

Abstract
Activating mutations in the luteinizing hormone receptor (LHR) gene are one of the most common mutations found in the gonadotropin receptor genes. Human males with these mutations exhibit precocious puberty while females do not have an obvious phenotype. To better understand the pathophysiology of premature LHR activation, transgenic mice have been generated with an activating mutation in LHR and a genetically engineered ligand-activated LHR. This review will summarize the major findings obtained with these two genetically modified mouse models and briefly discuss the similarities and differences between them and with the human phenotype.
Keywords: Luteinizing hormone receptor, transgenic, constitutive activation, yoked hormone receptor
1. Introduction
The gonadotropins and their receptors play a critical role in male and female reproduction. The hormones are heterodimers and share a common α subunit and a hormone-specific β subunit (Hearn and Gomme, 2000). The receptors belong to the rhodopsin family of seven transmembrane G protein-coupled receptors and are characterized by a large ectodomain comprising about half of the receptor and sufficient for high affinity ligand binding. The luteinizing hormone (LH) receptor (LHR) binds both pituitary-derived LH and the placental hormone, chorionic gonadotropin (CG), because of the high degree of sequence homology that exists between the two β-subunits, while the follicle stimulating hormone (FSH) receptor (FSHR) binds pituitary-derived FSH (Fanelli et al., 2001; Ascoli et al., 2002). The primary signaling pathway activated by these receptors is the Gαs mediated cAMP pathway. LHR, the focus of this review, also activates phospholipase C with the subsequent induction of inositol phosphates as well as the MAPK pathway in heterologous cell types (HEK 293 and Cos-7) (Ascoli et al., 2002) and in the mouse Leydig tumor cell line, MA-10 (Hirakawa et al., 2002).
A number of developmental and reproductive abnormalities can be attributed to activating and inactivating mutations in the gonadotropins and their receptors (Huhtaniemi and Themmen, 2005; Themmen, 2005). Consequently, genetically modified mouse models with loss-of-function and gain-of-function phenotypes have been generated to elucidate the molecular mechanisms of gonadotropin action in vivo (Risma et al., 1995; Kumar et al., 1997; Dierich et al., 1998; Kumar et al., 1998; Lei et al., 2001; Zhang et al., 2001; Rulli et al., 2002; Matzuk et al., 2003; Rulli et al., 2003; Ma et al., 2004). Of all the human mutations that are known for gonadotropins and their receptors, the most reported are the activating and inactivating LHR mutations. The inactivating mutations are spread throughout the molecule whereas the activating mutations tend to be restricted more to the transmembrane helices (TMH) and intracellular loops encoded by exon 11, with the largest number of activating mutations in TMH 6 (Shenker, 2002; Huhtaniemi and Themmen, 2005; Themmen, 2005). Although there are no reports of naturally occurring activating mutations in the extracellular domain, engineered mutations of a serine residue in the hinge region of LHR result in constitutive activity (Nakabayashi et al., 2003).
Inactivating mutations in humans result in male pseudohermaphroditism, the severity of which depends on whether the mutation partially or completely inhibits receptor activity, and impairs follicle maturation in females (Huhtaniemi and Themmen, 2005; Themmen, 2005). The corresponding mouse model, the LHR knock- out mouse, is infertile but the males do not exhibit pseudohermaphroditism (Lei et al., 2001; Zhang et al., 2001). A large number of activating mutations in LHR have been identified in boys presenting with sporadic or the more common familial male-limited precocious puberty (FMPP) (Shenker, 2002; Huhtaniemi and Themmen, 2005; Themmen, 2005). These mutations result in elevated levels of testosterone and Leydig cell hyperplasia. One of the most common activating mutations is the missense mutation in TMH 6 resulting in the replacement of aspartic acid with glycine (D578G). Females with the activating mutations do not have an apparent phenotype. A particularly potent activating somatic mutation (D578H) has been identified in boys with testicular adenomas wherein the mutation is limited to the adenoma and not the surrounding normal tissue (Liu et al., 1999; Canto et al., 2001; Richter-Unruh et al., 2002).
We have genetically engineered a ligand-mediated constitutively active LHR (YHR) by covalently linking the ligand human (h) CG to LHR in a single polypeptide chain (Fig. 1A) and tested its activity in vitro in cell culture (Wu et al., 1996) and in vivo in transgenic mice (Meehan et al., 2005). We also generated mice expressing rat LHR with a D556H mutation (equivalent to the human D578H mutation) as an example of ligand-independent receptor activation (Meehan et al., 2005). The key findings obtained with these transgenic mouse models, including the similarities and differences with the human phenotype of constitutive LHR activation, will be reviewed.
Fig. 1.
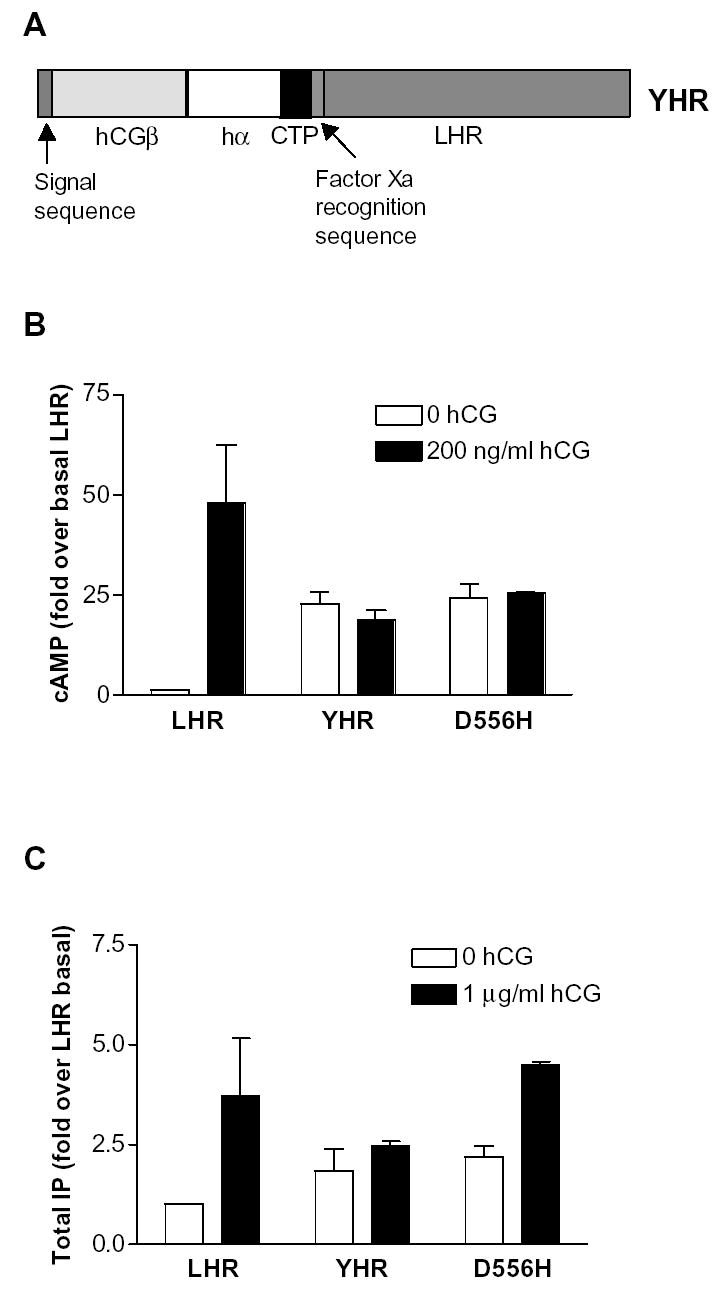
Schematic of YHR and its signaling properties. (A) YHR consists of hCGβ with its signal sequence linked to human α-subunit through the C-terminal peptide (CTP) of hCGβ. This yoked hormone, in turn, is covalently attached to the extracellular domain of rLHR via an additional CTP and a factor Xa cleavage sequence. Constitutive activation of YHR and the D556H LHR mutant showing increases in basal cAMP (B) and IP (C) in HEK 293 cells in the absence and presence of additional hCG. The data are presented as fold-increase over LHR in the absence of exogenous hCG.
2. In vitro bioactivity of D556H LHR and YHR
Analysis of the major signaling pathways in HEK and COS-7 cells demonstrated that YHR, rat D556H LHR and human D578H LHR increased the basal levels of cAMP and were unresponsive to exogenous hCG (Wu et al., 1996; Liu et al., 1999; Angelova et al., 2002). The level of constitutive activity exhibited by YHR and D556H LHR are similar (Fig. 1B) and neither were further activated by additional hCG. In addition, rat D556H LHR and YHR also showed an increase in the basal level of inositol phosphate (IP) (Fig. 1C) similar to that demonstrated for human D578H LHR (Liu et al., 1999). However, D556H was responsive to exogenous hCG whereas YHR was not. Recent studies have shown that the D578H mutation can also cause a constitutive activation of the MAPK pathway in the mouse Leydig tumor cell line, MA-10 (Hirakawa et al., 2002).
3. In vivo bioactivity of D556H LHR and YHR
Transgenes containing the complete coding sequence of D556H LHR with a N-terminal myc tag and YHR were cloned under the control of the inhibin α-subunit promoter and transgenic mice were generated by pronuclear injection (Meehan et al., 2005).
3.1. Phenotype of D556H LHR transgenic mice
Of the 5 female and 3 male founder mice, only one male and one female founder were fertile. However, neither of these fertile mice showed gonadal specific expression of D556H. Therefore, gonadal expression of the D556H correlated with infertility (Meehan et al., 2005). A limited analysis of the founder mice was performed at 8 months of age. The average testicular and seminal vesicle weights of the two founders with gonadal transgene expression were lower than the testis weight of non-transgenic controls (Table 1). The average serum testosterone level in the two D556H mice was reduced compared to control mice. Serum LH levels in D556H mice were similar to controls while serum FSH levels were reduced by more than 50%. The statistical significance of these apparent differences could not be determined due to the small sample size of the D556H mice (n=2). However, overall these results are similar to that obtained with transgenic YHR adult male mice (see below and Table 2).
Table 1.
Analysis of male D556H mice
| Genotype | Testis Weight (mg) | Seminal Vesicle Weight (mg) | Serum Testosterone (ng/ml) | Serum LH (ng/ml) | Serum FSH (ng/ml) |
|---|---|---|---|---|---|
| Control | 223±8 (7) | 409.5±2.7 (7) | 1.51±0.4 (6) | 0.51±0.03 (7) | 73±6 (4) |
| D556H | 184±5 (2) | 181±32 (2) | 0.61±0.3 (2) | 0.43±0.16 (2) | 32±8 (2) |
Data are mean ± SEM (n) for control animals and mean ± range (n) for D556H
Table 2.
Hormonal profile of male YHR+ mice
| Age (weeks) | Genotype | Testis Weight (mg) | SV weight (mg) | Testicular Testosterone (ng/g testis) | Serum Testosterone (ng/ml) | LH (ng/ml) | FSH (ng/ml) |
|---|---|---|---|---|---|---|---|
| 3 | WT | 54.2±4.1 | 3.1±0.4 | 1.5±0.4 | 0.22±0.02 | 0.50±0.18 | 19.1±2.0 |
| YHR+ | 35.1±1.7* | 5.8±0.6* | 18±2.9* | 0.76±0.47* | 0.1±0 *a | 1.0±0.1* | |
| 5 | WT | 120.0±7.9 | 16.4±3.0 | 4.8±0.9 | 0.37±0.09 | 0.41±0.22 | 33.5±3.3 |
| YHR+ | 65.2±2.2* | 31.1±5.1* | 69.9±5.1* | 1.35±0.12* | 0.13±0.02* | 2.8±0.2* | |
| 8 | WT | 177.9±4.0 | 113.3±5.3 | 100.8±49.1 | 7.0±2.5 | 0.26±0.03 | 38.7±2.3 |
| YHR+ | 106.7±4.0* | 99.0±10.8 | 72.6±11.9 | 1.7±0.5 | 0.35±0.05 | 16.6±3.8* | |
| 12 | WT | 200.1±2.4 | 191.6±10.2 | 78.1±50.5 | 0.87±0.27 | 0.91±0.50 | 42.6±3.1 |
| YHR+ | 156.1±3.7* | 143.5±5.6* | 102.3±47.5 | 3.8±1.9 | 0.26±0.03 | 19.6±1.5* |
Data are mean ±SEM (n=5–10)
significantly different from WT controls, P<0.05
values were below the standard curve and were assigned the value of the lowest standard (0.1 ng/ml).
Histological examination of the testes at 8 months of age did not reveal any apparent differences in the D556H expressing testes compared to control (Fig. 2, A and B). It was surprising that Leydig cell adenomas were not observed in these testes as all known cases of humans with the analogous D578H mutation exhibit adenomas (Liu et al., 1999; Canto et al., 2001; Richter-Unruh et al., 2002). Recent studies indicate that in hCG overexpressing mice, Leydig cell adenomas only occur in prepubertal mice and are restricted to fetal Leydig cells (Ahtiainen et al., 2005). Since the founders were infertile, we were unable to examine young mice for precocious puberty and premature testosterone synthesis or determine the reason for the infertility. It is possible that although spermatogenesis appears normal in the testis, sperm maturation in the epididymis is altered.
Fig. 2.
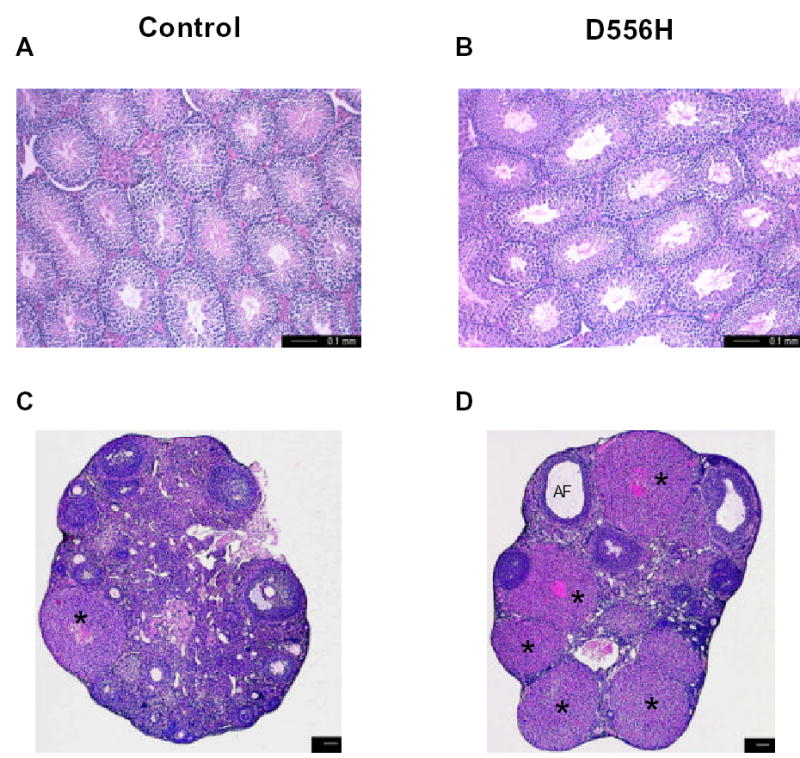
Photomicrographs of testicular and ovarian sections of control (A and C, respectively) and D556H founder (B and D) mice at 8 months of age. *, corpus luteum, AF, degenerating follicles. Scale bars represent 100 μm (C, D).
At 8 months of age, female transgenic founders were in a state of persistent diestrus compared to control animals that were cycling. Serum progesterone levels of the four D556H founders exhibiting gonadal specific transgene expression averaged 1.56 ± 0.5 ng/ml compared to 0.35 ± 0.15 ng/ml for control mice (n=4). This elevation in progesterone levels is consistent with the highly luteinized ovary of the D556H mice (Fig. 2D). Serum testosterone levels also appeared slightly elevated in the D556H founders (0.29 ± 0.04 ng/ml) compared to controls (0.17 ± 0.01 ng/ml). Ovarian sections of D556H mice showed an increase in the number of corpora lutea, a decrease in the number of developing follicles, interstitial cell hypertrophy and degenerating follicles compared to non-transgenic controls or founders that did not express the transgene (Fig. 2, C and D). Hemorrhagic cysts were evident in some of the transgenic ovaries similar that observed in the ovaries of the transgenic YHR mice discussed below. To date, there is no report of women with a D578H mutation; this mutation has appeared only as a somatic mutation restricted to Leydig cell tumors in boys ranging in age from 5 to 8 years, suggesting that this particular mutation is incompatible with germ line transmission.
3.2. Phenotype of the YHR transgenic mice
Male mice expressing the YHR transgene (YHR+) were fertile while female mice were subfertile. RT-PCR analysis of RNA from several tissues showed high levels of expression in the gonads of prepubertal and adult animals (Meehan et al., 2005). Expression was also detected in brain and low levels were detected in the heart (Meehan et al., 2005).
In male mice, most of the phenotypic and hormonal changes occurred at the pre-pubertal ages of 3 and 5 weeks. Serum and testicular testosterone levels were significantly elevated (Table 2) and, as expected, accompanied by an increase in seminal vesicle weight. Interestingly, seminal vesicle weights were reduced in adult animals (Meehan et al., 2005). The elevated testosterone suppressed LH and FSH levels, the latter remaining suppressed even in 1 year old YHR+ mice when testosterone levels were no longer elevated (unpublished observation). Testicular weights were reduced in YHR+ mice at prepubertal and adult ages most likely as a result of reduced FSH levels. The reason for the persistent reduction in FSH levels is unclear, but suggests that the premature increase in testosterone levels permanently alters the regulation of the hypothalamic-pituitary-gonadal axis. Preliminary studies suggest that FSH mRNA levels are decreased in the pituitaries of YHR+ mice, although the mechanism is unknown. Surprisingly, although testosterone levels were elevated, there was no evidence that the mice underwent precocious puberty as observed in humans. For example, spermatogenesis was not initiated earlier in YHR+ mice as spermatids were not detected in testicular sections of 3-week-old mice. There was also no evidence of Leydig cell hyperplasia as is observed in humans with constitutively active LHR.
Analogous transgenic mouse models, wherein LHR is activated by an overexpression of ligand, produced either ubiquitously (hCG) or in a pituitary-specific manner (LH) have been generated. In mouse models of ectopic overexpression of hCG producing very high levels of testosterone, infertility and Leydig cell hyperplasia were observed in older animals (Rulli et al., 2002; Matzuk et al., 2003; Rulli et al., 2003). Futher studies showed that although there was a significant increase in testosterone levels in hCG expressing mice as early as postnatal day 5, there was no evidence of early puberty (Ahtiainen et al., 2005). LHβ expressing male mice were not well characterized, but were reported to be subfertile (Risma et al., 1995). It is worth noting that in vitro studies have shown that at very high levels (similar to that produced in the hCG overexpressing mice), hCG is not specific for LHR, but can also activate the thyroid stimulating hormone receptor and, to a lesser extent, FSHR (Schubert et al., 2003).
The female phenotype of YHR+ mice was more distinct than that of the male and similar to that observed in the hCG and LH overexpressing mice (Risma et al., 1995; Rulli et al., 2002; Matzuk et al., 2003). YHR+ female mice exhibited precocious puberty as evidenced by vaginal opening occurred at 23.5 days compared to an average of 31.3 days for WT littermates (Meehan et al., 2005). Consistent with this observation was an increase in the ovarian estrogen levels (Fig. 3A) and a 10-fold increase in the uterine weight of 5-week-old YHR+ mice. There was also a 4-fold increase in progesterone levels in the YHR+ mice (Fig. 3B), with these increases in the steroid hormone levels resulting in a decrease in the serum levels of LH and FSH (Figs. 3C and D). These hormonal changes were no longer apparent in 12-week-old adult mice. Surprisingly, ovarian testosterone levels were not elevated in YHR+ mice (Meehan et al., 2005). The subfertility in the YHR+ female mice can be attributed to the abnormalities observed in the ovaries. At 5 weeks of age, increased folliculogenesis and luteinization were apparent and some of the corpora lutea retained the oocyte (Fig. 4A and B). Chronic YHR activity likely prevented the LH surge and resulted in anovulation. Degenerative changes including follicular cysts, interstitial cell hypertrophy and luteinzation and follicular atresia were apparent in mice at 12 weeks of age and in some mice as early as 8 weeks of age (Fig. 4C and D). Many of the YHR+ mice with multiple hemorrhagic cysts were acyclic and remained in a state of prolonged estrus with highly elevated estradiol and testosterone levels. The lack of normal follicular development in the YHR+ female mice suggests that premature ovarian failure may occur in these mice.
Fig. 3.
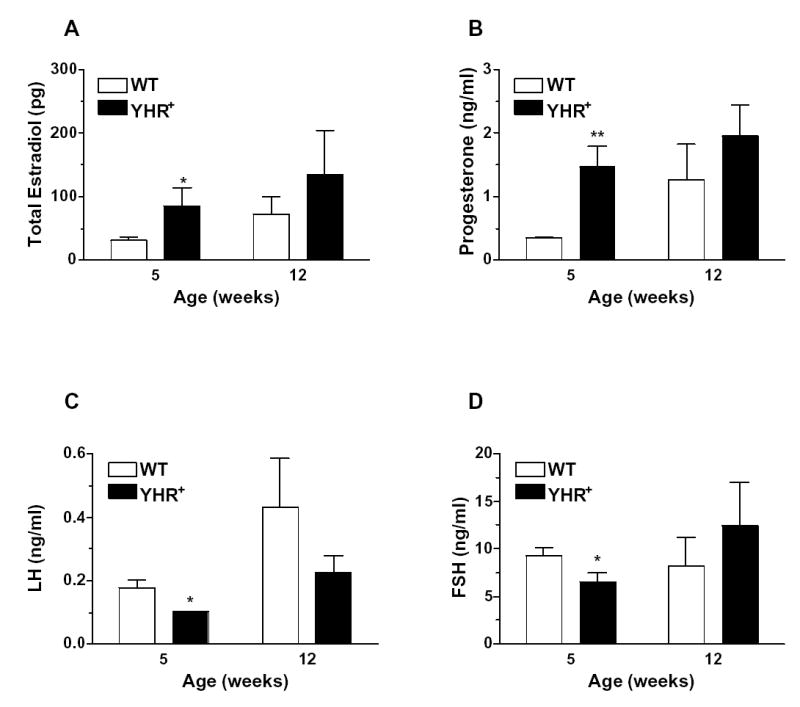
Hormonal profile of female YHR+ transgenic mice at prepubertal and adult ages. (A) Estradiol levels were measured in ovarian extracts and the data represent amount of hormone from both ovaries. (B, C and D) serum progesterone, LH and FSH respectively. Data are presented as mean ± SEM. n=5–8. * P<0.05, **P < 0.01 compared to WT.
Fig. 4.
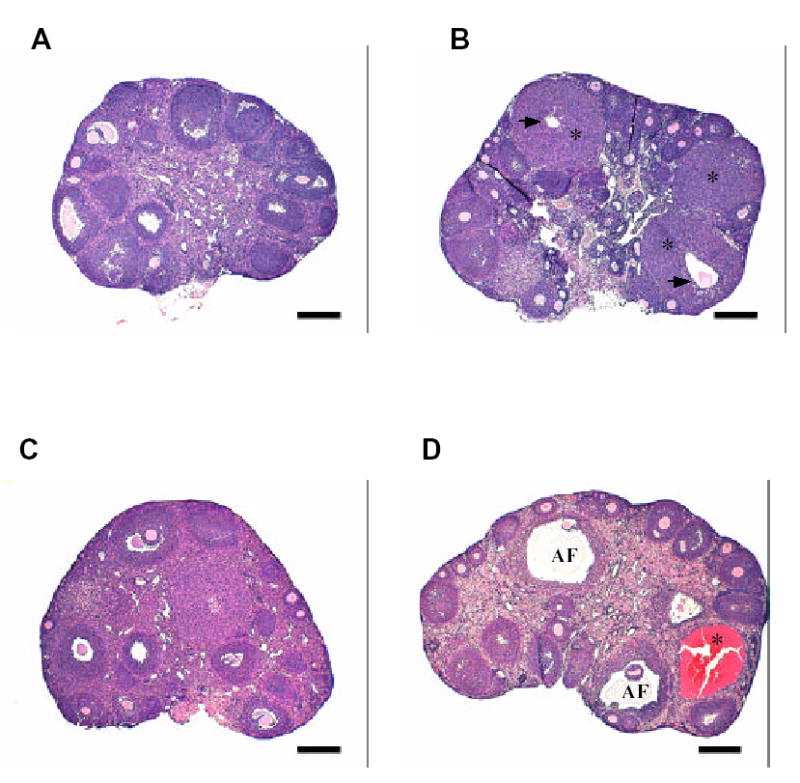
Photomicrographs of WT and YHR+ ovary. (A) WT ovary at 5 weeks and (C) 12 weeks of age. (B) YHR+ ovary at 5 weeks showing the presence of corpora lutea (*) with retained oocyte (arrow). (D) YHR+ ovary at 12 weeks demonstrating the presence of degenerating follicles (AF) and hemorrhagic cysts (*). Scale bars represent 250 μm.
The difference in fertility between the D556H and YHR mouse models is intriguing, considering the similarities in their basic signaling pathways and in their hormonal profile and gonadal histology, at least in the older mice. However, it is important to note that our analysis of the D556H mice was limited and a detailed analysis of prepubertal D556H mice might provide important insights into the disparate fertility phenotypes. The YHR+ mouse model offers a novel system for defining the signaling pathways mediated by constitutively active LHR in the gonads and for molecular studies on premature ovarian failure.
4. Conclusions
Mouse models have been developed to either mimic the human genetic mutations in gonadotropins and receptors or to better understand the hypothalamic-pituitary-gonadal axis, the major system regulating mammalian reproduction. While several models have provided information on new regulatory mechanisms, many do not completely mimic the human condition resulting from genetic changes as can be seen from the mouse models described in this article. In the D556H and YHR+ mouse models, the female phenotype of precocious puberty and ovarian degeneration is more obvious than that in the male, this being the opposite of the human condition. Women with constitutively active LHR do not undergo precocious puberty and their ovarian function appears normal (Rosenthal et al., 1996; Latronico et al., 2000). To date, there have been no reports of females with a D578H mutation; rather it has appeared only as a somatic mutation restricted to Leydig cell tumors. Although YHR+ male mice produce testosterone at 3 weeks, there is no evidence of accelerated spermatogenesis in testicular sections. Therefore, it appears that in mice, testosterone alone is not sufficient for pubertal development.
One possibility for the difference between the phenotype of mice expressing YHR or D556H LHR and humans with a constitutively active LHR is that the expression of the transgene under the control of the inhibin α-subunit promoter does not mimic the spatial and temporal expression of LHR. As a result, some of the signaling pathways activated in the transgenic mice may be different from that in humans. Attempts to detect cell specific expression of YHR and myc D556H LHR in the gonads by immunohistochemistry were unsuccessful. Although the inhibin α-subunit mRNA can be detected in Leydig cells of immature and adult rats (Roberts et al., 1989) and the inhibin α-subunit promoter can target transgene expression to mouse Leydig cells (Kananen et al., 1996) it also targets expression to Sertoli cells (Hsu et al., 1995). In the rat testis, the temporal expression of LHR and inhibin α-subunit protein appears to be similar as both can be detected in fetal Leydig cells as early as embryonic day 14 (Huhtaniemi, 1995; Majdic G et al., 1997; O’Shaughnessy et al., 1997). In the ovaries, the inhibin α-subunit promoter has been shown to direct transgene expression to theca and granulosa cells of secondary and preovulatory follicles (Meunier et al., 1988) whereas LHR is only present in the granulosa cells of preovulatory follicles (Richards, 1994). Therefore, in order to obtain a mouse model that is exactly like the human condition with the activating LHR mutations it will be necessary to faithfully mimic the cell specific and temporal expression of human LHR. Knock-in mice with germ-line LHR activating mutations would allow determination of whether the differences in the phenotype of the mouse models described herein and those in the human condition are due to the experimental system or represent important and interesting species differences in reproductive development.
Acknowledgments
The authors wish to thank Dr. David Puett for his support and critical reading of the manuscript. This work was supported by NIH grants HD 044119 and DK 33973.
References
- Ahtiainen P, Rulli SB, Shariatmadari R, Pelliniemi LJ, Toppari J, Poutanen M, Huhtaniemi IT. Fetal but not adult Leydig cells are susceptible to adenoma formation in response to persistently high hCG level: a study on hCG overexpressing transgenic mice. Oncogene. 2005;24:7301–7309. doi: 10.1038/sj.onc.1208893. [DOI] [PubMed] [Google Scholar]
- Angelova K, Fanelli F, Puett, D. A model for constitutive lutropin receptor activation based on molecular simulation and engineered mutations in transmembrane helices 6 and 7. J Biol Chem. 2002;277:32202–32213. doi: 10.1074/jbc.M203272200. [DOI] [PubMed] [Google Scholar]
- Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev. 2002;23:141–174. doi: 10.1210/edrv.23.2.0462. [DOI] [PubMed] [Google Scholar]
- Canto P, Soderlund D, Ramon G, Nishimura E, Mendex JP. Mutational analysis of the luteinizing hormone receptor gene in two individuals with leydig cell tumors. Am J Med Genet. 2001;108:148–152. doi: 10.1002/ajmg.10218. [DOI] [PubMed] [Google Scholar]
- Dierich A, Sairam MR, Monaco L, Fimia GM, Gansmuller A, LeMeur M, Sassone-Corsi P. Impairing follicle-stimulating hormone (FSH) signaling in vivo: targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc Natl Acad Sci USA. 1998;95:13612–13617. doi: 10.1073/pnas.95.23.13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanelli F, Themmen APN, Puett D. Lutropin receptor function: insights from natural, engineered and computer-simulated mutations. IUBMB Life. 2001;51:149–155. doi: 10.1080/152165401753544214. [DOI] [PubMed] [Google Scholar]
- Hearn MT, Gomme PT. Molecular architecture and biorecognition process of the cysteine knot protein superfamily: part I. The glycoprotein hormones. J Mol Recog. 2000;13:223–278. doi: 10.1002/1099-1352(200009/10)13:5<223::AID-JMR501>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Hirakawa T, Galet C, Ascoli M. MA-10 cells transfected with the human lutropin/choriogonadotropin receptor (hLHR): a novel experimental paradigm to study the functional properties of the hLHR. Endocrinology. 2002;143:1026–1035. doi: 10.1210/endo.143.3.8702. [DOI] [PubMed] [Google Scholar]
- Hsu SY, Lai RJ, Nanuel D, Hsueh AJW. Different 5′-flanking regions of the inhibin-α gene target transgenes to the gonad and adrenal in an age-dependent manner in transgenic mice. Endocrinology. 1995;136:5577–5586. doi: 10.1210/endo.136.12.7588311. [DOI] [PubMed] [Google Scholar]
- Huhtaniemi I. Molecular aspects of the ontogeny of the pituitary-gonadal axis. Reproduction Fertility and Development. 1995;7:1025–1035. doi: 10.1071/rd9951025. [DOI] [PubMed] [Google Scholar]
- Huhtaniemi IT, Themmen AP. Mutations in human gonadotropin and gonadotropin-receptor genes. Endocrine. 2005;26:207–217. doi: 10.1385/ENDO:26:3:207. [DOI] [PubMed] [Google Scholar]
- Kananen K, Markkula M, El-Hefnawy T, Zhang FP, Paukku T, Su JGJ, Hsueh AJW, Huhtaniemi I. The mouse inhibin α-subunit promoter directs SV40 T-antigen to leydig cells in transgenic mice. Molecular and Cellular Endocrinology. 1996;119:135–146. doi: 10.1016/0303-7207(96)03802-6. [DOI] [PubMed] [Google Scholar]
- Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nature Genetics. 1997;15:201–204. doi: 10.1038/ng0297-201. [DOI] [PubMed] [Google Scholar]
- Kumar TR, Low MJ, Matzuk MM. Genetic rescue of follicle-stimulating hormone β-deficient mice. Endocrinology. 1998;139:3289–3295. doi: 10.1210/endo.139.7.6111. [DOI] [PubMed] [Google Scholar]
- Latronico AC, Lins TSS, Brito VN, Arnhold IJP, Mendonca BB. The effect of distinct activating mutations of the luteinizing hormone receptor gene on the pituitary–gonadal axis in both sexes. Clinical Endocrinology. 2000;53:609–613. doi: 10.1046/j.1365-2265.2000.01135.x. [DOI] [PubMed] [Google Scholar]
- Lei ZM, Mishra S, Zou W, Xu B, Foltz M, Li X, Rao CV. Targeted disruption of luteinizing hormone/human chorionic gonadotropin receptor gene. Mol Endocrinol. 2001;15:184–200. doi: 10.1210/mend.15.1.0586. [DOI] [PubMed] [Google Scholar]
- Liu G, Duranteau L, Carel JC, Monroe J, Doyle DA, Shenker A. Leydig-cell tumors caused by an activating mutation of the gene encoding the luteinizing hormone receptor. N Engl J Med. 1999;341:1731–1736. doi: 10.1056/NEJM199912023412304. [DOI] [PubMed] [Google Scholar]
- Ma X, Dong Y, Matzuk MM, Kumar TR. Targeted disruption of luteinizing hormone beta-subunit leads to hypogonadism, defects in gonadal steroidogenesis, and infertility. Proc Natl Acad Sci USA. 2004;101:17294–17299. doi: 10.1073/pnas.0404743101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdic GASM, Sharpe R, Evans L, Groome N, Saunders P. Testicular expression of inhibin and activin subunits and follistatin in the rat and human fetus and enonate and during postnatal development in the rat. Endocrinology. 1997;138:2136–2147. doi: 10.1210/endo.138.5.5135. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, DeMayo FJ, Hadsell LA, Kumar TR. Overexpression of human chorionic gonadotropin causes multiple reproductive defects in transgenic mice. Biol Reprod. 2003;69:338–346. doi: 10.1095/biolreprod.102.013953. [DOI] [PubMed] [Google Scholar]
- Meehan TP, Harmon BG, Overcast ME, Yu KK, Camper SA, Puett D, Narayan P. Gonadal defects and hormonal alterations in transgenic mice expressing a single chain human chorionic gonadotropin-lutropin receptor complex. J Mol Endocrinol. 2005;34:489–503. doi: 10.1677/jme.1.01669. [DOI] [PubMed] [Google Scholar]
- Meunier H, Cajander S, Roberts V, Rivier C, Sawchenko P, Hsueh A, Vale W. Rapid changes in the expression of inhibin a-, bA-, and bB-subunits in ovarian cell types during the rat estrous cycle. Mol Endocrinol. 1988;2:1352–1363. doi: 10.1210/mend-2-12-1352. [DOI] [PubMed] [Google Scholar]
- Nakabayashi K, Kudo M, Hsueh AJW, Maruo T. Activation of the lutenizing hormone receptor in the extracellular domain. Mol Cell Endocrinol. 2003;202:139–144. doi: 10.1016/s0303-7207(03)00075-3. [DOI] [PubMed] [Google Scholar]
- O’Shaughnessy PJ, McLelland DMWM. Regulation of luteinizing hormone-receptor and follicle-stimulating hormone-receptor messenger ribonucleic acid levels during development in the neonatal mouse ovary. Biol Reprod. 1997;57:602–608. doi: 10.1095/biolreprod57.3.602. [DOI] [PubMed] [Google Scholar]
- Richards JS. Hormonal control of gene expression in the ovary. Endocrine Reviews. 1994;15:725–751. doi: 10.1210/edrv-15-6-725. [DOI] [PubMed] [Google Scholar]
- Richter-Unruh A, Wessels HT, Menken U, Bergmann M, Schmittmann-Ohters K, Schaper J, Tappeser S, Hauffa BP. Male LH-Independent Sexual Precocity in a 3.5-Year-Old Boy Caused by a Somatic Activating Mutation of the LH Receptor in a Leydig Cell Tumor. J Clin Endocrinol Metab. 2002;87:1052–1056. doi: 10.1210/jcem.87.3.8294. [DOI] [PubMed] [Google Scholar]
- Risma KA, Clay CM, Nett TM, Wagner T, Yun J, Nilson JH. Targeted overexpression of luteinizing hormone in transgenic mice leads to infertility, polycystic ovaries, and ovarian tumors. Proc Natl Acad Sci USA. 1995;92:1322–1326. doi: 10.1073/pnas.92.5.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts V, Meunier H, Sawchenko P, Vale W. Differential production and regulation of inhibin subunits in rat testicular cell types. Endocrinology. 1989;125:2350–2359. doi: 10.1210/endo-125-5-2350. [DOI] [PubMed] [Google Scholar]
- Rosenthal IM, Refetoff S, Rich B, Barnes RB, Sunthornthepvarakul T, Parma J, Rosenfield RL. Response to challenge with gonadotropin-releasing hormone agonist in a mother and her two sons with a constitutively activating mutation of the luteinizing hormone receptor--a clinical research center study. J of Clini Endocrinol Metab. 1996;81:3802–3806. doi: 10.1210/jcem.81.10.8855841. [DOI] [PubMed] [Google Scholar]
- Rulli SB, Kuorelahti A, Karaer O, Pelliniemi LJ, Poutanen M, Huhtaniemi I. Reproductive disturbances, pituitary lactotrope adenomas, and mammary gland tumors in transgenic female mice producing high levels of human chorionic gonadotropin. Endocrinology. 2002;143:4084–4095. doi: 10.1210/en.2002-220490. [DOI] [PubMed] [Google Scholar]
- Rulli SB, Ahtiainen P, Makela S, Toppari J, Poutanen M, Huhtaniemi I. Elevated steroidogenesis, defective reproductive organs, and infertility in transgenic male mice overexpressing human chorionic gonadotropin. Endocrinology. 2003;144:4980–4990. doi: 10.1210/en.2003-0403. [DOI] [PubMed] [Google Scholar]
- Schubert RL, Narayan P, Puett D. Specificity of Cognate Ligand-Receptor Interactions: Fusion Proteins of Human Chorionic Gonadotropin and the Heptahelical Receptors for Human Luteinizing Hormone, Thyroid-Stimulating Hormone, and Follicle-Stimulating Hormone. Endocrinology. 2003;144:129–137. doi: 10.1210/en.2002-220829. [DOI] [PubMed] [Google Scholar]
- Shenker A. Activating mutations of the lutropin/choriogonadotropin receptor in precocious puberty. Receptors and Channels. 2002;8:3–18. [PubMed] [Google Scholar]
- Themmen APN. An update of the pathophysiology of human gonadotrophin subunit and receptor gene mutations and polymorphisms. Reproduction. 2005;130:263–274. doi: 10.1530/rep.1.00663. [DOI] [PubMed] [Google Scholar]
- Wu C, Narayan P, Puett D. Protein engineering of a novel constitutively active hormone-receptor complex. J Biol Chem. 1996;271:31638–31642. doi: 10.1074/jbc.271.49.31638. [DOI] [PubMed] [Google Scholar]
- Zhang F-P, Poutanen M, Wilbertz J, Huhtaniemi I. Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LuRKO) mice. Mol Endocrinol. 2001;15:172–183. doi: 10.1210/mend.15.1.0582. [DOI] [PubMed] [Google Scholar]