ(x⃗) denote the dielectric displacement field and electrostatic potential as functions of spatial position (we assume a linear, osotropic dielectric medium). The self energy of atom i, Wi, is taken to be the electrostatic free energy with atom i charged and all other atoms of the solute neutral. The interaction potential, Iij, of atoms i and j is defined Iij = Wij − Wi − Wj.
Overview.
Solute atoms are modeled as shells of charge (total charge qi) characterized by a Born radius (bi). The electrostatic free energy, WElec, is expressed as a sum over interactions between shells: WElec = 1/2 Σi,j qiΦj. To determine qtΦj, charges i and j are mapped from the solute into the LDS, where Φj is approximated by the potentials of a set of suitably chosen image charge shells (of charge qjim and radii bjim). If neither shell i nor j crosses the LDS boundary, then qiΦj = Σshellsimage qiqjim/αijim, where
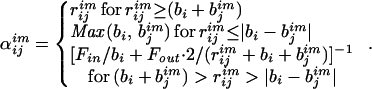 |
1 |
Fin/Fout denotes the surface fraction of image shell j that lies inside/outside of shell i.
Shell charges crossing the LDS boundary are treated as transparent to the two dielectric media (22) and are divided by the boundary into two independent charge caps. Each cap is replaced by a set of image caps, whose electrostatic potentials are evaluated according to Jackson (page 128 of ref. 19; available as supplemental data on the PNAS web site, www.pnas.org). If shells i and j both cross the dielectric boundary with rij < (bi + bj + 2 Å), the potential of one shell is sampled over the surface of the other shell. To accelerate calculations, all interactions that occur at rij ≥ (bi + bj + 2 Å) are treated as though between point charges, regardless of the proximity of either i or j to the dielectric interface.
Charge Mapping.
For an atom i in a solute molecule, we seek a position in the LDS that preserves electrostatic self energy expressed as a spatial integral over energy density:
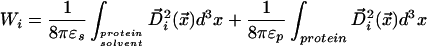 |
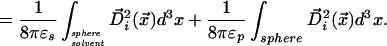 |
2 |
We replace the electric displacement with the displacement field of a point charge in a uniform dielectric medium appropriate to the domain of integration. This simplification is known as the Coulomb field approximation; implications of its use are discussed in refs. 17 and 18.
The integrals in Eq. 2 are approximated by discretely sampling points on a series of shells external to and concentric with atom i (Fig. 1) (supplementary material to ref. 15). Each point may be defined to be located in solvent or solute by using the prescription of ref. 23. Because of the spherical symmetry of the Coulomb field, knowledge of the fraction of each shell that is within the solute is sufficient to determine the self energy:
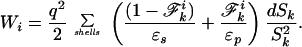 |
3 |
This approximate Wi is used only for mapping; a more accurate self-energy is derived by using the image charge solution described below.
We define two methods for mapping atoms in proteins. Fine mapping is used for interactions within amino-acid residues whereas coarse mapping is used for interactions between residues.
Fine mapping.
To map a single atom into the LDS, we find the di and R, which minimize the quantity
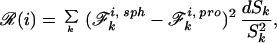 |
4 |
by using a golden section search (24). This is equivalent to demanding that the Coulomb field integral be preserved between geometries. Pairs of charges i j are mapped into the LDS by minimizing at fixed rij the quantity (ℛ(i) + ℛ(j)) with respect to di, dj, and R. We accomplish this via a Powell search (24). For pairs of charges, ℱ ki, sph is defined to be the fraction of shell k inside either the LDS or atom j (see supplemental data at www.pnas.org). Equations for computing this three-body ℱki, sph have been described by Richmond (25).
Coarse mapping.
We fix R such that 4/3πR3 gives the Connolly volume of the protein (26). For each atom, we minimize the quantity
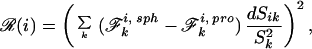 |
5 |
yielding a complete set of dis. The mapping of pairs of charges i j is geometrically specified by di, dj, R, and rij. There exist rare instances in which rij is incompatible with di, dj, and R. On these occasions, when rij < |dt − dj| or rij > (2R − di − dj), we adjust di by D⋅|di|/(|di| + |dj|) and dj by D⋅|dj|/(|di|+|dj|), where D = |di −dj| − rij in the former case and D = rij − 2R + di + dj in the latter. This method of partitioning D preferentially modifies the depths of atoms furthest from the dielectric boundary.
Image-Charge Solution.
Let xq, xobs denote the radial coordinates of a real charge and an observation point relative to the LDS center. The potential inside the LDS (xobs < R) due to a charge inside the dielectric interface (xq < R) is expressed exactly as a series of Legendre polynomials, which may be decomposed into Coulombic and solvent reaction contributions. It has been shown that the reaction potential can be approximated by an image point charge (20), either alone or in conjunction with the image shell charge at the dielectric boundary (21). Following Abagyan and Totrov (21), we approximate the electric potential at xobs < R due to a charge at xq < R with two point charges and a shell charge at the dielectric boundary:
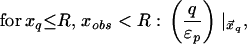 |
6a |
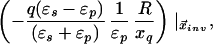 |
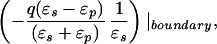 |
where xinv denotes the inverse point, colinear with xq and the LDS center, at radius R2/xq. If either xq > R or xobs > R, the series solution for the potential takes on a different form. Analogous manipulations yield the following image charge sets, which define over all space the potential of arbitrarily placed charges:
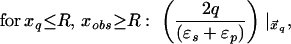 |
6b |
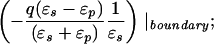 |
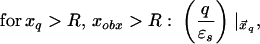 |
6c |
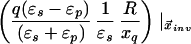 |
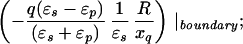 |
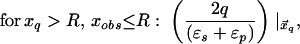 |
6d |
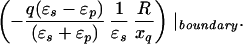 |
Application of Eqs. 6a–6d to charge shells and caps gives rise to image shells and caps (see supplementary material). The electrostatic interaction of atoms i and j is obtained by summing in vacuo the interactions of atom i with the appropriate charge set representing the potential generated by atom j.
The image charge method does not account for salt effects. In principle, these effects could be incorporated in the MTK model by recourse to the original polynomial expressions for the electrostatic free energy derived by Kirkwood (16).
Small Molecules and Bovine Pancreatic Trypsin Inhibitor (BPTI).
All small-molecule coordinates were kindly provided by D. Sitkoff (Bristol-Myers Squibb). Free energies of cavity formation in water for the small molecule were taken directly from ref. 27. For BPTI studies, the 4PTI structure (28) with no bound water molecules was used. Side-chain self energies were computed as the electrostatic free energy of the charged side chain in an otherwise neutral protein. Side-chain interaction energies were computed as the difference between the electrostatic free energy with both side chains charged and the sum of two side-chain self energies. DelPhi calculations were carried out on a 0.34-Å grid at 0 ionic strength with full Coulombic boundary conditions and a 1.4-Å probe sphere. The electrostatic free energy was evaluated as 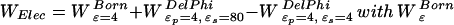 = 1/2Σ i,jqiqj/ɛαijand αijdefined by Eq. 1. DelPhi reaction-field energies were used for the test-charge calculations. Grid focusing and the DelPhi grid energies (29) were used for calculation of side-chain self energies and interaction energies in BPTI.
= 1/2Σ i,jqiqj/ɛαijand αijdefined by Eq. 1. DelPhi reaction-field energies were used for the test-charge calculations. Grid focusing and the DelPhi grid energies (29) were used for calculation of side-chain self energies and interaction energies in BPTI.
Timings.
The time (Ttotal) required to obtain electrostatic energies for a modeling calculation can be written in two terms that grow linearly and quadratically with the total number of rotamers n:
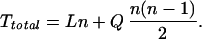 |
7 |
At a minimum, L and Q represent the average times required to calculate rotamer self and interaction energies respectively. L also includes the time spent obtaining depths for the MTK model. From the BPTI calculations, we have for the MTK model L = 11 sec/residue and Q = 0.13 sec/pair. For DelPhi, we find L = 111 sec/residue and Q = 106 sec/pair. The quadratic term dominates in most multicopy sampling problems (n > 170). In this regime, the MTK potential is evaluated nearly three orders of magnitude faster than the DelPhi potential.
pKa Values.
For pKa calculations, the protein was described by a conformational ensemble consisting of a fixed backbone and a probability-weighted set of allowable rotamers at each position. Backbone coordinates were taken from 1TUR (30) or 1PPF (31), as were side-chain coordinates in calculations using fixed structures. The rotamer library of Tuffery et al. (32), as modified by Koehl and Delarue (6), was used to represent side-chain conformational freedom in calculations incorporating side-chain relaxation. Side-chain flexibility was modeled for all amino acids except alanine, glycine, isoleucine, leucine, proline, and valine. Side chain coordinates were built by using charmm19 geometric parameters (33). Protons for acidic side chains were placed trans to the preceding methylene group. One deprotonated rotamer was included for each protonated rotamer to prevent entropic bias for the protonated state (34).
The energy function for all calculations consisted of the MTK electrostatic potential, W Elec, the charmm19 Lennard–Jones potential (33), W LJ, and a protonation potential, W H,p.
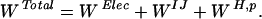 |
8 |
The protonation potential is derived from a thermodynamic cycle connecting a titrating site in a protein to an equivalent site in an isolated N-formyl, N-methyl amino amide (FMAA) of known pKa (35, 36) (see supplemental data). The protonation potential consists of the transfer free energy of the deprotonated FMAA from the protein into aqueous solution, W deprotonatedp→aq, the free energy of protonation of the FMAA in aqueous solution, W H,aq, and the transfer free energy of the protonated FMAA back into the protein, W protonatedaq→p:
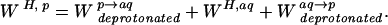 |
9 |
W H,aq is evaluated as 2.3⋅RT(pH − IpKa), where IpKa is the intrinsic pKa of the appropriate FMAA. Transfer free energies were calculated as the difference in the potential energy of the FMAA in the protein and free in solution. In all cases, the φ and ψ dihedral angles of the isolated FMAA were set to the values in the protein, and the side-chain dihedral angles were set to the values of the most commonly occurring rotamer. Intrinsic pKa values for the formulated amino amides were assigned as Glu, 4.4; Asp, 4.0; His, 6.3; peptide C terminus, 3.8 (37).
A mean-field method (6) was used to refine the rotamer probabilities such that the free energy of the ensemble was minimized. Because of the inclusion of a protonation potential, the rotamer distributions that minimized the free energy varied with pH. Each titrating site’s pKa was assigned as the pH at which protonated and deprotonated states were equally populated. These values represent averages over many rotamer states of the protein, each with a likelihood equal to the product of the individual rotamer probabilities (see ref. 6).
For all calculations, experimentally determined atomic coordinates were used to define the shape of the low-dielectric region. Consequently, the effects of rotamer changes on the dielectric boundary were not treated, and the pairwise factorization of the electrostatic potential was preserved.
RESULTS
Tests of the Modified TK Potential.
We have introduced modifications into the TK electrostatic model that improve charge representation, charge mapping, and the speed with which the electric potential can be evaluated. Our first test of the MTK potential was to calculate small molecule vacuum-to-water transfer free energies (Table 1). Using the parameters for solvation energy (PARSE) set of Born radii and partial charges (27), the calculated transfer free energies for a set of 53 canonical compounds (the training set of ref. 27) ranged over 80 kcal/mol with rms and (maximum) errors from experimentally measured values of 1.05 (3.54) kcal/mol.
Table 1.
Vacuum to water transfer free energies for small-molecule side-chain analogues
| Residue | Small molecule | ΔGexp | ΔGDelPhi | ΔGMTK | ΔGerror |
|---|---|---|---|---|---|
| Arg+ | N-propyl guanidinium | – | −66.07 | −64.49 | – |
| Lys+ | N-butyl ammonium | −69.24 | −69.44 | −69.11 | 0.13 |
| His+ | Methyl imidazolium | −64.13 | −64.36 | −64.81 | −0.68 |
| His0 | Methyl imidazole | −10.25 | −10.22 | −9.32 | 0.93 |
| Asp− | Acetate | −80.65 | −80.44 | −79.05 | 1.60 |
| Asp0 | Acetic acid | −6.70 | −6.63 | −5.57 | 1.13 |
| Asn | Acetamide | −9.72 | −9.76 | −8.85 | 0.87 |
| Glu− | Propionate | −79.12 | −79.21 | −77.53 | 1.59 |
| Glu0 | Propionic acid | −6.47 | −6.41 | −5.25 | 1.22 |
| Gln | Propionamide | −9.42 | −9.42 | −8.55 | 0.87 |
| Cys− | Methylthiol ion | −76.79 | −76.66 | −75.85 | 0.94 |
| Cys0 | Methylthiol | −1.24 | −1.35 | −1.08 | 0.16 |
| Tyr− | p-cresol ion | −75.01 | −74.88 | −71.90 | 3.11 |
| Tyr0 | p-cresol | −6.13 | −6.11 | −4.74 | 1.39 |
| Ser | Methanol | −5.08 | −5.44 | −4.83 | 0.25 |
| Thr | Ethanol | −4.90 | −5.02 | −4.55 | 0.35 |
| Met | Methylethyl sulfide | −1.49 | −1.46 | −1.07 | 0.42 |
| Trp | Methyl indole | −5.91 | −5.84 | −4.76 | 1.15 |
| Phe | Toluene | −0.76 | −0.77 | −0.37 | 0.39 |
| BB | N-methyl acetamide | −10.08 | −10.00 | −9.53 | 0.55 |
Shown are experimental and calculated vacuum (ɛp = 2, ɛs = 1) to water (ɛp = 2, ɛs = 80) transfer free energies (kcal/mol) for small-molecule analogs of the amino-acid side chains. ΔGexp indicates the experimental transfer free energy. ΔGDelPhi indicates the transfer free energy reported by Sitkoff et al. (27) using the DelPhi program (29). ΔGMTK and ΔGerror indicate the MTK transfer free energy and its error with respect to the experimentally measured value.
We next tested the accuracy of the MTK model in the protein BPTI. All energies calculated by the MTK method were compared with values derived from the program DelPhi (29), which is based on a finite-difference solution to the Poisson–Boltzmann equation (FDPB). The protein dielectric constant was set to 4 and the solvent dielectric constant to 80. In Fig. 2B, the self-energy of a 1-Å shell charge passing along a line through the center of mass of BPTI is plotted against position. The MTK potential of mean force exhibits close agreement with the FDPB potential, even in the rapidly varying region near the molecular surface.
Figure 2.

The MTK potential in BPTI. (A) To test the MTK electrostatic potential, a grid of 1-Å unit test charges was superimposed onto the protein BPTI. The edges of the grid (green spheres and lines) are aligned along BPTI’s principal axes of inertia. The grid originates at the center of mass of BPTI. Two points located on the axis of inertia with the largest moment also are shown (magenta spheres). (B) The self energy of a 1-Å unit test charge is plotted at positions along a principal axis of inertia of BPTI. The DelPhi self energy is indicated by a solid line. Self energies calculated by MTK (solid circles), the Gaussian-atom Coulomb field integral of ace (18) (open circles), and mimel (21) (cross hatch) are also shown. The fractional solvent-accessible surface area (SAS) of the test charge (dashed line) is plotted against the vertical axis on the right.
As a further test, we superimposed a 4-Å grid of test charges onto BPTI originating at the center of mass and aligned along the principle axes of inertia of the protein (Fig. 2A). A 1-Å shell with one electron unit of charge was placed at each grid point. The MTK self energies of the 441 test charges give an R-factor with respect to the DelPhi self energies of 8% (R-factor = Σi|WiMTK − WiDelPhi|/Σi|WiDelPhi|). Thus, the summed absolute values of self-energy errors represented 8% of the cumulative DelPhi self-energy. By comparison, the Coulomb field integral as implemented in the analytical algorithm ace (18) gives a self-energy R-factor with respect to DelPhi of 25%. As illustrated in Fig. 2B, the Coulomb field integral tends to underestimate self-energies for buried charges and overestimate them for surface charges.
The interaction energies of each test charge with a test charge at the center of mass also were evaluated. The MTK interaction energies give an R-factor with respect to DelPhi of 15% (i.e., the cumulative unsigned error is 15% of the summed unsigned interaction energies). By comparison, the generalized Born equation (14) (based on MTK self-energies) gives an R-factor of 42%. The generalized Born equation reliably describes the interaction of charges separated by long distances in proteins, but it incurs large errors for nearby charges.
Finally, we measured the electrostatic self- and interaction energies of the entire set of 37 polar side chains in BPTI. These values represent the electrostatic inputs required for multicopy sampling algorithms. Relative to DelPhi, the MTK potential of mean force gives rms and (maximum) errors of 0.6 (1.27) kcal/mol for the 37 self energies (Fig. 3A) and rms and (maximum) errors of 0.08 (1.21) kcal/mol for the 666 side-chain interaction energies (Fig. 3B).
Figure 3.

Comparison of electrostatic energies calculated by using the FDPB solver of DelPhi (29) (x axis) with energies calculated by using the MTK potential (y axis). The diagonal is shown as a solid line. (A) Electrostatic self energies for the 37 side chains of BPTI. (B) Electrostatic interaction energies for the 666 pairs of polar side chains in BPTI.
pKa Calculations.
As a functional application of the MTK model, we calculated pKa values for titratable groups in turkey ovomucoid third domain, which have been measured experimentally at low ionic strength (38, 39). Protonated and deprotonated forms of each titratable side-chain were treated as rotamers of the same amino acid. The free energy for side-chain protonation in the protein was calculated relative to the free energy for protonation of the corresponding N-formyl N-methyl amide free in solution (40) (see Materials and Methods). At pH values between 1 and 10, the protonation states of the interacting titratable groups were relaxed by a conventional mean-field optimization algorithm (6). The pH at which each titratable group became 50% deprotonated was designated as its pKa. Although the mean-field approximation is known to give imperfect titration curves for strongly interacting sites of degenerate pKa (36, 41), we wished to evaluate the electrostatic potential under conditions used for protein side-chain repacking studies.
Following the convention of ref. 37, the protein conformation was represented by fixed coordinates, the protein dielectric constant was evaluated as 20, and atoms were assigned Born radii and partial charges according to the PARSE parameter set (27). The pKa values for ovomucoid third domain calculated with the modified TK model give an rms deviation from the experimental values of 0.62 pH units and a maximum error of 1.0 pH unit. These values compare favorably (Table 2) with the best results obtained by conventional FDPB methods [rms error 0.59 pH units (37)]. The MTK pKa values are significantly better than the null model, which assumes no pKa shifts from intrinsic values (rms error 1.1 pH units).
Table 2.
Calculated pKa values for turkey ovomucoid third domain
| Site
|
rms Error | ||||||
|---|---|---|---|---|---|---|---|
| Asp 7 | Glu 10 | Glu 19 | Asp 27 | Glu 43 | CTER 56 | ||
| Experimental* | 2.7 | 4.1 | 3.2 | 2.3 | 4.8 | ≤2.7 | – |
| Null model† | 4.0 | 4.4 | 4.4 | 4.0 | 4.4 | 3.8 | 1.12 |
| FDPB‡ (NMR§, ɛp¶ = 20) | 3.5 | 3.3 | 2.9 | 3.1 | 4.5 | 2.3 | 0.59 |
| MTK‖ (NMR, ɛp = 20) | 3.7 | 3.4 | 2.9 | 2.8 | 4.8 | 3.4 | 0.62 |
| FDPB (X-ray**, ɛp = 20) | 2.9 | 3.4 | 2.6 | 3.6 | 4.4 | 2.4 | 0.69 |
| MTK (X-ray, ,ɛp = 20) | 3.0 | 3.5 | 2.7 | 3.7 | 4.7 | 3.2 | 0.70 |
| MTK (Relaxed‡‡, ɛp = 20) | 2.0 | 3.8 | 2.1 | 3.0 | 4.6 | 2.7 | 0.61 |
| FDPB (X-ray, ɛp = 4) | – | – | – | – | – | – | 1.60 |
| MTK (X-ray, ɛp = 4) | 2.7 | 3.3 | −0.2 | −0.7 | 5.0 | 3.3 | 1.90 |
| MTK (Relaxed, ɛp = 4) | 2.1 | 4.0 | 3.1 | 2.9 | 5.6 | 2.6 | 0.48 |
Experimentally measured pKa values (38).
Intrinsic model compound pKa values (37).
FDPB: literature pKa values calculated by using an FDPB electrostatic potential of mean force (37).
NMR: pKa values were determined for 12 NMR structures [1 TUR (30)] and were averaged.
ɛp: the protein dielectric constant used for the calculation.
MTK: pKa values calculated by using the MTK electrostatic potential with PARSE charge parameters (27).
X-ray: pKa values determined by using the X-ray crystal structure of turkey ovomucoid third domain [1PPF (31)].
Relaxed: pKa values determined by using backbone coordinates from the X-ray crystal structure of turkey ovomucoid third domain [1 PPF (31)]. Rotamer conformations for all polar side-chains (32) were relaxed to their free-energy minima by a self-consistent mean-field algorithm (7). The conformational potential consisted of the param19 Lennard–Jones potential (33), the MTK potential, and a protonation potential (see Materials and Methods).
Side-Chain Conformational Freedom.
By coupling the speed of the MTK methodology with a rotamer repacking algorithm, we have examined how global side-chain motions perturb the calculated pKa values of titratable sites in ovomucoid third domain (Table 2). All polar side chains were allowed full rotamer freedom, giving rise to >2 × 1023 possible rotamer conformations of the protein. At pH values between 1 and 10, the rotamer distribution was refined to the free-energy minimum defined by the MTK potential, the Lennard–Jones potential, and a protonation potential (see Materials and Methods). The pH at which each titratable group became 50% deprotonated was designated as its pKa. With a protein dielectric constant of 20, side-chain conformational sampling produced modest improvement in the agreement between calculated and observed pKa values (Table 2) (Δrms = 0.09 pH units). However, when the protein interior was assigned a dielectric constant of 4, the pKa values calculated with side-chain flexibility were dramatically improved relative to pKa’s calculated with a static structure (Δrms = 1.42 pH units), yielding computed values of unprecedented accuracy (rms error 0.48 pH units). In conjunction with explicitly modeled side-chain motion, the dielectric response of the protein is better represented by a constant of 4 than by a constant of 20.
DISCUSSION
To fully represent the electrostatic properties of proteins, it is necessary to model protein structural plasticity. For example, although pKa calculations on proteins have generally been carried out with static structures, charge changes at titratable sites are known to induce substantial rearrangements in protein side-chain conformation (42). These structural adjustments presumably diminish the energetic penalties associated with protonation and deprotonation. Unfortunately, the computation times required by conventional electrostatic potentials prohibit modeling of such global side-chain motions. Consequently, fixed-structure calculations have been performed. The dielectric response that would result from side-chain rearrangements has been represented implicitly by assigning an artificially large dielectric constant to the protein. Whereas the bulk dielectric constant of crystalline acetamide (a molecular analogue of the protein backbone) is 4, a protein dielectric constant of 20 is conventionally used for fixed-structure pKa calculations (37). Because it subsumes the effects of structural plasticity, the protein dielectric constant has come to be viewed as an adjustable parameter that depends on spatial position within the protein and on the electrostatic property being studied (43). Efforts to incorporate limited side-chain flexibility into electrostatic calculations have been reported [including allowance for hydroxyl rotamer relaxation (34), inclusion of multiple dihedral conformers at titrating sites (35, 44), and sampling of multiple protein conformations along a molecular dynamics trajectory (45–49)], but no methods exist to model the global rearrangements in protein structure that accompany pH changes.
The MTK potential was developed specifically to allow an accurate treatment of electrostatics in multicopy sampling calculations that treat global variation in side-chain chemistry and conformation. Using the MTK potential in conjunction with a side-chain repacking algorithm, we demonstrate that changes in the rotamer populations of polar residues contribute substantially to the dielectric response of a protein. The accuracy of calculated pKa values for ovomucoid third domain are improved by modeling these rearrangements. Moreover, when structural reorganization is treated microscopically, the remaining dielectric response of the protein is well represented by the dielectric constant of bulk crystalline acetamide (ɛp = 4). Macroscopic and microscopic estimates of the protein dielectric constant are thus reconciled. When the effects of protein structural reorganization are factored out of the electrostatic model, the protein dielectric constant can be treated as a uniform transferable property of the protein solute, as envisioned in the classical theory of dielectrics. We expect that the side-chain repacking approach presented here will prove generally useful for the computational analysis of sequence mutations in proteins. Indeed, fixed-structure calculations predict the destabilization resulting from single charge-to-neutral residue substitutions to be much larger than is measured experimentally (50).
Current protein design methods sidestep the consideration of large electrostatic energies by restricting polar residues to the surface of designed proteins (9, 10, 51). This simplification would appear to be justified by the argument that buried polar interactions universally destabilize proteins (52). In at least two situations, however, buried charges and hence accurate electrostatic potentials are required. First, buried polar residues are essential for enforcing structural specificity in some protein folds, even if they are also destabilizing. For example, core asparagine residues have been shown to control the strand number (53), helix orientation (54, 55), and specificity of helix association (56) in dimeric coiled-coils. Furthermore, buried polar residues play a central role in catalysis by protein enzymes (57). As the breadth of computational protein design grows to encompass protein function and the specificity of protein-protein interactions, the electrostatic potential will lie at the heart of the problem.
Supplementary Material
Acknowledgments
The authors thank P. Koehl and R. L. Baldwin for stimulating discussion and criticism throughout the course of this work and D. Sitkoff for generously providing small-molecule coordinates. This research was supported by a junior faculty award from the Howard Hughes Medical Institute and a grant from the Chicago Community Trust to P.B.H. P.B.H. is a Searle scholar and a Terman fellow.
ABBREVIATIONS
- BPTI
bovine pancreatic trypsin inhibitor
- TK
Tanford–Kirkwood
- MTK
modified TK
- LDS
low-dielectric sphere
- FDPB
finite-difference solution to the Poisson–Boltzmann equation
- FMAA
N-formyl, N-methyl amino amide
References
- 1.Desjarlais J R, Handel T M. Protein Sci. 1995;4:2006–2018. doi: 10.1002/pro.5560041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dahiyat B I, Mayo S L. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 3.Saven J G, Wolynes P G. J Phys Chem. 1997;101:8375–8389. [Google Scholar]
- 4.Desmet J, Maeyer M D, Hazes B, Lasters I. Nature (London) 1992;356:539–542. doi: 10.1038/356539a0. [DOI] [PubMed] [Google Scholar]
- 5.Lee C. J Mol Biol. 1994;236:918–939. doi: 10.1006/jmbi.1994.1198. [DOI] [PubMed] [Google Scholar]
- 6.Koehl P, Delarue M. J Mol Biol. 1994;239:249–275. doi: 10.1006/jmbi.1994.1366. [DOI] [PubMed] [Google Scholar]
- 7.Koehl P, Delarue M. Nat Struct Biol. 1995;2:163–170. doi: 10.1038/nsb0295-163. [DOI] [PubMed] [Google Scholar]
- 8.Elber R, Karplus M. J Am Chem Soc. 1990;112:9161–9175. [Google Scholar]
- 9.Harbury P B, Plecs J J, Tidor B, Alber T, Kim P S. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 10.Dahiyat B I, Sarisky C A, Mayo S L. J Mol Biol. 1997;273:789–796. doi: 10.1006/jmbi.1997.1341. [DOI] [PubMed] [Google Scholar]
- 11.Warshel A, Russell S T. Q Rev Biophys. 1984;17:283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- 12.Allen M P, Tildesley D J. Computer Simulation of Liquids. Oxford: Oxford Univ. Press; 1989. [Google Scholar]
- 13.Honig B, Nicholls A. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 14.Still W C, Tempczyk A, Hawley R C, Hendrickson T. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 15.Tanford C, Kirkwood J G. J Am Chem Soc. 1957;79:5333–5339. [Google Scholar]
- 16.Kirkwood J G. J Chem Phys. 1934;2:351–361. [Google Scholar]
- 17.Schaefer M, Froemmel C. J Mol Biol. 1990;216:1045–1066. doi: 10.1016/S0022-2836(99)80019-9. [DOI] [PubMed] [Google Scholar]
- 18.Schaefer M, Karplus M. J Phys Chem. 1996;100:1578–1599. [Google Scholar]
- 19.Jackson J D. Classical Electrodynamics. New York: Wiley; 1975. [Google Scholar]
- 20.Friedman H L. Mol Phys. 1975;29:1533–1543. [Google Scholar]
- 21.Abagyan R, Totrov M. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 22.Gilson M K, Rashin A, Fine R, Honig B. J Mol Biol. 1985;184:503–516. doi: 10.1016/0022-2836(85)90297-9. [DOI] [PubMed] [Google Scholar]
- 23.Beroza P, Fredkin D R. J Comput Chem. 1996;17:1229–1244. [Google Scholar]
- 24.Press W H, Teukolsky S A, Vetterling W T, Flannery B P. Numerical Recipes in C: The Art of Scientific Computing. New York: Cambridge Univ. Press; 1992. [Google Scholar]
- 25.Richmond T J. J Mol Biol. 1984;178:63–89. doi: 10.1016/0022-2836(84)90231-6. [DOI] [PubMed] [Google Scholar]
- 26.Connolly M L. J Am Chem Soc. 1985;107:1118–1124. [Google Scholar]
- 27.Sitkoff D, Sharp K A, Honig B. J Phys Chem. 1994;98:1978–1988. [Google Scholar]
- 28.Marquart M, Walter J, Deisenhofer J, Bode W, Huber R. Acta Crystallogr B. 1983;39:480–490. [Google Scholar]
- 29.Nicholls A, Honig B. J Comput Chem. 1991;12:435–445. [Google Scholar]
- 30.Krezel A M, Darba P, Robertson A D, Fejzo J, Macura S, Markley J L. J Mol Biol. 1994;242:203–214. doi: 10.1006/jmbi.1994.1573. [DOI] [PubMed] [Google Scholar]
- 31.Bode W, Wei A Z, Huber R, Meyer E, Travis J, Neumann S. Embo J. 1986;5:2453–2458. doi: 10.1002/j.1460-2075.1986.tb04521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tuffery P, Etchebest C, Hazout S, Lavery R. J Biomol Struct Dyn. 1991;8:1267–1289. doi: 10.1080/07391102.1991.10507882. [DOI] [PubMed] [Google Scholar]
- 33.Brooks B R, Bruccoleri R E, Olafson B D, States D J, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 34.Alexov E G, Gunner M R. Biophys. 1997;72:2075–2093. doi: 10.1016/S0006-3495(97)78851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.You T J, Bashford D. Biophys J. 1995;69:1721–1733. doi: 10.1016/S0006-3495(95)80042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bashford D, Karplus M. J Phys Chem. 1991;95:9556–9561. [Google Scholar]
- 37.Antosiewicz J, McCammon J A, Gilson M K. Biochemistry. 1996;35:7819–7833. doi: 10.1021/bi9601565. [DOI] [PubMed] [Google Scholar]
- 38.Schaller W, Robertson A D. Biochemistry. 1995;34:4714–4723. doi: 10.1021/bi00014a028. [DOI] [PubMed] [Google Scholar]
- 39.Swint-Kruse L, Robertson A D. Biochemistry. 1995;34:4724–4732. doi: 10.1021/bi00014a029. [DOI] [PubMed] [Google Scholar]
- 40.Yang A S, Gunner M R, Sampogna R, Sharp K, Honig B. Proteins. 1993;15:252–265. doi: 10.1002/prot.340150304. [DOI] [PubMed] [Google Scholar]
- 41.Gilson M K. Proteins. 1993;15:266–282. doi: 10.1002/prot.340150305. [DOI] [PubMed] [Google Scholar]
- 42.Gursky O, Badger J, Li Y L, Caspar DLD. Biophys J. 1992;63:1210–1220. doi: 10.1016/S0006-3495(92)81697-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warshel A, Papazyan A. Curr Opin Struct Biol. 1998;8:211–217. doi: 10.1016/s0959-440x(98)80041-9. [DOI] [PubMed] [Google Scholar]
- 44.Beroza P, Case D A. J Phys Chem. 1996;100:20156–20163. [Google Scholar]
- 45.Bashford D, Gerwert K. J Mol Biol. 1992;224:473–486. doi: 10.1016/0022-2836(92)91009-e. [DOI] [PubMed] [Google Scholar]
- 46.Zhou H X, Vijayakumar M. J Mol Biol. 1997;267:1002–1011. doi: 10.1006/jmbi.1997.0895. [DOI] [PubMed] [Google Scholar]
- 47.Sham Y Y, Chu Z T, Warshel A. J Phys Chem. 1997;101:4458–4472. [Google Scholar]
- 48.Sham Y Y, Muegge I, Warshel A. Biophys J. 1998;74:1744–1753. doi: 10.1016/S0006-3495(98)77885-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.vanVlijmen H W T, Schaefer M, Karplus M. Proteins. 1998;33:145–158. doi: 10.1002/(sici)1097-0134(19981101)33:2<145::aid-prot1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 50.Meeker A K, Garcia-Moreno B, Shortle D. Biochemistry. 1996;35:6443–6449. doi: 10.1021/bi960171+. [DOI] [PubMed] [Google Scholar]
- 51.Bryson J W, Desjarlais J R, Handel T M, DeGrado W F. Protein Sci. 1998;7:1404–1414. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hendsch Z S, Tidor B. Protein Sci. 1994;3:211–226. doi: 10.1002/pro.5560030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harbury P B, Zhang T, Kim P S, Alber T. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 54.Lumb K J, Kim P S. Biochemistry. 1995;34:8642–8648. doi: 10.1021/bi00027a013. [DOI] [PubMed] [Google Scholar]
- 55.Oakley M G, Kim P S. Biochemistry. 1998;37:12603–12610. doi: 10.1021/bi981269m. [DOI] [PubMed] [Google Scholar]
- 56.Zeng X, Herndon A M, Hu J C. Proc Natl Acad Sci USA. 1997;94:3673–3678. doi: 10.1073/pnas.94.8.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shan, S. O. & Herschlag, D. (1999) Methods Enzymol, in press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.