

Abstract
The success of mapping genes involved in complex diseases, using association or linkage disequilibrium methods, depends heavily on the number and frequency of susceptibility alleles of these genes. These methods will be economically and statistically feasible if common diseases are usually influenced by one or a few susceptibility alleles at each locus (common disease–common variant, CDCV, hypothesis), but not so if there is a high degree of allelic heterogeneity. Here, we use forward-time population simulations to investigate the impact of various genetic and demographic factors on the allelic spectra of human diseases, on the basis of two models proposed by Reich and Lander and by Pritchard. Factors considered are more complex demographies, a finite-allele mutation model, population structure and migration, and interaction between disease susceptibility loci. The conclusion is that the CDCV hypothesis holds and that the phenomenon is caused by transient effects of demography (population expansion). As a result, we devise a multilocus generalization of the Reich and Lander model and demonstrate how interaction between loci with respect to their response to selection may lead to complex effects. We discuss the implications for mapping of complex diseases.
ALLELIC spectra, i.e., the number and frequency of susceptibility alleles, of genes involved in common diseases are crucial for the success of mapping these genes using association or linkage disequilibrium (LD) methods. There are two main reasons for this: First, a critical assumption of both association and LD mapping is that there is little allelic heterogeneity within loci. Statistical power of these methods will be greatly reduced if a gene contains many rare alleles, as is often the case for rare Mendelian diseases (Terwilliger and Weiss 1998). Second, the cost of identifying a large number of rare alleles is much higher than that for a few common alleles. The gene typing cost alone may deter such studies, especially when there are a large number of disease susceptibility loci (DSL) for the disease. For a further analysis, see Yang et al. (2005).
The common disease–common variant (CDCV) hypothesis proposes that common diseases usually are caused by one or a few common disease susceptibility alleles at each DSL (Lander 1996). Due to the difficulties in mapping common disease genes, we have a limited amount of empirical data. The best-known examples include the ApoE ɛ4 allele in Alzheimer's disease (Saunders et al. 1993), Factor V (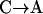 at 1691) allele in deep-venous thrombosis (Bertina et al. 1994), and CKR5Δ32 in resistance to human immunodeficiency virus infection (Dean et al. 1996). Although this limited evidence is generally in favor of the CDCV hypothesis, it might be argued that these genes are found exactly because they have common alleles, so their abundance is an artifact of the ascertainment bias (Smith and Lusis 2002).
at 1691) allele in deep-venous thrombosis (Bertina et al. 1994), and CKR5Δ32 in resistance to human immunodeficiency virus infection (Dean et al. 1996). Although this limited evidence is generally in favor of the CDCV hypothesis, it might be argued that these genes are found exactly because they have common alleles, so their abundance is an artifact of the ascertainment bias (Smith and Lusis 2002).
Two theoretical models of human genetic disease were proposed by Reich and Lander (2001) and Pritchard (2001). They are called model RL and model P, respectively. Their features are summarized in Table 1. Both models RL and P use the “effective number of alleles” (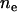 ) to measure allelic spectrum diversity. Model RL concerns the evolution of the effective number of alleles of a monogenic disease in a human population that undergoes rapid population expansion, whereas model P concerns the distribution of the equilibrium total disease allele frequency (f0) and the effective number of alleles of polygenic diseases in the current human population.
) to measure allelic spectrum diversity. Model RL concerns the evolution of the effective number of alleles of a monogenic disease in a human population that undergoes rapid population expansion, whereas model P concerns the distribution of the equilibrium total disease allele frequency (f0) and the effective number of alleles of polygenic diseases in the current human population.
TABLE 1.
Summary of models RL and P
| Reich and Lander's model RL | Pritchard's model P |
|---|---|
| Assumptions | |
| Single-locus model. | Multilocus model. |
| Instant population growth from N0 = 104 to N1 = 6 × 109 at ∼100,000 years ago. | Constant population size (N = 104). |
| Current population not in equilibrium state. | Current population in equilibrium state. |
| Selection acts independently at each locus. | |
| Methods | |
| Population-genetics analytic approach. | Coalescent-like simulation. |
| Modeling the evolution of allelic diversity from founder to current generation. | Using the MCMC method to explore the joint distribution of model parameters. |
| Major results | |
| Both rare and common diseases in the founder population have simple spectra. | The majority of potential DSL will have essentially no genetic variation unless the disease alleles are under weak purifying selection. |
| Both rare and common diseases in the current population will have diverse spectra, if the current population is in equilibrium state. | At loci where the mutation rate is low, the susceptibility classes usually are dominated by a single major mutation. |
| Rare diseases reach equilibrium faster than common diseases so rare diseases have more diverse spectra than common diseases. | Loci where the mutation rate is high contribute disproportionately to the genetic variance but introduce more allelic heterogeneity at the same time. |
MCMC, Markov chain Monte Carlo.
These different approaches lead to different conclusions. Reich and Lander's (2001) model RL leads to the conclusion that the CDCV hypothesis holds and that the phenomenon is caused by transient effects of demography (population expansion). In the long run, when a human population reaches mutation, selection, and drift equilibrium, all diseases will have diverse spectra. However, common diseases diversify their spectra slower than rare diseases so they tend to have simpler spectra before the equilibrium state is reached. Pritchard's (2001) model P, on the contrary, concludes that loci that contribute substantially to genetic variance are more polymorphic than average, and such loci are the result of high mutation rate, rather than of the commonness of the disease. Although Pritchard and Cox (2002) argue that the differences between these two models can be partly explained by the use of different mutation rates, their respective levels of support for the CDCV hypothesis cannot be easily reconciled.
In this article, we first simulate models RL and P, following their original assumptions, and then try to reconcile the two models or choose one over the other. We also want to investigate the robustness of the models by studying the impact of alternative or additional genetic features on the models. Features considered are more complex demographies, a finite-allele mutation model, population structure and migration, and choices of effective population size of the human population. In addition, we explore the effects of interaction of loci modifying the disease susceptibility locus. The support for the CDCV hypothesis is discussed on the basis of these studies.
METHODS
We simulate the evolution of human diseases using a forward-time population genetics simulation environment simuPOP (Peng and Kimmel 2005). The reason why we take a forward-time approach instead of a backward-time (coalescent) approach is that we want to observe the evolution of allelic spectra from the founder to the current generation. The coalescent approach, though successful in other such studies (Kruglyak 1999; Pritchard 2001), does not suit this purpose due to the fact that it discards ancestral information that is irrelevant to the coalescent tree. Another important reason is that it is more convenient to use a forward-time approach when selection is modeled.
Simulated diseases are characterized by the number of DSL, the mutation pattern and rate, and single- and multilocus selection models. To reproduce the results of the single-locus model RL and the multilocus model P, we use different genetic models and parameter values consistent with those used in the original publications. These assumptions are summarized in Table 2. Following the notations in Reich and Lander (2001), we let f be the total disease allele frequency at a given generation, f0 be the equilibrium total disease allele frequency—that is, the frequency expected under the balance between mutation and selection— and fexp be the total disease allele frequency just before population expansion. Note that we frequently use f0 instead of f to simplify discussions.
TABLE 2.
Summary of simulation assumptions
| Single-locus model RL | Multilocus model P |
|---|---|
| Genotypic structure | |
| One disease-susceptibility locus on one chromosome. There are one wild-type (N) allele and k − 1 disease (S) alleles. | Multiple (L) disease susceptibility loci on different chromosomes, with the same maximum number of allelic states (k). |
| Selection model | |
| Fitness of an individual follows either an additive or a recessive model. Fitness of an individual with genotype NN, NS, or SS is 1, 1 − s/2, 1 − s for an additive model and 1, 1, 1 − s for a recessive model. | Each DSL follows either an additive or a recessive fitness model as in the case of the single-locus model. The overall fitness of an individual with fitness gi, i = 1,…, L at each DSL follows either a multiplicative (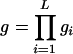 ) or an additive ( ) or an additive ( ) multilocus model. ) multilocus model. |
| Mutation model | |
k-allele (Jukes and Cantor 1969) model with k > 105 to approximate the infinite allele model. Given mutation rate μ, an allele will mutate to any other state with equal probability 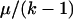 , regardless of its current allelic state. , regardless of its current allelic state. |
k-allele model with smaller k (e.g., k = 200) to be closer to a bidirectional mutation model (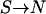 and and 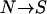 ). The mutation rate may vary from locus to locus. ). The mutation rate may vary from locus to locus. |
| Recombination | |
| NA | NA because all DSL are physically unlinked. |
| Demographic | |
| Instant, linear, or exponential population growth model. N0 = 104, N1 = 106 or 107. | Constant population size at N = 104 or 105. |
| Population structure | |
| Population may be split into equally sized subpopulations (m = 10 or 100) before population expansion. During population expansion, these subpopulations may evolve with or without migration. | NA |
| Migration | |
| Cyclic stepping-stone model at rate 10−3. | NA |
| Penetrance | |
| Indirectly modeled, see explanation in the text. | Indirectly modeled, see explanation in the text. |
Several features of the models are not directly simulated, the most significant one being penetrance. Penetrance and fitness are different but related concepts. Our simulations follow the allelic composition of a population, which is the result of mutation, selection, and genetic drift. The affection status is assumed to reflect the same underlying genotype as fitness, and its impact on the allelic composition of the next generation is represented by fitness. Therefore, we bypass modeling penetrance in our simulations.
Our selection model is a stochastic model based on individual genotype. It might be more reasonable to assume a more complicated model that involves affection. For example, we might select against an affected individual at a given probability. However, since penetrance is also a stochastic process based on genotype, this two-step penetrance/selection model can be replaced by an equivalent selection model that works directly on the genotype. For example, if individuals with genotype AA, Aa, or aa are affected with probability 0, 0.2, or 0.8, and affected individuals have probability 0.5 to be removed or not produce offspring, the corresponding selection model has fitness 1, 0.9, or 0.6 for genotype AA, Aa, or aa. Therefore, at least for our simulations, a two-step model would not make any difference.
Our selection model is more general than model P. Model P assumes that selection acts independently at each DSL and there is no interaction among loci. In our model, the fitness value of an individual is the joint force of fitness at all DSL. In this context, it is important to examine interactions between loci. The basic models of multilocus selection that we use are defined in Table 2. An example of interaction effects is provided in results.
The overall mutation rate μ in our k-allele mutation model (Table 2) differs from the mutation rates in models RL and P. More specifically, the mutation rate in model RL is equal to the forward mutation rate μS in model P and is denoted by μS instead of μ to avoid confusion. The relationships between μ, μS, and the reverse mutation rate μN are
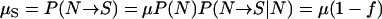 |
(1) |
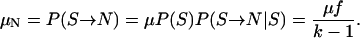 |
(2) |
For given μ and f, μN is determined by the maximum allele state k. μN is extremely small if a large k is used to mimic the infinite-allele model. μS and μ are close to each other in the cases of rare diseases, but can differ substantially for common diseases. Furthermore, since total disease allele frequency (f) is changing during evolution, μN and μS are not constant in our simulations. We do not attempt to mimic these mutation rates exactly since we believe that our mutation model is closer to reality. We use large k (k > 104) to approximate the infinite-allele model used in model RL and use smaller k for a better approximation to model P.
Simulations are run under a variety of demographic models and parameter settings. At the beginning, we create a founder population of N0 individuals, each having L chromosomes with one DSL on each of them. We use L = 1 for the simulations of monogenic diseases and L = 50 for polygenic cases. This founder population is initialized with a given initial allelic spectrum (usually with 90% wild-type alleles and five equally frequent disease alleles) and it is then evolved for G0 generations until it reaches mutation, selection, and drift equilibrium. The population is then expanded to size N1 after G1 generations. Before expansion, the founder population can be split into a given number of equally sized subpopulations. During population expansion, these subpopulations may evolve independently (without migration) or with a varying level of migration among them.
Allelic spectra and their summary measure, the effective number of alleles, are recorded throughout the simulations. Since there is no direct measurement of ne from the population, we estimate it from population allele frequencies using
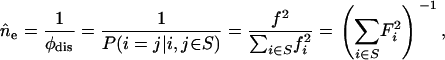 |
(3) |
where φdis is the expected allelic identity among disease alleles, 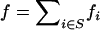 is the total disease allele frequency of the disease, fi is the allele frequency of allele i, and Fi = fi/f is the proportion of allele i in the S class. ne reaches its maximum value of k − 1 when all disease alleles have the same frequency (
is the total disease allele frequency of the disease, fi is the allele frequency of allele i, and Fi = fi/f is the proportion of allele i in the S class. ne reaches its maximum value of k − 1 when all disease alleles have the same frequency (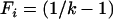 ) under a k-allele mutation model, regardless of the value of f.
) under a k-allele mutation model, regardless of the value of f.
RESULTS
Single-locus model:
Reich and Lander's (2001) model RL for monogenic diseases is studied. We first verify this model using forward-time simulations and then discuss the impact of various genetic and demographic factors on the model. Although the theoretical mutation rate (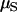 ) and simulation mutation rate (μ) differ by a factor of 1 − f0 (see Equation 1), it is safe to treat them as equal since f0 will not exceed 0.04 in this section.
) and simulation mutation rate (μ) differ by a factor of 1 − f0 (see Equation 1), it is safe to treat them as equal since f0 will not exceed 0.04 in this section.
Verification of the basic model:
Reich and Lander (2001) employ theoretical estimates of the evolution of the effective number of disease alleles (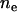 ), using an instant population-growth model. These estimates are used as a baseline for later sections. Assuming that the total disease allele frequency (f) is not far from its equilibrium value f0 during evolution, the estimated ne, under the infinite-allele model, is
), using an instant population-growth model. These estimates are used as a baseline for later sections. Assuming that the total disease allele frequency (f) is not far from its equilibrium value f0 during evolution, the estimated ne, under the infinite-allele model, is
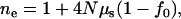 |
(4) |
where N is the effective population size and μS is the mutation rate. f0 is determined by the nature of the disease. For example, the equilibrium value of the total disease allele frequency of rare recessive diseases can be approximated by 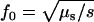 , where s is the selection coefficient, provided that
, where s is the selection coefficient, provided that 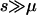 .
.
Although the allelic spectra of both rare and common diseases are similar in equilibrium states, the rates at which these are approached differ greatly. For a population that has been expanded instantly from size N0 to N1, the proportion of alleles derived from before expansion will decay exponentially with rate 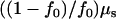 . The effective number of alleles will increase with expectation
. The effective number of alleles will increase with expectation
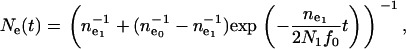 |
(5) |
where 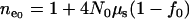 and
and 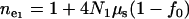 (Reich and Lander 2001). Since
(Reich and Lander 2001). Since 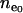 and
and 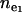 are similar for rare and common diseases, the rate of reaching equilibrium is determined by the total disease allele frequency (f0) within the exponential term of Equation 5.
are similar for rare and common diseases, the rate of reaching equilibrium is determined by the total disease allele frequency (f0) within the exponential term of Equation 5.
To verify the above theoretical estimates, we ran an extensive array of simulations using recessive diseases with different combinations of parameters μ, s, N0, and N1. A summary of the results is shown in supplemental Figure 1 at http://www.genetics.org/supplemental/. These simulation studies are in remarkable agreement with the theory.
Impact of demographic models:
Different human populations have different demographic histories. Some populations like the Scandinavian Saami isolate have approximately constant population size, but most of them have undergone a rapid population expansion (Laan and Pääbo 1997). Among many population expansion models, the exponential population-growth model is a simple, yet reasonably realistic one. It is widely assumed that the general human population had constant size N0 = 10,000 until G0 = 5000 generations before the present and then expanded exponentially to its present-day size of N1 = 6 billion (Harpending et al. 1998; Kruglyak 1999; Reich and Lander 2001). G0 = 5000 corresponds to 100,000 years given a 20-year generation time. Note that N1 = 6 billion is the census population size and should not be used as the effective population size in model RL. We use N1 = 106 or 107 here and discuss the impact of this parameter on the model later. Besides the exponential growth model, we also simulated the instant population-growth model to reproduce the theoretical results in Reich and Lander (2001) and a linear population-growth model for comparison purposes.
Choice of demographic models can have a large impact on model RL. As pointed out in Reich and Lander (2001), slower population expansion would result in slower growth in allelic diversity. If we use an exponential population-growth model, the human population increases rather slowly most of the time. This has two consequences: During the slow-growing period, small population size tends to limit the growth of the effective number of alleles so that ne will increase slower than in a faster-growth model; the “large population” stage is effectively shorter than in the instant growth model and gives diseases less time to reach equilibrium.
Figure 1a plots the dynamics of ne in six simulations, which use the same basic parameters (N0 = 104, N1 = 107, μ = 10−5) but different selection coefficients (s = 0.99 for rare disease and s = 0.01 for common disease) and demographic models (instantaneous, linear, or exponential). Although equilibrium ne is close to 400 for all six cases, the kinetics are quite different. The effective number of alleles of common disease increases slower than that of the rare disease, but the difference is smaller for linear and exponential growth models than that for the instant growth model. Due to the demographic difference, ne of a rare disease under the exponential growth model at generation 5000 is at the same level as that under the instant population-growth model at generation 1000.
Figure 1.—


Impact of demographic and mutation models. (a) Evolution of ne of a rare (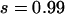 ) and a common disease (
) and a common disease (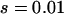 ) under instant, linear, and exponential population-growth models. N0 = 104, N1 = 107, μ = 10−5. The two solid curves are theoretical estimates under the infinite-allele model and instant growth. (b) Change of ne with number of allelic states 100, 500, 1000, 2000, using an instant population-growth model. N0 = 104, N1 = 107, μ = 10−5, s = 0.1. The solid curve is the theoretical estimate under the infinite-allele model.
) under instant, linear, and exponential population-growth models. N0 = 104, N1 = 107, μ = 10−5. The two solid curves are theoretical estimates under the infinite-allele model and instant growth. (b) Change of ne with number of allelic states 100, 500, 1000, 2000, using an instant population-growth model. N0 = 104, N1 = 107, μ = 10−5, s = 0.1. The solid curve is the theoretical estimate under the infinite-allele model.
Impact of the mutation model:
Model RL uses the infinite-allele model. When the effective population size is large, this model leads to an unrealistically large ne. For example, when N = 109 and μ = 10−5, the equilibrium ne = 3.2 × 104 for a common disease of size 0.2. However, due to the constraints on gene length, silent or recurrent mutations, effective number of alleles for real human diseases is usually smaller than this number.
In the previous simulations, we used a k-allele model with a large number of alleles (k > 104) to mimic the infinite-allele model. Recurrent mutations do occur but at such a small rate that they have almost no impact on the proportion of alleles derived from before population expansion or the equilibrium effective number of alleles.
The probability of recurrent mutation increases with decreasing k. This leads to smaller observed equilibrium ne, compared to ne under the infinite-allele model. To verify this, we simulate the evolution of ne of a disease with s = 0.1 in a population that has grown instantly from N0 = 104 to N1 = 107 at 6000 generations ago, using k-allele models with k = 100, 500, 1000, or 2000. Among these k-values, only k = 2000 reaches the ne expected under the infinite-allele model (Figure 1b). Although it is not clear how exactly ne will change with k when k > ne, when k < ne and there are enough disease alleles to fill every allelic state (high N and μ or small s), ne will be close to 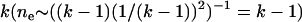 regardless of the exact values of N, μ, or s (k = 100 in Figure 1b).
regardless of the exact values of N, μ, or s (k = 100 in Figure 1b).
Although equilibrium ne with a small maximum number of allele states is smaller than that of the infinite-allele model, at the beginning the former increases faster than the latter. This takes place because μN, the mutation rate from susceptibility to normal alleles, is no longer negligible and accelerates the dissolution of the dominant disease allele when k is small.
Impact of subpopulation structure and migration:
The human population went through complex migration patterns that might affect the allelic spectra of human diseases. We start from the simplest cases when no migration is allowed among subpopulations.
Suppose that the population after instant expansion is split into m equally sized subpopulations, which then evolve independently without migration afterward. In each subpopulation, the equilibrium effective number of alleles is 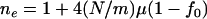 (Equation 4 with population size replaced by
(Equation 4 with population size replaced by 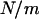 ), where f0 is assumed to be the same in all subpopulations because it is determined by the nature of disease. The allelic spectrum of the whole population is the composition of these subpopulation spectra. The equilibrium ne in the whole population is located between
), where f0 is assumed to be the same in all subpopulations because it is determined by the nature of disease. The allelic spectrum of the whole population is the composition of these subpopulation spectra. The equilibrium ne in the whole population is located between 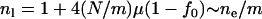 (when the allelic spectra are identical in all subpopulations) and
(when the allelic spectra are identical in all subpopulations) and
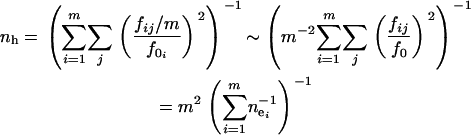 |
(6) |
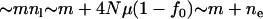 |
(7) |
(when disease alleles are totally different among subpopulations. Here fij and fi0 are the frequency of allele j and all disease alleles in subpopulation i, respectively, and we assume that 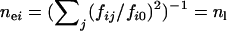 , i = 1,…, m are identical in all subpopulations.) Assuming a split-and-grow demographic model, allelic spectra in subpopulations are similar at the beginning and become increasingly distinct over time. Therefore, ne will approach nh in the long run when the differences between allelic spectra in subpopulations increase with time. The difference between nh and ne in a single population is determined by the number of subpopulations m. For example, in the case of a rare disease in many small tribes, each tribe may be dominated by one or a few tribe-specific mutants. The overall ne will be close to the number of tribes, larger than the small ne in individual tribes. However, numeric experiments show that variations of fi0 will significantly reduce nh and make it difficult to reach m + ne.
, i = 1,…, m are identical in all subpopulations.) Assuming a split-and-grow demographic model, allelic spectra in subpopulations are similar at the beginning and become increasingly distinct over time. Therefore, ne will approach nh in the long run when the differences between allelic spectra in subpopulations increase with time. The difference between nh and ne in a single population is determined by the number of subpopulations m. For example, in the case of a rare disease in many small tribes, each tribe may be dominated by one or a few tribe-specific mutants. The overall ne will be close to the number of tribes, larger than the small ne in individual tribes. However, numeric experiments show that variations of fi0 will significantly reduce nh and make it difficult to reach m + ne.
To confirm these analyses, we evolve a rare (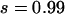 ) and a common (
) and a common (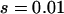 ) disease, using a demography where a founder population is instantly expanded from N0 = 104 to N1 = 0.5 × 107 and at the same time splits into m (m = 10 or 100) subpopulations. The equilibrium effective number of alleles of the whole population is ∼200 for both diseases if we ignore population structure. Figure 2a plots the case when m = 10. We see that ne in each subpopulation evolves roughly as expected, and the overall ne is very close to the theoretical value estimated from a single uniform population. The impact of subpopulation structure becomes obvious when m = 100 (Figure 2b). While ne in subpopulations evolves as expected, the overall ne, as the result of composition of allelic spectra in 100 subpopulations, increases faster and arrives at a larger equilibrium ne than that expected theoretically using a single population.
) disease, using a demography where a founder population is instantly expanded from N0 = 104 to N1 = 0.5 × 107 and at the same time splits into m (m = 10 or 100) subpopulations. The equilibrium effective number of alleles of the whole population is ∼200 for both diseases if we ignore population structure. Figure 2a plots the case when m = 10. We see that ne in each subpopulation evolves roughly as expected, and the overall ne is very close to the theoretical value estimated from a single uniform population. The impact of subpopulation structure becomes obvious when m = 100 (Figure 2b). While ne in subpopulations evolves as expected, the overall ne, as the result of composition of allelic spectra in 100 subpopulations, increases faster and arrives at a larger equilibrium ne than that expected theoretically using a single population.
Figure 2.—




Impact of population structure and migration: evolution of ne of a rare (s = 0.99, solid lines) and a common disease (s = 0.01, dotted lines) after instant population expansion from N0 = 104 to N1 = 0.5 × 107 with μ = 10−5. The population is split into 10 (a and c) or 100 (b and d) subpopulations after expansion. Migration rate is 0 for a and b and 0.1% for c and d. Thick lines are total ne, and thin lines are average ne of all the subpopulations. The top dashed lines correspond to nh = m + ne ∼ 300. Note that the y-axes are in log scale.
Although ne in a structured population tends to be larger than that in a single population, ne in each subpopulation evolves as expected, unless new mutants are introduced by migration. From a single subpopulation point of view, migration is a way to introduce new mutants, usually at a higher intensity than mutation. Consequently, in a subpopulation with migration, ne is larger than that in an isolated subpopulation. On the other hand, while the homogenizing effect of migration is not obvious soon after the population split, when allelic spectra in subpopulations are similar to each other, it mixes alleles from subpopulations and keeps ne of a structured population away from nh. In an extreme scenario when migration is so strong that all subpopulations have the same allelic spectra, ne of the whole population is the same as that of a single subpopulation.
Migration does not have the same impact on common and rare diseases. When a disease is common, a significant proportion of migrants are affected. The impact of migration on the allelic spectrum is strong compared to weak mutation and selection. When a disease is rare, there are few affected migrants, so disease alleles tend to remain private in their own subpopulation. Since selection is strong in this case, migration is no longer a dominating force.
These analyses are confirmed by Figures 2c and 2d, which are similar to Figures 2a and 2b, except that migration is allowed between subpopulations. In these simulations, 0.1% of individuals in a subpopulation migrate to the adjacent subpopulations at each generation. When a disease is common, migration is strong enough to make allelic spectra more similar in all subpopulations. The allelic spectrum of the whole population is therefore closer to those of the subpopulations (compare common diseases in Figure 2b and 2d). The impact is, as expected, most evident in common diseases in a population with 10 subpopulations (Figure 2c), followed by common diseases in a population with 100 subpopulations (Figure 2d) and rare diseases (Figure 2, c and d).
In conclusion, the allelic structure is more diverse in a subpopulation with new mutants introduced as a result of migration than that in an isolated subpopulation. However, from the whole-population point of view, the homogenizing effect of migration decreases ne, which otherwise is greater than ne in a single population. The impact depends on the number of subpopulations, the level of migration, and the commonness of the disease.
Impact of effective population size:
Reich and Lander (2001) use current census population size N1 = 6 × 109 as the effective population size of the current human population. This is apparently inappropriate since the human population has a complex population structure and is far from random mating. However, none of the popular definitions (inbreeding, variance, and eigenvalue) of effective population size can be used. They are all at a magnitude of 104 and would lead to small estimates of ne, which is incompatible with empirical data (ne ≤ 5 when N = 104 and μ ≤ 10−4 and even smaller when μ ≤ 10−5). The best definition in this context may be the size of an ideal population in which a disease allele has the same probability to be fixed/extinct as in real human population. However, we are not aware of such a definition being used.
Fortunately, an accurate estimate of effective population size is not essential in the study of the allelic spectrum of the current human population. Since the human population expanded not long ago (∼3000–10,000 generations), ne's of most diseases are still small (Reich and Lander 2001; see Table 1 or Figure 2 for ne estimates of real diseases). The effective population size, though determining the future equilibrium ne, has far less impact on ne of the current human population than that of mutation rate.
This can be confirmed by empirical data. Figure 3 of Reich and Lander (2001) gives an almost perfect match to empirical data using N1 = 6 × 109 and μ = 3.2 × 10−6. However, this result is insensitive to the value of N1. From a pure regression point of view, any N1 > 1 × 106 would fit reasonably well and the best fit of the data is obtained using N1 = 4.85 × 106. The population size we used (106 or 107) should serve our simulations well.
Figure 3.—


Distribution of total disease allele frequency: ne and f0 at each DSL of polygenic diseases. N = 104 (a) or 105 (b); μ = 10−5; k = 200; and s = 0, 0.5 × 10−4, 10−4, 0.5 × 10−3 (from top to bottom). Solid curves are densities given by Equation 8. Dashed lines are estimated effective number of alleles. The mean f0 of all DSL at the last generation is used to estimate μS and μN.
Multilocus model:
Here we study the allelic spectra of DSL responsible for polygenic diseases, relating these predictions to model P. The main questions we want to answer are how well our simulations mimic model P, how DSL interact with each other, especially between DSL with different mutation rates or selection coefficients, and whether or not we can treat the multilocus model as a set of independent single-locus models.
Verification of the basic model:
We assume that (1) the disease has L DSL located on different chromosomes, (2) all DSL of a polygenic disease contribute equally to the disease, (3) the fitness value at each DSL fits an additive model with selection coefficient s, (4) the overall fitness fits a multiplicative or additive multilocus model, (5) the effective population size N is constant at 104 or 105, and (6) the mutation rate (μ = 10−5) is the same at all DSL following a k-allele mutation model with k = 200. We run the simulation for extended number of generations to let the population reach mutation, selection, and drift equilibrium. Since hypotheses 4 and 5 are different from model P (as in Pritchard 2001), we are interested in how well our simulations mimic model P.
Simple formulas (e.g., 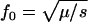 for recessive diseases or
for recessive diseases or 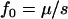 for additive diseases) can be used to estimate the equilibrium total disease allele frequencies of DSL when
for additive diseases) can be used to estimate the equilibrium total disease allele frequencies of DSL when 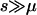 but not for the cases of common diseases with s ∼ μ. Assuming forward and reverse mutation rates μS and μN and selection coefficient s, the distribution of equilibrium overall frequency f0 of susceptibility alleles in the population is given by Wright's formula
but not for the cases of common diseases with s ∼ μ. Assuming forward and reverse mutation rates μS and μN and selection coefficient s, the distribution of equilibrium overall frequency f0 of susceptibility alleles in the population is given by Wright's formula
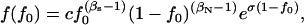 |
(8) |
where βS = 4NμS, βN = 4NμN, and σ = 2Ns (s in this article is twice that of Pritchard 2001) are scaled parameters. The normalization constant c can be obtained by numerical integration. This formula works best in the cases of weak selection (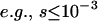 ). For larger s (
). For larger s (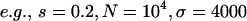 ), the exponential term will dominate
), the exponential term will dominate  and make it essentially a δ-function at 0. We simulate populations with N = 105, in addition to N = 104 used in Pritchard (2001), because larger N leads to cases with βS > 1 that have significantly different distributions of f0 from the cases with βS < 1, due to the
and make it essentially a δ-function at 0. We simulate populations with N = 105, in addition to N = 104 used in Pritchard (2001), because larger N leads to cases with βS > 1 that have significantly different distributions of f0 from the cases with βS < 1, due to the 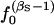 term of Equation 8.
term of Equation 8.
Figure 3, a (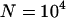 ) and b (
) and b (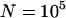 ), plots the relationship between f0 and ne for all DSL of some of the simulations, along with densities of f0 estimated from Equation 8 and estimated effective number of alleles. Example 1 in the supplemental material at http://www.genetics.org/supplemental/ plots two similar simulations with additive and multiplicative multilocus selection models, along with 50 single-locus models mimicking the independent selection model of model P. Despite the fact that μS and μN can vary from locus to locus due to the variation of f0 (Equations 1 and 2), we use μN and μS estimated from the mean of f0 at the last generation for theoretical estimates. These simulations show that additive and multiplicative multilocus selection models mimic the independent selection model in model P quite well (further discussed below), with ne close to theoretical estimates. The distributions of f0 follow Equation 8 in the cases with relative large s, but not so when s is close to 0. In the cases when s = 0, Equation 8 predicts high density around zero (with estimated 54 or 15% DSL having <0.1 total disease allele frequency, for cases with N = 104 or N = 105, respectively). This is not confirmed by our simulations.
), plots the relationship between f0 and ne for all DSL of some of the simulations, along with densities of f0 estimated from Equation 8 and estimated effective number of alleles. Example 1 in the supplemental material at http://www.genetics.org/supplemental/ plots two similar simulations with additive and multiplicative multilocus selection models, along with 50 single-locus models mimicking the independent selection model of model P. Despite the fact that μS and μN can vary from locus to locus due to the variation of f0 (Equations 1 and 2), we use μN and μS estimated from the mean of f0 at the last generation for theoretical estimates. These simulations show that additive and multiplicative multilocus selection models mimic the independent selection model in model P quite well (further discussed below), with ne close to theoretical estimates. The distributions of f0 follow Equation 8 in the cases with relative large s, but not so when s is close to 0. In the cases when s = 0, Equation 8 predicts high density around zero (with estimated 54 or 15% DSL having <0.1 total disease allele frequency, for cases with N = 104 or N = 105, respectively). This is not confirmed by our simulations.
At DSL with no or very weak selection, the total disease allele frequencies approach 1 in our simulations. This seems understandable: At such DSL, mutation generates new variants at random. Since there is no selection against any particular allele, the allelic spectra eventually are flat. The total disease allele frequencies at these DSL therefore equal 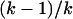 for a k-allele mutation model and 1 for the infinite-allele model, provided that the population is large enough to hold all alleles. We do not know what caused Equation 8 to fail, but this discrepancy might challenge some of the conclusions drawn from it in Pritchard (2001, p. 129), such as “in the absence of selection, a DSL is much more likely to be near fixation,” which considers both extinction and fixation.
for a k-allele mutation model and 1 for the infinite-allele model, provided that the population is large enough to hold all alleles. We do not know what caused Equation 8 to fail, but this discrepancy might challenge some of the conclusions drawn from it in Pritchard (2001, p. 129), such as “in the absence of selection, a DSL is much more likely to be near fixation,” which considers both extinction and fixation.
We also consider the effective number of alleles at each DSL (Figure 3). They are generally scattered around theoretical estimates given by Equation 4. Since ne < 1 + 4Nμ is small (ne < 5 when N = 104 and μ < 10−4), the allelic structure is simple for both rare and common diseases if these parameters are appropriate. Either larger effective population size or higher mutation rate is needed to explain high allelic diversity of some real human diseases under this model. For example, mutants causing the rare disease (prevalence rate at birth is 2.4 × 10−4) Duchenne muscular dystrophy have a highly diverse spectrum. With an estimated mutation rate 7.9 × 10−5, it requires a population with effective population size 3 × 105 to accommodate these mutants with ne > 100 (van Essen et al. 1992; Reich and Lander 2001).
Varying selection and mutation coefficient:
DSL of polygenic diseases usually do not contribute equally to the diseases. There might be some DSL that are under strong selection and many other DSL that are only slightly deleterious. The same holds for mutation rates. These locus-by-locus differences may cause interactions among DSL and disallow the dissection of the multilocus model into several single-locus models.
Figure 4 shows the results of two simulations with varying selection coefficients (Figure 4a) or varying mutation rates (Figure 4b). The diseases are recessive at each DSL and the overall fitness is modeled by a multiplicative model. The population size and mutation rate are N = 105 and μ = 10−5, respectively. Simple estimates of f0 and ne assuming a single-locus model are given by the solid lines, which match f0 and ne at each DSL of the multilocus model almost perfectly. This is true for other simulations we have run using additive single- and multilocus models (results not shown). It is therefore safe to treat this multilocus model as a set of independent single-locus models.
Figure 4.—


Varying selection coefficient and mutation rate: total disease allele frequency (f0) and effective number of alleles (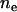 ) of a common disease caused by 50 loci with varying selection coefficient (a) or mutation rate (b), in a population of constant size 105. (a) Disease with varying selection coefficient at each DSL. DSL are equally spaced in the interval (
) of a common disease caused by 50 loci with varying selection coefficient (a) or mutation rate (b), in a population of constant size 105. (a) Disease with varying selection coefficient at each DSL. DSL are equally spaced in the interval (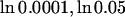 ) with μ = 10−5. (b) Disease with varying mutation rate at each DSL that is equally spaced in the interval (ln 6.5 × 10−6, ln 1.2 × 10−4) with μ = 0.01. Diseases are recessive at each DSL. Solid lines are theoretical estimates given by
) with μ = 10−5. (b) Disease with varying mutation rate at each DSL that is equally spaced in the interval (ln 6.5 × 10−6, ln 1.2 × 10−4) with μ = 0.01. Diseases are recessive at each DSL. Solid lines are theoretical estimates given by 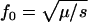 and Equation 4.
and Equation 4.
The CDCV hypothesis is not supported on the basis of Figure 4. The allelic structures of more common DSL (caused by less selection or higher mutation) are either the same (Figure 4a) or more diverse (Figure 4b) than that of a rarer DSL.
Evolution of DSL:
Although model P deals only with the equilibrium state of DSL, we are interested to see how the equilibrium state is attained. An evolutionary model is needed, so we borrow model RL with the instant population-growth model here.
Evolution of the allelic spectrum of a DSL is determined by population size, mutation rate, and most importantly by the total disease allele frequency of the DSL. In the context of a common disease, the distribution of f0 is quite dispersed, especially when N is small (see Figure 3). Consequently, we would expect highly dispersed fexp at the beginning of population expansion. This results in different evolutionary patterns between DSL with identical parameter settings. An example in the supplemental material at http://www.genetics.org/supplemental/ confirms this. In this example, four DSL of a polygenic disease evolve under identical parameter settings. The evolution of ne at these DSL differs greatly because their fexp deviate from f0 due to small initial population size and the resulting dispersed distribution of f0.
Interaction between DSL:
Another question that remains is whether or not the DSL in model P interact. The evolution of a DSL is determined by initial spectrum, mutation, selection, and the demographic model. Among these factors, selection is the source of interaction with other DSL, because the fitness of an individual is a joint effect of selection at all DSL.
The results we have shown all use additive or multiplicative multilocus selection models. From the above results, and the examples in the supplemental material at http://www.genetics.org/supplemental/, DSL in such models seem to evolve independently since the equilibrium total allele frequency and effective number of alleles all match those estimated from the single-locus model RL. This is not surprising since the essence of these two models is that the overall fitness can be written as a function of individual fitness values, without an interaction term. The lack of interaction between DSL using these two multilocus selection models is proved in the supplemental material, both mathematically and through a special set of simulations.
However, in general, disease susceptibility loci can interact with each other and lead to complex fitness functions. In these cases, there is no reason to believe that the multilocus model is a simple composition of single-locus models.
We demonstrate the interaction between DSL using a two-locus example. Assumed fitnesses of genotypes are as listed below:
As we can see from the table, locus B acts as a modifier to locus A. It does not cause the disease by itself, but decreases the fitness of individuals with allele a, from slightly (Aa or aa with heterozygote Bb) to highly deleterious (with homozygote bb). Therefore, the total disease allele frequency changes with respect to the frequency of allele b, and we may no longer have constant selection pressure over locus A. The equilibrium allele frequency is implied by the interaction of both loci.
Figure 5 displays the evolution of loci A and B in three simulations. In the first case, only wild-type allele B is presented and allele A evolves independently of locus B with fexp ∼ f0 = 0.1. In the next two simulations, allele b is present and this changes the total disease allele frequency of locus A. When these two loci reach an equilibrium state before population expansion, the total disease allele frequencies at both loci stay constant and the effective number of alleles evolves as expected with fexp being the result of interaction between the two loci. When these two loci are not at equilibrium before population expansion (the last two simulations), their total disease allele frequency gradually reaches equilibrium and results in changing selection pressure on both loci. As a result, the evolution of the effective number of alleles deviates from theoretical estimates, which reflect constant selection pressure. This is caused by interaction or, in this particular example, by modification of selection at locus A by the state of locus B.
Figure 5.—

An example of interaction between DSL: evolution of the total disease allele frequency (left) and effective number of alleles (right) in a two-locus model using an instant population-growth model, with N0 = 5000, N1 = 106, G0 = 2000 (burn-in), G1 = 4000, μ = 10−4. Dashed horizontal lines on the left side are estimated equilibrium total disease-allele frequencies. Solid curves on the right side are theoretical estimates of effective number of alleles, using the total allele frequency before expansion (fexp). Row 1, evolution of locus A, with only wild-type allele B. Rows 2 and 3, evolution of loci A and B when both loci reach equilibrium state before population expansion. Rows 4 and 5, evolution of loci A and B when they are not at equilibrium at population expansion.
DISCUSSION
We use different approaches to confirm the major results of model RL (Reich and Lander 2001) and model P (Pritchard 2001). For model RL, we mostly use large k to mimic the infinite-allele model and we simulate relatively rare diseases that have total disease allele frequency close to equilibrium. Most importantly, we focus on the evolution of allelic diversity rather than on the final population. For model P, we use smaller k to mimic the bidirectional mutation process ( and
and  ) and simulate more common diseases that have a more diverse total disease allele frequency. We also run the simulations for an extended number of generations to let the simulations reach equilibrium states. The results obtained from these two sets of simulations conform to their respective theoretical estimates developed in the original publications, but differ fundamentally regarding the support for the CDCV hypothesis.
) and simulate more common diseases that have a more diverse total disease allele frequency. We also run the simulations for an extended number of generations to let the simulations reach equilibrium states. The results obtained from these two sets of simulations conform to their respective theoretical estimates developed in the original publications, but differ fundamentally regarding the support for the CDCV hypothesis.
Model RL (Reich and Lander 2001) explains the high diversity of rare diseases well and gives support for the CDCV hypothesis. Because the effective numbers of alleles of common diseases increase slower than rare diseases after population expansion, common diseases usually have simpler spectra than rare diseases. Model RL, however, faces some difficulties in modeling almost neutral DSL. In this case, the total disease allele frequency of the disease is close to 1 so the 1 − f0 term in Equation 4 leads to an unrealistically small estimate of ne. However, in the context of mapping disease susceptibility genes, these DSL are of little interest to us.
Under model P, as can be seen in Figure 4, common diseases have more diverse spectra. Moreover, due to the assumption of a constant small population in equilibrium, the original version of model P (Pritchard 2001) fails to explain the high diversity of some Mendelian diseases. For common diseases, this model leads to the conclusion that the allelic diversity at a DSL is positively related to the mutation rate but that the selection pressure at this DSL is almost irrelevant. The results we obtained from this model are not consistent with the CDCV hypothesis.
The major difference between models RL and P is whether or not the current human population is in mutation, selection, and drift equilibrium. Model RL, while recognizing that both common and rare diseases in the current human population would have high allelic diversity under the equilibrium assumption, claims that the current human population has expanded from the recent past and has not reached equilibrium. On the other hand, model P is based on the equilibrium assumption of the human population.
The evidence for the equilibrium assumption of model P is, however, lacking both theoretically and empirically. According to model RL, a typical common disease would need 1 million years to reach an equilibrium state, much longer than the length of modern human history. Our simulations confirm this estimate. To obtain the equilibrium ne and f0 for common diseases, we usually need to run our simulations for >20,000 generations, ∼400,000 years if we assume 20 years per generation. The DNA level variation of the human genome is also far away from the equilibrium state, which, under assumption of neutrality, predicts equal frequency of nucleotides. The equilibrium assumption cannot be simply replaced or discarded in model P because this model concerns only the current human population. If we convert model P to an evolutionary model by evolving a constant population for a certain number of generations, the choice of initial allelic spectra will become a critical problem.
In conclusion, it seems that a better model for the evolution of allelic spectra of DSL responsible for a polygenic disease would be a multilocus model based on model RL, with more realistic demographies. As we have discussed in the previous section, if multiplicative or additive multilocus selection is assumed, this model can be studied locus by locus as a set of independent single-locus models. All results obtained in Single-locus model can be applied to this model. In the supplemental material at http://www.genetics.org/supplemental/, we demonstrate (example 3) how to use what we learned from model RL to predict the evolution of the DSL of a polygenic disease, under an exponential population-growth model, with varying mutation rate and single-locus selection pressure, and a multiplicative multilocus selection model.
Summarizing our simulations, we expect increased allelic diversity with:
Larger population size (or larger recent population size for varying demographic model).
Higher mutation rate and more allelic states (which may be the result of a longer gene).
Smaller total disease allele frequency at the beginning of population expansion. This may be the result of higher selection pressure, shorter evolution time before expansion, or chance (genetic drift).
Longer evolutionary history (older mutants), which provides a locus with more time to gain diversity.
More subpopulations and/or lower migration among subpopulations.
Conclusions 1–3 are qualitatively consistent with theoretical expression (4).
We have run many simulations to study the impact of each genetic feature on the allelic diversity. For example, we used k = 200, 500, 1000, 2000 to see the impact of the possible number of allelic states on the effective number of alleles. We also varied selection rate 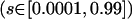 and mutation rate (
and mutation rate (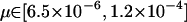 ) over a wide range of possible values. Although direct comparison is not possible since these simulations use a different population size, length of evolution, and so forth, we can conclude from all simulations that the allelic spectrum is most sensitive to the total disease allele frequency at the beginning of population expansion (fexp). For other genetic features, it is difficult to rank their relative importance. If two conflicting forces (such as higher mutation rate but faster population expansion) are involved, it seems necessary to resort to a simulation program.
) over a wide range of possible values. Although direct comparison is not possible since these simulations use a different population size, length of evolution, and so forth, we can conclude from all simulations that the allelic spectrum is most sensitive to the total disease allele frequency at the beginning of population expansion (fexp). For other genetic features, it is difficult to rank their relative importance. If two conflicting forces (such as higher mutation rate but faster population expansion) are involved, it seems necessary to resort to a simulation program.
The total disease allele frequency at the disease locus (or loci in a polygenic disease setting) at the beginning of population expansion has a great impact on the evolution of allelic diversity. To minimize the impact of initialization of total disease allele frequency, we use a burn-in period to let the population reach mutation, selection, and drift equilibrium before population expansion. This method works well in most cases but may fail when the disease is very rare so we may end up with no disease allele before population expansion. The variability of fexp may also cause problems. This is likely to happen when the disease is common or when the population size is small. In these cases, the distribution of fexp is dispersed so fexp may be far away from the equilibrium value f0. The value of fexp, instead of the equilibrium value f0, determines the evolution of the allelic spectrum at this DSL.
It is often questionable if we should use equilibrium f0 before population expansion, especially for DSL with almost neutral disease-susceptibility alleles, which have a high expected equilibrium f0 (f0 > 50%). The reason is that we rarely see this high level of disease-allele frequency in reality. A likely solution to this problem is using a short burn-in period with a well-chosen initial allelic spectrum to keep f0 small during evolution.
Evolution of real human diseases is likely to be more complex than models we use. For example, disease mutants are unlikely to be selectively equivalent and the fitness of an individual allele should affect its relative frequency with respect to other disease alleles. Also, we use a multiplicative or additive multilocus fitness model, which allows us to study the allelic spectrum locus by locus. However, disease loci may interact with each other in many ways. Interaction between DSL on the same chromosome should also be considered. This tends to be a complicated issue since recombination will then play a role in the model.
For the single-locus fitness model, we discussed only the strictly additive cases. Recessive, dominant, or codominant models should be explored, as well as the balancing selection and antagonistic pleiotropy models. Among these fitness models, balancing selection has been known to maintain stable frequencies of two or more phenotypic forms. Although our program can simulate these cases, the discussion of these factors is beyond the scope of this article.
We do not explicitly simulate the impact of a bottleneck effect on the allelic diversity of human diseases. However, a burn-in process with small population size followed by rapid population expansion approximates the evolution following a long-neck bottleneck (Harpending et al. 1998). Therefore, we can conclude that a severe bottleneck would remove most alleles of both rare and common diseases and result in simple spectra. If a bottleneck is recent, both rare and common diseases will have simple spectra while rare diseases will recover their diversity faster than common ones.
Our evolutionary model assumes constant mutation and selection pressure. Total disease-allele frequency will oscillate around equilibrium frequency (with a distribution following Equation 8) and the population will eventually reach mutation, selection, and drift equilibrium. Recently, Di Rienzo and Hudson (2005) proposed another evolutionary model that may also explain the CDCV hypothesis. In this model, an allele might have been initially deleterious, but has become advantageous because of the change of environment. This model is supported by some empirical evidence and can complement our model in explaining allelic diversity of common human diseases.
Our model is consistent with the CDCV hypothesis. In addition, on the basis of this model, under the assumption of multiplicative or additive multilocus selection models, the DSL responsible for a common polygenic disease can be studied as if they were DSL of independent single-locus diseases. The common disease–common variant hypothesis, confirmed by our analyses on the single-locus model, is therefore consistent with polygenic common diseases. If more complex multilocus selection schemes are assumed, the interaction effects will appear, as demonstrated by the example in Figure 5.
The above independence-based argument, however, is potentially incomplete, because the relationship between commonness and weak selection for a polygenic disease does not have to hold as in the case of monogenic diseases. If a common disease is caused by several loci under weak selection, then according to our model these DSL will be common and will have simple allelic spectra. This is the CDCV hypothesis in the cases of polygenic diseases (Smith and Lusis 2002). However, a common disease may well be caused by rare alleles at numerous DSL, if each one can single-handedly cause the disease. This is referred to as the genetic heterogeneity model. Our model suggests that these DSL will be rare and have highly diverse allelic spectra. Although theoretical studies and empirical data suggest that DSL for a complex disease are usually under weak selection, we cannot rule out the possibility of the heterogeneity model (Yang et al. 2005). As a matter of fact, a common disease may be caused by a few loci with common alleles and many more with rare alleles. These loci with common alleles are the ones most ready to be mapped.
FURTHER EXAMPLES:
Example 1: comparison between single and two multilocus selection models:
We simulate the evolution of polygenic diseases (L = 50) in a constant population of size N = 104 or 105, with μ = 10−5 and s = 0.5 × 10−3. To mimic independent selection pressure, we also run 50 single-locus simulations corresponding to some of the multilocus simulations. Supplemental Figure 2 at http://www.genetics.org/supplemental/ displays the total disease allele frequency and effective number of alleles at each DSL for some of these simulations.
Three simulations using different multilocus models [first three cases in supplemental Figure 2 (http://www.genetics.org/supplemental/), multiplicative multilocus model, additive multilocus model, and 50 single-locus models] yield indistinguishable results. This indicates that both additive and multiplicative models mimic independent selection originally assumed in model P well, at least in the simulated parameter range. The last case of supplemental Figure 2 is similar to case 1 but uses a smaller population size (N = 104). Genetic drift is stronger in this population than in other cases and results in a more dispersed distribution of f0.
Example 2: diversity and importance of fexp:
Supplemental Figure 3 at http://www.genetics.org/supplemental/ plots the dynamics of the total disease allele frequency (f) and the effective number of alleles (ne) of four DSL of a polygenic disease (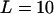 ), under an instant population-growth model with N0 = 104, N1 = 106, and G0 = 5000, with μ = 10−5 and s = 0.001 identical at all DSL. A multiplicative multilocus selection model is used. Although the equilibrium f0 of all DSL is 0.1, the distribution of f0 at generation 5000 is quite dispersed because of the small population size during the burn-in period. After population expansion, f0 of all DSL approaches 0.1 slowly, but the evolution of ne at each DSL is roughly determined by the total disease allele frequency of DSL at the beginning of the population expansion (see the theoretical curves in the right plots). Consequently, ne's of the DSL of a common disease may approach their equilibrium states at vastly different rates.
), under an instant population-growth model with N0 = 104, N1 = 106, and G0 = 5000, with μ = 10−5 and s = 0.001 identical at all DSL. A multiplicative multilocus selection model is used. Although the equilibrium f0 of all DSL is 0.1, the distribution of f0 at generation 5000 is quite dispersed because of the small population size during the burn-in period. After population expansion, f0 of all DSL approaches 0.1 slowly, but the evolution of ne at each DSL is roughly determined by the total disease allele frequency of DSL at the beginning of the population expansion (see the theoretical curves in the right plots). Consequently, ne's of the DSL of a common disease may approach their equilibrium states at vastly different rates.
Example 3: multilocus example using an exponential population-growth model:
Supplemental Figure 4 at http://www.genetics.org/supplemental/ plots the evolution of the four DSL of a polygenic disease, using an exponential population-growth model with N0 = 104, N1 = 106, and G0 = G1 = 5000. The mutation and selection rates at these four DSL are s = 10−3, μ = 10−4; s = 10−3, μ = 10−5; s = 10−5, μ = 10−4; and s = 10−5, μ = 10−5, respectively. Using what we have learned from model RL, we can conclude that equilibrium ne is ∼400 for cases 1 and 3 and ∼40 for cases 2 and 4 (depending on the values of N and μ). The rate at which ne approaches its equilibrium state is fastest for case 2, followed by cases 1, 4, and 3 (see the values of μ/s). Because of the exponential growth model and the commonness of these DSL (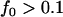 ), none of the DSL should have reached the equilibrium state. The allelic diversity at generations 10,000 should be highest for case 2 (relatively rarer disease) or case 1 (higher equilibrium ne).
), none of the DSL should have reached the equilibrium state. The allelic diversity at generations 10,000 should be highest for case 2 (relatively rarer disease) or case 1 (higher equilibrium ne).
Web resources:
All simulations are performed using a simuPOP (Peng and Kimmel 2005) script simuCDCV.py, which is distributed as part of simuPOP at http://simupop.sourceforge.net. It can be run using the provided graphical user interface or as a batch command, with or without R-assisted visualization.
Table 3.
| Fitness | BB | Bb | bb |
|---|---|---|---|
| AA | 1 | 1 | 1 |
| Aa | 0.999 | 0.99 | 0.9 |
| aa | 0.998 | 0.98 | 0.8 |
Acknowledgments
The authors acknowledge the exhaustive discussions and helpful comments of William Amos and Francois Balloux, the important points raised by anonymous referees, and the help and encouragement of Christopher Amos. B.P. was supported by a training fellowship from the W. M. Keck Foundation to the Gulf Coast Consortia through the Keck Center for Computational and Structural Biology, by the Rice Terascale Cluster funded by the National Science Foundation under grant EIA-0216467, by Intel, and by HP. M.K. was supported in part by grant CA75432 from the National Cancer Institute, by grant ES09912 from the National Institutes of Health, and by KBN (Polish Research Committee) grant 3T11F 01029.
APPENDIX
Verification of theoretical estimates of model RL:
To verify the estimate of the equilibrium effective number of alleles, we use different combinations of N, μs, and s and evolve a constant-size population for a long period of time until it reaches mutation, selection, and drift equilibrium. f0 and ne are observed and plotted in supplemental Figure 1, a and b, along with their theoretical expectations at http://www.genetics.org/supplemental/. Note that curves in supplemental Figure 1b are almost horizontal, indicating that the effective numbers of alleles for rare and common diseases are similar in equilibrium states.
We then run simulations using a demographic scenario in which the founder population grows instantly from N0 = 104 to N1 = 107. The dynamics of the percentage of ancestral alleles derived from before expansion and the effective number of disease alleles of three diseases are plotted in supplemental Figure 1, c and d, at http://www.genetics.org/supplemental/. The simulations and theoretical expectations agree reasonably well.
Lack of interaction caused by additive and multiplicative multilocus selection models:
Given fitness values gi at each DSL i, the overall fitness of an individual with L DSL is 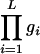 for a multiplicative and max
for a multiplicative and max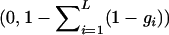 for an additive multilocus selection model. Using these models, we obtain that the evolution of a DSL does not depend on interaction with other loci, as we have shown in this article and in all the examples in the supplemental material at http://www.genetics.org/supplemental/.
for an additive multilocus selection model. Using these models, we obtain that the evolution of a DSL does not depend on interaction with other loci, as we have shown in this article and in all the examples in the supplemental material at http://www.genetics.org/supplemental/.
This can be proven rigorously. Assume that the marginal fitness values in a DSL are gNN, gNS, gSS for genotypes NN, NS, and SS, respectively [e.g., (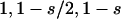 ) for an additive DSL with selection coefficient s], and the frequencies and fitnesses of genotypes at another DSL are pi and gi, respectively, where i is the index of possible genotypes. For an individual having genotype XY at locus A (XY can be NN, NS, or SS), the expected (average) overall fitness value is
) for an additive DSL with selection coefficient s], and the frequencies and fitnesses of genotypes at another DSL are pi and gi, respectively, where i is the index of possible genotypes. For an individual having genotype XY at locus A (XY can be NN, NS, or SS), the expected (average) overall fitness value is
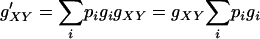 |
(A1) |
under the multiplicative multilocus fitness model and
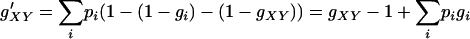 |
(A2) |
under the additive model. From the viewpoint of locus A, since XY can be any genotype at this locus, Equations A1 and A2 imply a systematic decrease (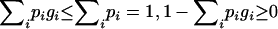 ) of fitness of every individual compared to a selection model with A as the only locus. Because gXY are relative fitness values, such changes of fitness will have very little, if any, impact on the selection process. For example, during random mating, simuPOP (Peng and Kimmel 2005) selects an individual with a probability that is proportional to his/her fitness value. The probability of being selected is the same for a systematic multiplicative change of fitness values (
) of fitness of every individual compared to a selection model with A as the only locus. Because gXY are relative fitness values, such changes of fitness will have very little, if any, impact on the selection process. For example, during random mating, simuPOP (Peng and Kimmel 2005) selects an individual with a probability that is proportional to his/her fitness value. The probability of being selected is the same for a systematic multiplicative change of fitness values (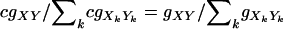 , where c is the multiplicative factor, and k iterates over the individuals in the population considered) and is only slightly different for an additive change when
, where c is the multiplicative factor, and k iterates over the individuals in the population considered) and is only slightly different for an additive change when 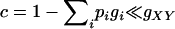 (usually the case for common diseases, proof not shown). This result can be easily extended to cases with more than two DSL.
(usually the case for common diseases, proof not shown). This result can be easily extended to cases with more than two DSL.
These analyses are confirmed by a special set of simulations, where the population is initialized with equilibrium f0 and ne for all DSL and starts evolving without a burn-in stage. We compare the evolution of these DSL with theoretical expectations and with simulations using the corresponding single-locus models and do not note a systematic difference. Two sample t-tests are used at each generation, and none of the P-values are <0.05.
References
- Bertina, R. M., B. P. Koeleman, T. Koster, F. R. Rosendaal, R. J. Dirven et al., 1994. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 369: 64–67. [DOI] [PubMed] [Google Scholar]
- Dean, M., M. Carrington, C. Winkler, G. A. Huttley, M. W. Smith et al., 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia growth and development study, multicenter AIDS cohort study, multicenter hemophilia cohort study, San Francisco city cohort, ALIVE study. Science 273: 1856–1862. [DOI] [PubMed] [Google Scholar]
- Di Rienzo, A., and R. R. Hudson, 2005. An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends Genet. 21: 596–601. [DOI] [PubMed] [Google Scholar]
- Harpending, H. C., M. A. Batzer, M. Gurven, L. B. Jorde, A. R. Rogers et al., 1998. Genetic traces of ancient demography. Proc. Natl. Acad. Sci. USA 95: 1961–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes, T. H., and C. R. Cantor, 1969. Evolution of protein molecules, pp. 21–132 in Mammalian Protein Metabolism, edited by M. N. Munro. Academic Press, New York.
- Kruglyak, L., 1999. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat. Genet. 22: 139–144. [DOI] [PubMed] [Google Scholar]
- Laan, M., and S. Pääbo, 1997. Demographic history and linkage disequilibrium in human populations. Nat. Genet. 17: 435–438. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., 1996. The new genomics: global views of biology. Science 274: 536–539. [DOI] [PubMed] [Google Scholar]
- Peng, B., and M. Kimmel, 2005. simuPOP: a forward-time population genetics simulation environment. Bioinformatics 21: 3686–3687. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., 2001. Are rare variants responsible for susceptibility to complex diseases. Am. J. Hum. Genet. 69: 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., and N. J. Cox, 2002. The allelic architecture of human disease genes: Common disease-common variant … or not? Hum. Mol. Genet. 11: 2417–2423. [DOI] [PubMed] [Google Scholar]
- Reich, D. E., and E. S. Lander, 2001. On the allelic spectrum of human disease. Trends Genet. 17: 502–510. [DOI] [PubMed] [Google Scholar]
- Saunders, A. M., W. J. Strittmatter, D. Schmechel, P. H. George-Hyslop, M. A. Pericak-Vance et al., 1993. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43: 1467–1472. [DOI] [PubMed] [Google Scholar]
- Smith, D. J., and A. J. Lusis, 2002. The allelic structure of common disease. Hum. Mol. Genet. 11: 2455–2461. [DOI] [PubMed] [Google Scholar]
- Terwilliger, J. D., and K. M. Weiss, 1998. Linkage disequilibrium mapping of complex disease: Fantasy or reality? Curr. Opin. Biotechnol. 9: 578–594. [DOI] [PubMed] [Google Scholar]
- van Essen, A. J., H. F. Busch, G. J. te Meerman and L. P. ten Kate, 1992. Birth and population prevalence of Duchenne muscular dystrophy in the Netherlands. Hum. Genet. 88: 258–266. [DOI] [PubMed] [Google Scholar]
- Yang, Q., M. J. Khoury, J. Friedman, J. Little and W. D. Flanders, 2005. How many genes underlie the occurrence of common complex diseases in the population? Int. J. Epidemiol. 34: 1129–1137. [DOI] [PubMed] [Google Scholar]