

Abstract
Transcription factor-binding sites and the cis-regulatory modules they compose are central determinants of gene expression. We previously showed that binding site motifs and modules in proximal promoters can be used to predict a significant portion of mammalian tissue-specific transcription. Here, we report on a systematic analysis of promoters controlling tissue-specific expression in heart, kidney, liver, pancreas, skeletal muscle, testis and CD4 T cells, for both human and mouse. We integrated multiple sources of expression data to compile sets of transcripts with strong evidence for tissue-specific regulation. The analysis of the promoters corresponding to these sets produced a catalog of predicted tissue-specific motifs and modules, and cis-regulatory elements. Predicted regulatory interactions are supported by statistical evidence, and provide a foundation for targeted experiments that will improve our understanding of tissue-specific regulatory networks. In a broader context, methods used to construct the catalog provide a model for the analysis of genomic regions that regulate differentially expressed genes.
Keywords: cis-regulatory modules, tissue-specific regulation
Introduction
Reverse engineering mammalian transcriptional regulatory circuits can be achieved using systematic methodology that includes both computational and experimental techniques, working in tandem to generate, refine and verify hypotheses. Understanding tissue-specific transcription is a necessary step for extending regulatory circuit reverse-engineering efforts from single-cell eukaryotes to metazoans. We recently demonstrated that the information in proximal promoters can predict a significant portion of tissue-specific elevated or inhibited expression (Smith et al, 2006). Here, focusing on tissue-specific regulatory pattern identification and prediction accuracy instead of proof of existence, we use refined analysis and data curation methods to discover and catalog high-confidence regulatory interactions and sites. This catalog will assist experimental efforts to reverse engineer tissue-specific transcriptional regulatory networks from the bottom up.
Numerous techniques for analysis of regulatory sequences have been proposed, and the problem of module identification is now receiving due attention (Zhou and Wong, 2004; Gupta and Liu, 2005; Zhu et al, 2005). Previously characterized binding site motifs have been used to infer transcription factor function in certain tissues (Nelander et al, 2005). Xie et al (2005) identified conserved motifs across ortholog promoters of four mammalian genomes. Robertson et al (2006) describe cisRed, a database that integrates genome annotation data, homology data and genome alignments to identify motifs with conserved sites across mammals. We analyzed proximal promoters with evidence for tissue-specific regulation in order to identify tissue-specific motifs, modules and their sites in proximal promoters. We developed a new technique for characterizing tissue-specific modules that ensures that each module component significantly improves tissue-specific module enrichment.
We integrated multiple sources of expression data to identify reliable sets of transcripts that are under tissue-specific regulation in human and mouse. Using transcription start site (TSS) annotation in Cold Spring Harbor Mammalian Promoter Database (CSHLmpd) (Xuan et al, 2005), we compiled sets of proximal promoters corresponding to transcripts with evidence for specific regulation in the selected tissues. Our analysis was based on motifs discovered de novo (called novel motifs) using DME (Smith et al, 2005a) and DME-B (Smith et al, 2006), as well as previously characterized vertebrate binding-site motifs (called known motifs) from TRANSFAC (Matys et al, 2003) and JASPAR (Sandelin et al, 2004). We evaluated motifs according to enrichment in tissue-specific promoters relative to other promoters from the same species. We showed that motifs associated with factors with known tissue-specific roles rank high for enrichment, that motif ranks are significantly correlated between human and mouse and that this same set of motifs and their corresponding cis-elements are unlikely to be identified using traditional, order-preserving alignments of ortholog promoter sequences. We constructed modules of interacting motifs (both novel and known motifs), ensuring that each component contributed significantly to the enrichment of the whole module. We annotated tissue-specific promoters with predicted tissue-specific regulatory elements and demonstrated that these sites are in excellent agreement with experimentally annotated liver-specific sites in the human albumin promoter and skeletal-muscle-specific sites in the human α-actin promoter. Both promoters are particularly well annotated with experimentally verified tissue-specific regulatory elements and permit an informative comparison. In other tissues, we gave predicted sites for tissue-specific motifs in representative promoters. The complete data and analysis are available in TCat: The Catalog of Tissue-Specific Regulatory Motifs (http://rulai.cshl.edu/tcat).
Results
We describe first steps toward cataloging high-confidence tissue-specific motifs, modules and their sites. We first collected and integrated expression and function data from various sources, and identified transcripts that are likely to be under tissue specific regulation. We demonstrated that transcripts with evidence for tissue-specific regulation from multiple expression sources in one species (human or mouse) are significantly more likely to have evidence for tissue-specificity in the other species. We analyzed and annotated proximal-promoter sets in seven representative tissues from both human and mouse, demonstrating that motifs and predicted binding sites are in agreement with experimentally verified data and that analyses in human and mouse are significantly correlated. We also showed that the top-scoring sites in orthologous tissue-specific promoters from human and mouse rarely have significant conservation of site order, suggesting that comparative genomics alone may not be sufficient to decode the regulatory signals in these proximal promoters.
Transcripts under tissue-specific regulation
Few transcripts have expression restricted to a single tissue, but many transcripts appear to be regulated in a tissue-specific manner (Su et al, 2004), and the corresponding promoters are likely to contain tissue-specific regulatory elements. To circumvent problems associated with individual sources of information, we used a voting system that combined information about expression, function and tissue specificity from different sources. Table I gives the number of transcripts with single and multiple sources of evidence (votes) for tissue-specific regulation in each tissue. Orthologs of transcripts with multiple votes for tissue-specific regulation were more likely to have evidence for specific regulation in that tissue, suggesting that the false-positive rate for calling a transcript tissue specific is lower when based on multiple votes. The number of ortholog transcript pairs with multiple votes for tissue-specific regulation in both species ranged from 1 in CD4 T cells to 69 in liver. Table II lists genes and orthologous transcripts with votes for skeletal-muscle-specific regulation in both human and mouse. Gene and transcript lists for other tissues are given in Supplementary Section 1.5.
Table 1.
Ability of single versus multiple votes to predict tissue-specificity of a transcript's ortholog
| Human |
Mouse |
Common evidence |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Tissue | Multiple | Single | Multiple | Single | Multiple | Single | P-value | ||
| CD4 T-cells | 2 | 247 | 6 | 212 | 3/7 | 42.9% | 36/435 | 8.3% | 1.79E−02 |
| Heart | 28 | 260 | 105 | 560 | 35/122 | 28.7% | 102/766 | 13.3% | 3.78E−05 |
| Kidney | 43 | 188 | 172 | 540 | 42/200 | 21.0% | 66/706 | 9.3% | 1.74E−05 |
| Liver | 152 | 411 | 271 | 651 | 148/354 | 41.8% | 184/982 | 18.7% | 6.46E−17 |
| Pancreas | 31 | 186 | 47 | 313 | 26/61 | 42.6% | 75/450 | 16.7% | 9.93E−06 |
| Skeletal muscle | 49 | 394 | 141 | 681 | 52/174 | 29.9% | 137/1000 | 13.7% | 4.47E−07 |
| Testis | 38 | 287 | 446 | 668 | 67/471 | 14.2% | 86/923 | 9.3% | 4.09E−03 |
| Columns labeled ‘Multiple' and ‘Single' give the number of transcripts with multiple and single votes for specificity, respectively. Columns under ‘Common evidence' show the proportion of transcripts with single and multiple votes for tissue-specificity that have an ortholog with at least one vote for specificity in the same tissue. Excluding CD4 T-cells (which represents a small sample), human and mouse transcripts with multiple votes for tissue-specificity are significantly more likely to have an ortholog with at least one vote for specificity in the same tissue (P<0.01; Fisher's exact test). | |||||||||
Table 2.
Transcripts with multiple votes for tissue-specificity in both human and mouse skeletal muscle
| Symbol | Name | Human RefSeq | Mouse RefSeq | Votes |
|---|---|---|---|---|
| MYH2 | Myosin, heavy polypeptide 2 | NM_017534 | NM_144961 | 7 |
| TTID | Myotilin | NM_006790 | NM_021484 | 6 |
| TNNT3 | Troponin T type 3 | NM_006757 | NM_011620 | 6 |
| TNNC2 | Troponin C type 2 | NM_003279 | NM_009394 | 6 |
| MYBPC2 | Myosin binding protein C | NM_004533 | NM_146189 | 6 |
| HUMMLC2B | Fast skeletal myosin light chain 2 | NM_013292 | NM_016754 | 6 |
| ACTN2 | Actinin α2 | NM_001103 | NM_033268 | 5 |
| VAMP5 | Vesicle-associated membrane protein 5 | NM_006634 | NM_016872 | 4 |
| TRIP10 | Thyroid hormone receptor interactor 10 | NM_004240 | NM_134125 | 4 |
| TPM3 | Tropomyosin 3 | NM_153649 | NM_022314 | 4 |
| SGCG | Sarcoglycan γ | NM_000231 | NM_011892 | 4 |
| MYOD1 | Myogenic differentiation 1 | NM_002478 | NM_010866 | 4 |
| MYF6 | Myogenic factor 6 (herculin) | NM_002469 | NM_008657 | 4 |
| CKM | Creatine kinase, muscle | NM_001824 | NM_007710 | 4 |
| CACNG1 | Calcium channel, voltage-dependent γ1 | NM_000727 | NM_007582 | 4 |
| The ‘Votes' column gives the total number of votes for skeletal muscle-specificity in both human and mouse. Our analysis used promoter sets of size 100 for both tissues, including promoters that correspond to transcripts with a single vote for tissue-specific regulation. Tables for the remaining 6 tissues are given in supplementary material. |
Enrichment of known tissue-specific motifs
Knowledge of factors and corresponding binding sites that regulate tissue-specific transcription can be used to evaluate motif ranking. We measured motif enrichment in tissue-specific promoter sets using balanced error rates, evaluating motifs for their ability to distinguish tissue-specific sets (foreground sets) from background sets that are composed of non-tissue-specific promoter samples from CSHLmpd. Balanced error rates measure proportions of misclassified promoters after normalization of foreground and background sizes. We ranked motifs according to enrichment and determined whether the ranks assigned to binding-site motifs for factors with known tissue-specific roles are significantly elevated in the corresponding tissues. The results presented in Table III demonstrate that binding-site motifs for these factors ranked significantly high (P<0.01) according to a Wilcoxon signed-ranks test in almost all tissues tested. Excluding DBP in human liver and HNF-3 in mouse liver, these factors had evidence for expression in their respective tissues, and their binding-site motifs were highly enriched in our foreground sets. Results are summarized in Table IV, and the motifs with greatest enrichment in each tissue are given in TCat. Table IV also includes information for HNF-6 in liver. HNF-6 is a known liver regulator, but there is no evidence for its expression in liver based on our data, and its binding-site motif was not enriched in our liver foreground sets. In addition, C/G- or A/T-rich motifs are likely to be enriched in foreground sets that are C/G or A/T rich relative to promoter base composition. To eliminate this potential bias, we adjusted the GC content in background sets to match foreground sets. Some known tissue-specific motifs were identified as enriched only after GC content correction.
Table 3.
Significance of elevated ranks for motifs associated with important factors in liver, skeletal muscle and testis
| Tissue | Factors | Motifs | Human P-value | Mouse P-value |
|---|---|---|---|---|
| Liver | HNF-1, HNF-3, HNF-4, C/EBP, DBP | 68 | 2.72E−18 | 4.19E−12 |
| Skeletal muscle | MEF-2, SRF, Myogenin, Sp1 | 45 | 1.33E−14 | 2.29E−5 |
| Testis | SRY, CREM, RFX | 30 | 0.087 | 1.89E−4 |
| Motifs give the total number of motifs associated with the listed factors. |
Table 4.
Evidence for expression and classification quality of binding-site motifs for factors with known tissue-specific regulatory roles
| Expressed |
Classifies |
|||||||
|---|---|---|---|---|---|---|---|---|
| Tissue | Factor | Similarmotifs | Hs | Mm | Hs | Mm | Comment | |
| Liver | HNF-1 | Hepatocyte nuclear factor 1; Member of the homeodomain factor family. | Yes | Yes | Yes | Yes | Ranks in the top 3 in both species. | |
| HNF-3 | Hepatocyte nuclear factor 3; Member of the fork-head factor family. | HFH, XFD and certain other fork-head family members. | Yes | No | Yes | Yes | Unranked in mouse or mouse orthologs of human liver-specific promoters. | |
| HNF-4 | Hepatocyte nuclear factor 4; Member of thyroid hormone receptor-like family. | PPAR, COUP; Steroid/thyroid hormone receptor-like super-family. | Yes | Yes | Yes | Yes | Ranks 1st in both species. | |
| HNF-6 | Hepatocyte nuclear factor 6; A homeodomain factor from the CUT subfamily. | No | No | No | No | The known motif in TRANSFAC (v9.4) may be poorly characterized. | ||
| C/EBP | CCAAT/enhancer binding protein; Variants form a subfamily of basic region leucine zipper family. | Yes | Yes | Yes | Yes | Ranks in the top 10 in both species and is known to interact with other high-ranking motifs. | ||
| DBP | Albumin D-site binding protein; A member of PAR family of b-ZIP factors. | PAR family: HLF, TEF and VBP. | No | Yes | Yes | Yes | All three sources call DBP present in mouse, none of the three call DBP present in human. | |
| Skeletal Muscle | MEF-2 | Myocyte-specific enhancer factor 2; A member of the MADS domain family. | Splice variant RSRFC4. | Yes | Yes | Yes | Yes | Mouse skeletal muscle promoters are enriched with C/G-rich motifs. The A/T-rich MEF-2 ranks 2nd after GC-content correction. |
| SRF | Serum response factor; a member of the MADS domain family. | Yes | Yes | Yes | No | The A/T-rich SRF is not identified in mouse even after GC-content correction. | ||
| MyoD | Myogenic factor 3; Member of the myogenin family. | Myogenin family, E-box binding factors. | Yes | Yes | Yes | Yes | Ranks in the top 5 in both species and predicted to interact with SRF and MEF-2. | |
| Sp1 | Stimulating protein 1; A ubiquitous factor with a Cys2His2 zinc finger domain. | Yes | Yes | Yes | Yes | The C/G-rich motif is higly ranked with and without GC-content correction. | ||
| Testis | SRY | Sex-determining region on Y chromosome; Member of the high-mobility group (HMG) class of factors. | SOX factors. | Yes | Yes | Yes | No | The A/T-rich motif is ranked 2nd in human after GC-content correction. |
| CREM | Cyclic AMP-responsive element modulator; Member of the CREB/ATF subfamily of the bZIP factors. | CREB/ATF family. | Yes | Yes | Yes | Yes | C/G-balanced and synergistic, ranks 1st in mouse with and without GC-content correction. | |
| RFX | Regulatory factor X; subfamily of the forkhead factors with winged-helix binding domains. | Yes | Yes | No | Yes | The core of the RFX motif (GTTGCCA) is highly similar to the reverse of the core MYB motif (CCGTTG), ranking top in human. | ||
| A motif classifies foreground from background if it is ranked in the top 20 distinct motif classifiers. | ||||||||
Nuclear receptor binding-site motifs and E-box motifs are among the top enriched motifs in 11 and 10 of the 14 human and mouse tissues, respectively. A Wilcoxon signed-ranks test showed that nuclear receptor and E-box motifs (represented by 54 and 39 TRANSFAC motifs, respectively) are enriched in the union of our foreground sets with P-values below 6.07E−14 and 2.02E−13 (nuclear-receptor motifs) and 2.22E−10 and 4.57E−03 (E-box) in human and mouse, respectively. These results suggest a diversity of tissue-specific roles for nuclear receptors and E-box binders, likely mediated by tissue-specific cofactors.
Tissue-specific cis-regulatory elements
Highly enriched motifs, and the associated score thresholds identified by our methods, provide a starting point for targeted experimental annotation of tissue-specific promoters. Figure 1 shows known and predicted sites mapped on the −500 to +100 region of the human albumin promoter and the −250 to +50 region of the human skeletal muscle α-actin promoter. Human albumin has known functional binding sites for HNF-1, C/EBP, AFP and NF-Y (Paonessa et al, 1988; Sawadaishi et al, 1988; Frain et al, 1990; Li et al, 1990), all of which were identified among the top predicted motifs or included in top modules for liver. Locations of known sites for C/EBP (at −437) and NF-Y (at −125) do not perfectly align with the corresponding predicted sites (at −462 and −143, respectively), but current knowledge about binding sites for those factors raises the possibility that the predicted locations are more accurate. The only predicted binding site for these factors in the human albumin promoter that is not depicted in the figure is a C/EBP site at −956. Human skeletal muscle α-actin has known sites for SRF, TEF and a known TATA box (Boxer et al, 1989; MacLellan et al, 1994), all among top predicted motifs for human skeletal muscle (TATA box motifs have high similarity with MEF-2 motifs). Figure 2 gives predictions in a representative human promoter in each of the remaining tissues.
Figure 1.

Verified and predicted binding sites in human albumin and skeletal muscle α-actin promoters. Predicted sites are represented by horizontal bars and verified sites by vertical bars. Verified sites for albumin (Paonessa et al, 1988; Sawadaishi et al, 1988; Frain et al, 1990; Li et al, 1990) and for α-actin (Boxer et al, 1989; MacLellan et al, 1994) were mapped to the promoter from CSHLmpd to obtain their correct locations relative to the TSS.
Figure 2.

Predicted binding sites for selected factors in promoters from the human tissue-specific sets. The selected factors are among the top ranked in the corresponding tissues.
Comparison to previous results
Previous analysis of tissue-specific patterns in regulatory regions includes analysis based on cross-species conservation (Xie et al, 2005) and coexpression (Smith et al, 2006).
Xie et al (2005) identified conserved elements in orthologous promoters of four mammalian genomes. They found 59 experimentally validated motifs that are significantly conserved and enriched in at least one human tissue. These include E-box, ETS, MEF2, MEIS1 and NF-1 in skeletal muscle; Chx10 in kidney; NRF-1, ELK-1, GABP and E12 in CD4 T cells; AP-4 and MEF-2 in heart; and NRF-1 in testis. Our results agree with Xie et al (2005) on the enrichment of E-Box and MEF-2 in skeletal muscle, ETS in CD4 T-cells and E-box in pancreas.
In previous work (Smith et al, 2006), we tested the hypothesis that proximal promoters contain information that can be used to predict tissue-specific expression. We were not concerned with identifying the most significant tissue-specific motifs, modules and sites. Considering the difference between the goals of the two projects, it is not surprising that the predictive models described by Smith et al (2006) have little similarity to our top motifs and modules. The most significant similarities between our top tissue-specific patterns and the predictive models of Smith et al (2006) include the enrichment of ETS in CD4 T-cell-specific promoters and the enrichment of Smith et al (2006) motifs Novel3 and Novel6 in mouse testis and Novel1 in human testis. The three novel testis motifs are very similar to motifs that rank in the top 100 in our analysis, but the enrichment of these motifs was not sufficiently high for inclusion in TCat.
Correlation between human and mouse regulatory regions
We compared motif enrichment ranks in each human foreground set to ranks in the corresponding mouse foreground set using Spearman's rank correlation test, and found that enrichment ranks across species are highly correlated (P<0.001) for all but CD4 T cells (Supplementary Table 15). In CD4 T cells, motif enrichment ranks are similar only for few highly enriched motifs. Despite the motif-enrichment ranks correlation, the order of the top predicted binding sites is not usually conserved between orthologous promoters. Fewer than 10% of orthologous pairs showed significant (P<0.01) conservation of site order. Weak site-order conservation suggests that the top tissue-specific sites would be difficult to identify using traditional cross-species alignment alone, and methods that rely on co-linear promoter alignment may have high false-negative detection rates. This evidence is in agreement with Frith et al (2006), who found that homologous transcription start sites can be separated by more than 100 nucleotides. A list of the nine genes (out of 102 candidates) with significant conservation of site order is given in Supplementary Section 2.3.
Materials and methods
The steps used in creating the catalog include (1) identifying tissue-specific transcripts, (2) identifying factors that are expressed in each tissue, (3) obtaining promoter sequences for tissue-specific transcript, and (4) identifying individual motifs and modules (i.e. sets of interacting motifs) that characterize tissue-specific promoter sets.
Identifying tissue-specific transcripts
To identify motifs and modules that regulate tissue-specific transcription, we analyzed promoters of transcripts that appear to be regulated in a tissue-specific manner. If an information source indicated that a transcript has restricted expression, unusually high expression, or a specific function in the tissue, that source voted for tissue specificity of the transcript. For each tissue, we sorted the transcripts according to the number of votes received, retaining the top 100 with distinct TSS as tissue specific. Ties in the ranking were broken according to intensity values from the GNF SymAtlas expression data (discussed below), which we have found to be the most complete and the most reliable source of tissue-based expression information. We used the same number of transcripts for each tissue to facilitate comparison across tissues, and 100 sequences provided sufficient information for our analysis while allowing identification of well-known tissue-specific motifs.
Microarray data
The GNF SymAtlas microarray data were generated using Affymetrix HG-U133A array and the custom GNF1H and GNF1M Affymetrix arrays, and include expression profiles for 79 human and 61 mouse tissues (Su et al, 2004). Among these are the seven tissues we selected to include in the catalog. Tissues were selected with consideration to data availability in GNF and other sources and interest from Zhang lab members and collaborators. A transcript received a vote for tissue specificity from this information source if it was called present and its intensity exceeded its mean across all tissues by 3 standard deviations.
The Hughes Toronto microarray data (Zhang et al, 2004), which was generated using custom-built oligonucleotide arrays, provide mouse expression profiles for 55 tissues, including all of our tissues but CD4 T cells. A transcript received a vote for tissue specificity if it was called present in the tissue, and had intensity at least 10 standard deviations above its mean across all 55 tissues. This large number of standard deviations was required to limit the number of transcripts receiving positive votes.
The GeneNote expression profiles (Shmueli et al, 2003), which were generated using the Affymetrix GeneChip HG-U95, provide human expression data for 12 tissues, including all of our tissues but CD4 T cells and testis. The GeneNote data were used in the same way as the GNF SymAtlas data, with a transcript being called tissue specific if it is present in that tissue and has intensity at least 3 standard deviations above its mean across all 12 tissues.
EST data
dbEST is a database of expressed sequence tags (Boguski et al, 1993), and contains source information, such as the tissue of origin, for each EST. This information is used to annotate UniGene clusters with the source data, and a UniGene is said to have restricted expression in a tissue if more than half of the ESTs contributing to that UniGene have the same source tissue. A transcript received one vote for specificity in a particular tissue if the corresponding UniGene cluster is annotated as having expression restricted to that tissue.
GO terms
We associated a set of GO Terms with each tissue. This was performed by compiling a set of keywords for each tissue (e.g. ‘renal' was associated with kidney; ‘sperm' was associated with testis), and searching GO Term names and definitions for those keywords. This produced, for each tissue, a set of GO Terms that were subsequently reviewed to ensure that the context of the keywords was appropriate. A transcript of a gene annotated with a GO Term that is associated with a tissue received a vote for specificity in that tissue.
Selecting promoter sequences
Although regulatory elements can exist almost anywhere in the genome, they are concentrated near the TSS (Cooper et al, 2006). We used the CSHLmpd to map transcripts to promoters, using experimentally confirmed promoters from EPD (Perier et al, 1998), DBTSS (Suzuki et al, 2002) and GenBank, as well as computationally predicted promoters. For each promoter, we used the proximal sequence region of −1000 to +100 relative to the TSS.
Each part of our analysis is based on comparing the tissue-specific promoter sets to a background of random promoters from the same species. For each tissue, a background set was constructed by selecting 1000 transcripts uniformly at random from the set of RefSeqs for the corresponding species with TSS annotation in CSHLmpd. For each tissue, transcripts with at least one vote for specificity in that tissue were removed from consideration before selecting the background.
Because our analysis focused on proximal promoters, −1000 to +100 relative to the TSS, if the TSS annotation is off by several hundred base pairs, important promoter regions might be excluded. AKR1D1, identified as liver specific by our voting system, has two known TSSs within 500 bp of the first exon for the corresponding RefSeq (NM_005989) (Charbonneau and Luu-The, 1999). We used the TSS located upstream of the first exon, but could have chosen to use the other promoter, which was annotated by a generally more reliable source (DBTSS versus GenBank). Currently, in such situations, there is little information that identifies the promoter responsible for observed tissue-specific regulation, but comparative genomics and rapidly improving arrays promise better 5′ end identification, thus improving proximal promoter annotation and association between transcripts and promoters (Kim et al, 2005; Carninci et al, 2006). Negative promoter sets can be used to cancel out patterns that are not related to tissue-specific transcription regulation. We use random negative promoter sets with and without GC-content correction; this correction cancels the inuence of genomic GC-content isochore variability.
Identifying and evaluating motifs
Given a motif M (represented as a position–frequency matrix) and a sequence S, the max-score of M in S, max-score (M, S) is the score of the top scoring subsequence of S when aligned against the scoring matrix for M. Details on constructing and using scoring matrices can be found in Stormo (2000). For a fixed threshold λ, the max-score classification method classifies S as belonging to the foreground if max-score (M,S)⩾λ, and the background otherwise. Given a set of foreground sequences FG (i.e. the tissue-specific promoters) and a set of background sequences BG, the sensitivity of M and λ under max-score classification is
and the specificity is
The balanced error rate for M and λ under max-score classification is then
The quantity of interest in our analysis corresponds to the optimal value of λ for M in distinguishing FG from BG:
Many known motifs are similar to each other, usually owing to similar binding specificities for distinct factors or distinct origins for mofits associated with a single factor. We used MATCOMPARE (Schones et al, 2005) to eliminate redundancies in the sets of known and novel motifs.
Identifying and evaluating modules
Modules are used to classify sequences based on the max-score values of the motifs they contain. Let ℳ={M1, …, Mk} be a module and Λ={λ1, …, λk} be an associated set of thresholds. The max-score classification for modules assigns sequence S to the foreground if and only if max-score (Mi, S)⩾λi holds for all i (1⩽i⩽k). Given sets of sequences FG and BG, the sensitivity of ℳ and Λ under max-score classification is
and the specificity is
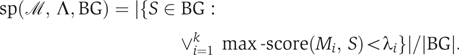 |
The balanced-error rate for ℳ and Λ under the max-score classification is
As with mofits, we are interested in the optimal value of Λ and define
Because modules are intended to describe synergistic function of a set of motifs, we are interested in modules whose performance is better than expected given the performance of the individual motifs composing the module. For a module ℳ composed of k motifs, let ℳ′⊂ℳ minimize B(ℳ′, FG, BG) over all size k−1 modules built from motifs in ℳ, and let ℳ′=ℳ\ℳ′. To assess whether ℳ, with balanced-error rate u, significantly improves over ℳ′ and ℳ′, we use the probability
We estimated this probability empirically, by sampling from the distribution of balanced-error rates resulting from intersections of sets with balanced-error rates B(ℳ′, FG, BG) and B(ℳ′, FG, BG).
We used MODULATOR, which is available in CREAD (Smith et al, 2005b), to construct modules. Given a set of motifs, a set of foreground sequences and a set of background sequences, MODULATOR identifies those modules composed of the given motifs that have the best balanced-error rates. A branch-and-bound algorithm is used to simultaneously optimize the score thresholds for the motifs in a module. Modules are constructed by adding motifs to existing modules until a user-specified module size is reached or until motif addition does not significantly improve enrichment. Each time a motif is added to a module, the resulting larger module is retained only if the balanced-error rate of the larger module is improved significantly above expectation. The initial modules of size two are obtained by combining pairs of motifs.
For modules that are entirely composed of known motifs, the top 100 motifs (before eliminating redundancies) were used. Modules were allowed to contain up to four motifs for reasons of computational feasibility, but many top modules are smaller. Novel modules, which must contain at least one novel motif, were constructed using the top 100 novel motifs and the top 100 known motifs. Redundancies were removed from the lists of top modules using a procedure described in Supplementary Section 2.
Measuring the significance of motifs and modules
To measure significance of enrichment for top known motifs we used known motifs to classify randomly assembled promoter sets. We constructed 1000 foreground/background pairs for each species by selecting 100 sequences for each foreground and 1000 for each background uniformly at random from CSHLmpd. For each foreground/background pair we calculated the balanced-error rate of each known motif. The best balanced-error rates overall obtained on random samples for human and mouse were 0.364 and 0.368, respectively. We used the distribution of these error rates to identify the q-value (Storey and Tibshirani, 2003) significance of the error rate of each motif. Tissues whose highest ranking motifs fail the q<0.05 test include CD4 T cells and heart in human, and CD4 T cells in mouse. TCat includes q-value annotation for each ranked known motif. The full set of motifs for which q<0.05 is estimated to include five false leads per 100 predictions. We did not obtain statistical significance measures for novel motifs, because this will require running DME and DME-B more times than is computationally feasible.
Modules were identified by combining motifs whose cooccurrence was enriched in the foreground sets. We measured enrichment of modules using the balanced-error rate (analogous to that of motifs), and we required that each motif in a module contributes significantly to the enrichment of the module as a whole. To test significance, we randomly selected 100 of the 1000 foreground/background pairs used to evaluate individual known motifs, and performed the module identification procedure on each of the 100 selected pairs. The best balanced-error rates for modules that are entirely composed of known motifs (called known modules) in random human and mouse sets were 0.3145 and 0.304, respectively. We used these balanced-error rates as an estimate of the critical value for P<0.01. We opted for using P-value cutoffs instead of computing q-values because accurate q-value estimation for modules is computationally prohibitive. Top known modules in human kidney (0.2955), liver (0.3105), pancreas (0.3125) and testis (0.3) scored better than the cutoff, as did top known modules in mouse kidney (0.3005), liver (0.3025) and testis (0.2945).
Supplementary Material
Supplementary information
Acknowledgments
We thank our anonymous reviewers for suggestions that have greatly improved the quality of this paper, and BIOBASE for providing access to TRANSFAC. This work is supported by NIH grant HG001696 and NSF grants DBI-0306152 and EIA-0324292.
References
- Boguski MS, Lowe TM, Tolstoshev CM (1993) dbEST—database for expressed sequence tags. Nat Genet 4: 332–333 [DOI] [PubMed] [Google Scholar]
- Boxer L, Miwa T, Gustafson T, Kedes L (1989) Identification and characterization of a factor that binds to two human sarcomeric actin promoters. J Biol Chem 264: 1284–1292 [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CAM, Taylor MS, Engstrom PG, Frith MC, Forrest ARR, Alkema WB, Tan SL, Plessy C, Kodzius R, Ravasi T, Kasukawa T, Fukuda S, Kanamori-Katayama M, Kitazume Y, Kawaji H, Kai C, Nakamura M, Konno H, Nakano K, Mottagul-Tabar S, Arner P, Chesi A, Gustincich S, Persichetti F, Suzuki H, Grimmond SM, Wells CA, Orlando V, Wahlestedt C, Liu ET, Harbers M, Kawai J, Bajic VB, Hume DA, Hayashizaki Y (2006) Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38: 626–635 [DOI] [PubMed] [Google Scholar]
- Charbonneau A, Luu-The V (1999) Assignment of steroid 5beta-reductase (SRD5B1) and its pseudogene (SRD5BP1) to human chromosome bands 7q32 → q33 and 1q23 → q25, respectively, by in situ hybridization. Cytogenet Cell Genet 84: 105–106 [DOI] [PubMed] [Google Scholar]
- Cooper SJ, Trinklein ND, Anton ED, Nguyen L, Myers RM (2006) Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res 16: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frain M, Hardon E, Ciliberto G, Sala-Trepat JM (1990) Binding of a liver-specific factor to the human albumin gene promoter and enhancer. Mol Cell Biol 10: 991–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Ponjavic J, Fredman D, Kai C, Kawai J, Carninci P, Hayashizaki Y, Sandelin A (2006) Evolutionary turnover of mammalian transcription start sites. Genome Res 16: 713–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Liu JS (2005) De novo cis-regulatory module elicitation for eukaryotic genomes. Proc Natl Acad Sci USA 102: 7079–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B (2005) A high-resolution map of active promoters in the human genome. Nature 436: 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Huang JH, Rienhoff HY Jr, Liao WS-L (1990) Two adjacent c/ebp-binding sequences that participate in the cell-specific expression of the mouse serum amyloid a3 gene. Mol Cell Biol 10: 6624–6631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLellan W, Lee T, Schwartz R, Schneider M (1994) Transforming growth factor-beta response elements of the skeletal alpha-actin gene. Combinatorial action of serum response factor, YY1, and the SV40 enhancer-binding protein, TEF-1. J Biol Chem 269: 16754–16760 [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulls OV, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Munch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E (2003) TRANSFAC(R): transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31: 374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelander S, Larsson E, Kristiansson E, Mansson R, Nerman O, Sigvardsson M, Mostad P, Lin-dahl P (2005) Predictive screening for regulators of conserved functional gene modules (gene batteries) in mammals. BMC Genomics 68, doi:10.1168/1471-2164-6-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paonessa G, Gounari F, Frank R, Cortese R (1988) Purification of a NF1-like DNA-binding protein from rat liver and cloning of the corresponding cDNA. EMBO J 7: 3115–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier RC, Junier T, Bucher P (1998) The eukaryotic promoter database EPD. Nucleic Acids Res 26: 353–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Bilenky M, Lin K, He A, Yuen W, Dagpinar M, Varhol R, Teague K, Griffith OL, Zhang X, Pan Y, Hassel M, Sleumer MC, Pan W, Pleasance ED, Chuang M, Hao H, Li YY, Robertson N, Fjell C, Li B, Montgomery SB, Astakhova T, Zhou J, Sander J, Siddiqui AS, Jones SJM (2006) cisRED: a database system for genome-scale computational discovery of regulatory elements. Nucleic Acids Res 34 (Suppl 1): D68–D73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B (2004) JASPAR: an open access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 32: D91–D94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawadaishi K, Morinaga T, Tamaoki T (1988) Interaction of a hepatoma-specific nuclear factor with transcription-regulatory sequences of the human alpha-fetoprotein and albumin genes. Mol Cell Biol 8: 5179–5187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones D, Sumazin P, Zhang MQ (2005) Similarity of position frequency matrices for transcription factor binding sites. Bioinformatics 21: 307–313 [DOI] [PubMed] [Google Scholar]
- Shmueli O, Horn-Saban S, Chalifa-Caspi V, Shmoish M, Ophir R, Benjamin-Rodrig H, Safran M, Domany E, Lancet D (2003) GeneNote: whole genome expression profiles in normal human tissues. C R Biol 10–11: 1067–1072 [DOI] [PubMed] [Google Scholar]
- Smith AD, Sumazin P, Xuan Z, Zhang MQ (2006) DNA motifs in human and mouse proximal promoters predict tissue-specific expression. Proc Natl Acad Sci USA 104: 6275–6280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Sumazin P, Zhang MQ (2005a) Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc Natl Acad Sci USA 102: 1560–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Sumazin P, Zhang MQ (2005b) CREAD: Comprehensive regulatory element analysis and discovery. World Wide Web (http://cread.sf.net/)
- Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100: 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo GD (2000) DNA binding sites: representation and discovery. Bioinformatics 16: 16–23 [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 101: 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Yamashita R, Nakai K, Sugano S (2002) DBTSS: DataBase of human transcriptional start sites and full-length cDNAs. Nucleic Acids Res 30: 328–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M (2005) Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Z, Zhao F, Wang JH, Chen GX, Zhang MQ (2005) Genome-wide promoter extraction and analysis in human, mouse, and rat. Genome Biol 6: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Morris QD, Chang R, Shai O, Bakowski MA, Mitsakakis N, Mohammad N, Robinson MD, Zirngibl R, Somogyi E, Laurin N, Eftekharpour E, Sat E, Grigull J, Pan Q, Peng WT, Krogan N, Greenblatt J, Fehlings M, van der Kooy D, Aubin J, Bruneau BG, Rossant J, Blencowe BJ, Frey BJ, Hughes TR (2004) The functional landscape of mouse gene expression. J Biol 3: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Wong WH (2004) CisModule: De novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc Natl Acad Sci USA 101: 12114–12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Shendure J, Church GM (2005) Discovering functional transcription-factor combinations in the human cell cycle. Genome Res 15: 848–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information