

Abstract
Interleukin-18 (IL-18) plays an important role in host defense against microbial pathogens. Many poxviruses encode homologous IL-18 binding proteins (IL-18BP) that neutralize IL-18 activity. Here, we examined whether IL-18BP neutralizes IL-18 activity by binding to the same region of IL-18 where IL-18 receptor (IL-18R) binds. We introduced alanine substitutions to known receptor binding sites of human IL18, and found that only the substitution of Leu5 reduced the binding affinity of IL-18 with IL-18BP of variola virus (varvIL-18BP) by more than 4-fold. The substitutions of Lys53 and Ser55, which were not previously known to be part of the receptor binding site but that are spatially adjacent to Leu5, reduced the binding affinity to varvIL-18BP by approximately 100- and 7-fold, respectively. These two substitutions also reduced the binding affinity with human IL-18R alpha subunit (hIL-18Rα) by 4- and 2-fold, respectively. Altogether, our data shows that varvIL-18BP prevents IL-18 from binding to IL-18R by interacting with three residues that are part of the binding site for hIL-18Rα.
Keywords: poxvirus, variola virus, smallpox, IL-18, IL-18BP, biacore, surface plasmon resonance
Introduction
Interleukin-18 (IL-18) is a pro-inflammatory cytokine that belongs to the IL-1 superfamily (Dinarello, 2006; Gracie, Robertson, and McInnes, 2003). Similar to IL-1β, IL-18 is synthesized as an inactive precursor (proIL-18) and becomes active only after its cleavage by the IL-1β-converting enzyme/caspase-1. The recently determined human IL-18 (hIL-18) structure shows a β-trefoil fold that is similar to that of IL-1β (Kato et al., 2003). The receptor for IL-18 (IL-18R) is a heterodimer consisting of the ligand-binding α subunit (IL-18Rα) and the signal-transducting β subunit (IL-18Rβ). Mutagenesis studies of hIL-18 revealed the presence of three receptor binding sites: two of these (site I and II) are important for IL-18Rα binding and the third (site III) is probably involved in IL-18Rβ binding (Kato et al., 2003). The binding of IL-18 with IL-18R results in a ternary complex that triggers a signal cascade involving IL-1 receptor activating kinase (IRAK), TRAF6 and NF-κB (Kojima et al., 1998; Robinson et al., 1997).
IL-18 was formerly known as the interferon-γ-inducing factor. It acts synergistically with co-stimulants to induce interferon-γ production from T lymphocytes and macrophages and enhances the cytotoxicity of natural killer cells (Dinarello, 2006; Gracie, Robertson, and McInnes, 2003). Hence, it is an important regulator of both innate and acquired immune responses. However, it is also implicated in endotoxin-induced liver damage and some chronic inflammatory diseases (Gracie, Robertson, and McInnes, 2003; Tsutsui et al., 2000). Not surprisingly, its activity is modulated in vivo by a negative feedback mechanism involving a natural occurring IL-18 antagonist, the IL-18 binding protein (IL-18BP) (Novick et al., 1999), whose expression is induced by IFN-γ (Muhl et al., 2000; Paulukat et al., 2001). IL-18BP differs significantly in sequence from IL-18R, but it binds to IL-18 with a sub-nanomolar affinity and prevents IL-18 from binding to its receptor (Aizawa et al., 1999; Novick et al., 1999).
Functional homologs of IL-18BP have also been found in poxviruses, namely molluscum contagiosum virus, vaccinia virus, cowpox virus, variola virus and ectromelia virus (Born et al., 2000; Calderara, Xiang, and Moss, 2001; Esteban, Nuara, and Buller, 2004; Smith, Bryant, and Alcami, 2000; Xiang and Moss, 1999). Among them, variola virus and molluscum contagiosum virus infect humans exclusively (Moss, 2001). Variola virus is the causative agent for smallpox, a deadly disease that was eradicated after a global vaccination campaign using the closely related vaccinia virus. It is now considered as one of the most dangerous bioterrosim agents, as the majority of the population is not immunized and no antiviral or generally acceptable vaccine for smallpox is available. Molluscum contagiosum virus causes small, benign skin lesions that are usually self-limited but become more extensive in immunocompromised individuals (Gottlieb and Myskowski, 1994). The poxvirus-encoded IL-18BP represents an immune evasion mechanism for poxviruses. Viral infection induces IL-18 release and the administration of exogenous IL-18 in mice protects against herpes simplex virus and vaccinia virus infections (Fujioka et al., 1999; Pirhonen, 2001; Tanaka-Kataoka et al., 1999). Poxviral IL-18BP suppresses IL-18-mediated activity and contributes to the virulence of ectromelia virus and vaccinia virus (Born et al., 2000; Reading and Smith, 2003; Symons et al., 2002).
The IL-18 binding site was identified in human and several viral IL-18BP through site-directed mutagenesis and binding studies (Esteban and Buller, 2004; Xiang and Moss, 2001a; Xiang and Moss, 2001b). Despite a relatively low overall sequence identity between the human and viral IL-18BP, the residues that constitute the IL-18 binding site are mostly conserved in all IL-18BP. In the current study, we identified in hIL-18 the main binding site for IL-18BP of variola virus (varvIL-18BP) and found that it is also part of the binding site II for hIL-18Rα, suggesting that varvIL-18BP neutralizes hIL-18 activity by competing with hIL-18Rα for a common binding site.
RESULTS
Mutagenesis and purification of hIL-18
To determine whether variola virus IL-18BP (varvIL-18BP) competes with hIL18R for the same binding site in hIL-18, we set out to test whether the residues of hIL-18 that form the hIL18R binding sites are also important for the binding with varvIL-18BP. 14 hIL-18 residues were previously identified to form the binding site I, II for hIL18Rα and the binding site III for hIL18Rβ (Kato et al., 2003). They are Arg13, Asp17, Met33, Asp35 and Asp132 (Site I), Lys4, Leu5, Lys8, Arg58, Met60 and Arg104 (Site II), and Lys79, Lys84 and Asp98 (Site III) (Fig. 1A). In addition, Glu6, Lys53 and Ser55 were also chosen for substitution. Glu6 and Lys53 were previously suggested by molecular modeling to be part of the binding site for human IL-18BP, and hIL-18 mutant with K53A substitution is more resistant to neutralization by human IL-18BP (Kim et al., 2001; Kim et al., 2000). (Glu6 and Lys53 were previously referred to as Glu42 and Lys89 by Kim et al., whom numbered the residues from the first residue of IL-18 precursor). Ser55 was specifically chosen for substitution after examination of hIL-18 structure following our preliminary studies. It is a surface residue that is spatially adjacent to Lys53 (Fig. 1A). Altogether, 17 hIL-18 residues were individually substituted with alanine.
FIG. 1.
Mutagenesis and purification of hIL-18. (A) Locations of the mutated residues in hIL-18 structure (PDB# 1J0S). The backbone of hIL-18 and the side chains of 17 residues that were subjected to mutagenesis are shown. Residues at the known binding site I, II, and III for hIL-18R are colored in red, orange and blue, respectively. The remaining three are colored in yellow. The residues are numbered starting from the first residue of the mature hIL-18. (B). Purification of proIL-18 and the generation of mature IL-18. The schematic diagram of the recombinant proIL-18 protein with an N-terminal 6-His tag and an engineered Factor Xa cleavage site is shown. The proIL-18 proteins were over-expressed in E. coli. and captured with Ni-NTA resin. Mature IL-18 proteins were released from the resin by Factor Xa digestion. Coomassie-stained protein samples are shown with the positions of proIL-18 and mature IL-18 indicated. (C). Further purification of mature IL-18 by size exclusion chromatograph. Mature IL-18 proteins that were released from the Ni-NTA resin (starting material) were applied to sephacryl S-100HR column, and multiple protein fractions were collected. The silver-stained protein samples are shown.
All the hIL-18 proteins, wild type or mutated, were expressed in E. coli. as the precursor form (proIL-18), in which the caspase-1 cleavage site was substituted with the Factor Xa site (Fig. 1B), similar to a previous report (Liu et al., 2000). The proIL-18 proteins were bound to metal affinity resin via an N-terminal 6-His tag (Fig. 1B). The authentic, mature hIL-18 proteins were released from the resin by Factor Xa digestion and further purified by size-exclusion chromatography (Fig. 1B&C). A protein fraction consisted almost exclusively of the mature IL-18, such as fraction #2 or #3 in Fig. 1C, was used in subsequent binding studies.
Construction and expression of variola virus IL-18BP
The coding sequence for varvIL-18BP was constructed by mutagenizing the closely related IL-18BP of ectromelia virus (ectvIL-18BP), as variola virus genomic DNA is not readily available. Only six residues are different between IL-18BPs of variola virus and ectromelia virus naval strain (Fig. 2A), and the mutagenesis of ectvIL-18BP at these six residues resulted in varvIL-18BP. To express a recombinant varvIL-18BP protein with a C-terminal biotinylation site and a 6-His tag for subsequent binding analysis, the coding sequence for varvIL-18BP was cloned into a mammalian expression vector that encodes the tags and that was previously used for the study of human and molluscum contagiosum virus IL-18BP (Xiang and Moss, 2001a; Xiang and Moss, 2001b). The recombinant varvIL-18BP was transiently expressed in HEK293T cells, purified from the culture medium via the 6-His tag, and biotinylated at the C-terminal site by biotin holoenzyme synthetase. The biotinylated varvIL-18BP reacted specifically to streptavidin in Western blotting and migrated slightly faster than similarly biotinylated ectvIL-18BP on SDS-PAGE gel (Fig. 2B). The reason for the small change in gel mobility is unclear, but is not due to any un-intended mutation in coding sequence of varvIL-18BP, as it was confirmed by sequencing.
FIG. 2.
The construction and expression of variola virus IL-18BP (varvIL-18BP).
(A). The alignment of IL-18BPs of ectromelia virus and variola virus. Six amino acids that are different between the two proteins are indicated by their residue numbers. These six residues were mutated in ectromelia virus IL-18BP (ectvIL-18BP) to generate varvIL-18BP.
(B). Detection of in vitro biotinylated varvIL-18BP and ectvIL-18BP. Recombinant IL-18BPs were transiently expressed in 293T cells, bound to Ni-NTA resin and biotinylated on the resin by biotin holoenzyme synthetase. The proteins were eluted from the resin with buffer containing imidazole and the biotinylated species were detected by chemiluminescence with a streptavidin-horseradish peroxidase conjugate after Western blotting. The positions and masses in kDa of protein markers are shown on the left.
Alanine substitution of Leu5, Lys53 and Ser55 significantly decrease the binding affinity of hIL-18 with varvIL-18BP
Biotinylated varvIL-18BP was immobilized on a BIAcore sensor chip coated with streptavidin, and its binding with various mutated hIL-18 proteins was monitored in real time with a BIAcore 3000 sensor. The sensorgram of the wild type (WT) hIL-18 showed a quick increase in signal during the association phase and a slow decline in signal during the dissociation phase (Fig. 3), demonstrating a fast on rate and a slow off rate. While the on rate was proportional to the concentration of hIL-18 that was injected, the off rate was independent of the protein concentration and indicative of the affinity between hIL-18 and varvIL-18BP. The slow off rate observed with the WT hIL-18 was not altered significantly by mutations of the known IL-18R binding sites except for the substitution of Leu5 (L5A) (Fig. 3A, B, C), which increased the off rate significantly. The off rate was increased even more by the substitution of Ser55 (S55A) or Lys53 (K53A) but was not affected significantly by the substitution of Glu6 (E6A) (Fig. 3D).
FIG. 3.
The binding affinity of hIL-18 with varvIL-18BP is reduced by alanine substitution of Leu5, Lys53 or Ser55. The biotinylated varvIL-18BP was captured on a BIAcore streptavidin-coated SA chip, and its binding with hIL-18 proteins was monitored in real time with a BIAcore 3000 sensor. The colored lines are the responses obtained with different hIL-18 mutants. Shown in the same color are the name and protein concentration of the respective mutant. The injection of hIL-18 started at 100 sec and stopped at 400 sec.
To obtain the kinetic and affinity constants, sensorgrams were acquired with different concentrations of the same hIL-18 protein and globally fitted to a 1-to-1 binding model with BIAEVALUATION software (Fig. 4). The WT hIL-18 bound to varvIL-18BP with a KD of 3 nM, in agreement with a previous report (Esteban, Nuara, and Buller, 2004). K53A, S55A and L5A substitutions increased the KD 100-, 7- and 4-fold, respectively, while no other mutation altered the KD by more than 2-fold (Table I). Leu5, Lys53 and Ser55 form a contiguous surface patch (Fig. 1A), suggesting that they are part of a single binding site for varvIL-18BP.
FIG. 4.
Kinetic analyses of the binding to varvIL-18BP by L5A, K53A and S55A mutants. BIAcore sensorgrams were obtained similarly as described in Fig. 3 but with different concentrations of the same hIL-18 mutant. The response curves were globally fitted to a 1:1 binding model with the obtained kinetics constant shown. The colored and black lines are the actual responses in RU and globally fitted curves, respectively. The residual responses, below each set of curves, represent deviations of the actual responses from the fitted curves.
Table I.
Kinetics and affinity constants of hIL-18 mutants with varvIL-18BP
| hIL-18 mutantsa | KOn,104/Ms | Koff,10−4/s | KD, nM |
|---|---|---|---|
| WT | 6.9 ± 0.5 | 2.1 ± 0.1 | 3.1 ± 0.1 |
| K4A | 8.7 ± 0.1 | 2.8 ± 0.2 | 3.2 ± 0.2 |
| L5A | 6.8 ± 0.1 | 8.8 ± 0.1 | 12.9 ± 0.1 |
| E6A | 7.1 ± 0.1 | 3.7 ± 0.2 | 5.5 ± 0.2 |
| K8A | 6.5 ± 0.1 | 1.9 ± 0.1 | 2.9 ± 0.1 |
| R13A | 7.6 ± 0.1 | 3.8 ± 0.2 | 4.9 ± 0.2 |
| D17A | 7.8 ± 0.1 | 2.5 ± 0.2 | 3.2 ± 0.2 |
| M33A | 7.6 ± 0.2 | 2.6 ± 0.1 | 3.4 ± 0.1 |
| D35A | 7.8 ± 0.1 | 2.1 ± 0.1 | 2.7 ± 0.1 |
| K53A | 6.0 ± 0.1 | 230 ± 70 | 400 ± 100 |
| S55A | 4.6 ± 0.1 | 9.1 ± 0.5 | 20 ± 2 |
| R58A | 8.1 ± 0.4 | 4.5 ± 0.1 | 5.6 ± 0.1 |
| M60A | 6.5 ± 0.1 | 2.9 ± 0.1 | 4.4 ± 0.1 |
| K79A | 6.7 ± 0.1 | 2.0 ± 0.1 | 3.0 ± 0.3 |
| K84A | 6.3 ± 0.2 | 1.8 ± 0.1 | 2.8 ± 0.2 |
| D98A | 8.3 ± 0.8 | 2.9 ± 0.3 | 3.5 ± 0.1 |
| R104A | 7.9 ± 0.1 | 2.8 ± 0.1 | 3.5 ± 0.1 |
| D132A | 7 ± 1 | 4.6 ± 0.5 | 6 ± 2 |
The kinetics and affinity constants were derived from 2 independent experiments similar to those shown in Fig. 4. K values are means ± standard deviations.
Alanine substitution of Lys53 and Ser55 also decreased the binding affinity of hIL-18 with hIL-18Rα
Glu6, Lys53 and Ser55 are spatially adjacent to the binding site II for IL-18Rα (Fig. 1A), so they were also evaluated for their contribution towards IL-18Rα binding with BIAcore. Similar to varvIL-18BP, the ectodomain of human IL-18Rα (amino acids 1-229) was expressed in HEK293T cells with a C-terminal biotinylation site and 6-His tag, purified from the culture medium, biotinylated with biotin holoenzyme synthetase and immobilized on a streptavidin-coated BIAcore sensor chip (Fig. 5A). The WT hIL-18 bound to the immobilized hIL18Rα with fast on and off rates (Fig. 5B). The KD for the WT and the L5A mutant were 3 and 30 nM, respectively, in agreement with a previous report (Kato et al., 2003). The E6A substitution did not decrease the affinity of hIL-18 with hIL-18Rα, but K53A and S55A substitutions decreased the affinity by more than 4- and 2-fold, respectively (Fig. 5B), indicating that Lys53 and Ser55 are also part of the binding site for hIL-18Rα.
FIG. 5.

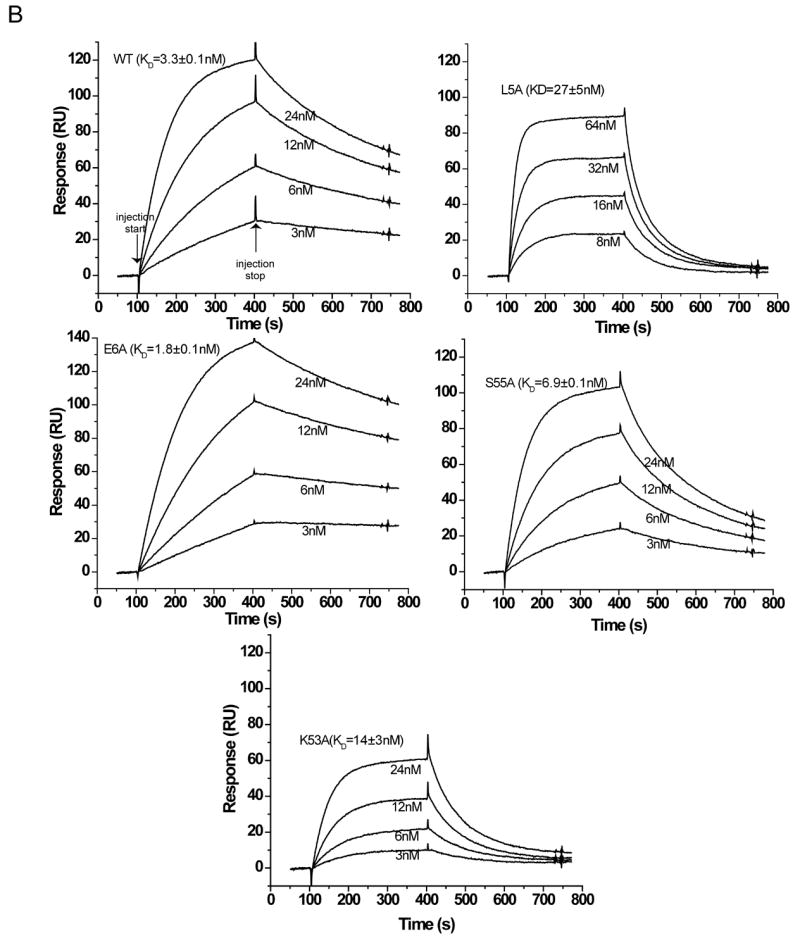
The binding affinity of hIL-18 with hIL-18Rα is reduced by alanine substitution of Leu5, Lys53 or Ser55. (A). Detection of in vitro biotinylated hIL-18Rα ectodomain. Recombinant hIL-18Rα ectodomain was purified and biotinylated similarly as describe in Fig. 2. The biotinylated species were detected with a streptavidin-horseradish peroxidase conjugate. (B). Kinetic analyses of hIL-18Rα binding by L5A, K53A and S55A mutants. The biotinylated hIL-18Rα ectodomain was captured on a BIAcore SA chip, and its binding with hIL-18 proteins was monitored with a BIAcore 3000 sensor. BIAcore sensorgrams obtained with different concentrations of the same hIL-18 mutant were globally fitted to a 1:1 binding model and the obtained kinetics constants are shown.
The finding that K53A substitution decreased the affinity of hIL-18 with hIL-18Rα was rather unexpected, as K53A substitution was previously reported to enhance IL-18 activity (Kim et al., 2001). Indeed, the K53A mutant induced the secretion of a higher level of IFN-γ from human macrophagic KG-1 cells than the WT hIL-18 (Fig. 6). In contrast, L5A mutant induced a lower level of IFN-γ secretion than the WT hIL-18 (Fig. 6), also in agreement with a previous report (Kato et al., 2003). The level of secreted IFN-γ induced by the S55A mutant was lower than that by the WT hIL-18 at high hIL-18 concentrations but was higher than that by the WT at low IL-18 concentrations (Fig. 6). These results suggest that IL-18 activity does not necessarily correlate with the affinity between hIL-18 and IL-18Rα.
FIG. 6.
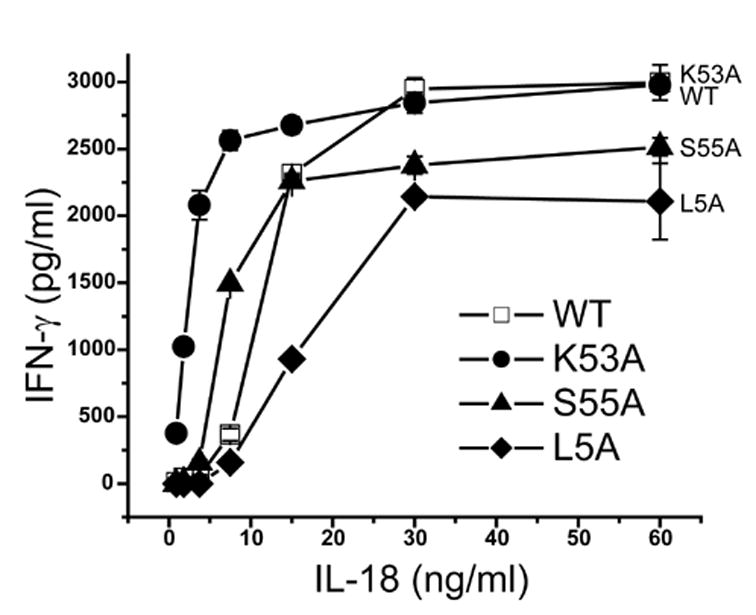
S55A substitution enhances IL-18 activity at low hIL18 concentrations. 1x106 KG-1 cells in 250 μl were stimulated with different amount of hIL-18 at 37°C. After 24 h, 100 μl of clarified supernatants were assayed for human IFN-γ with ELISA. The assay was done in duplicate with the standard deviation shown.
Taken altogether, the data suggests that varvIL-18BP neutralizes hIL-18 activity by competing with hIL-18Rα for the binding of Leu5, Lys53 and Ser55 (Fig. 7).
FIG. 7.
(A). Leu5, Lys53 and Ser55 of hIL-18 are located at a contiguous surface patch. The hIL- 18 residues at or near the known binding site II for IL-1Rα are shown in spacefill. Three residues (Leu5, Lys53 and Ser55) that contribute to binding with both varvIL-18BP and IL-18Rα are shown in red. Glu6, which does not contribute significantly to binding with either varvIL-18BP or IL-18Rα, is show in gray. The remaining site II residues, which do not contribute to binding with varvIL-18BP, are shown in orange. (B). Locations in a hIL-18BP structure model (Kim et al., 2000) of three residues that contribute most significantly to hIL-18 binding. The three hIL-18BP residues were identified previously (Xiang and Moss, 2001b) and are conserved between human and poxviral IL-18BPs. The side chains of the three residues are shown in spacefill with their residue numbers.
DISCUSSION
Variola virus is one of the most feared bioterrorism agents. However, an understanding of variola virus pathogenesis at the molecular level is not yet available for the development of a therapy for smallpox, as smallpox was eradicated before modern molecular techniques became available. In the current study, we focused on varvIL-18BP, a putative virulence factor for variola virus. We identified Leu5, Lys53 and Ser55 of hIL-18 to be the binding site for varvIL-18BP. In addition, we found that Lys53 and Ser55 were also part of a binding site for hIL-18Rα. This finding, together with the previous knowledge that Leu5 was part of the binding site II for hIL-18Rα (Kato et al., 2003), reveals that varvIL-18BP neutralizes hIL-18 activity by competing with hIL-18Rα for the binding of three common residues. Although the varvIL-18BP binding site in hIL-18 is only a small part of the much bigger IL-18R binding site, the binding of varvIL-18BP with IL-18 presumably will create steric interference and prevent IL-18R from interacting with the rest of site II residues. It is quite remarkable for varvIL-18BP to target the same residues that hIL-18Rα binds, as IL-18BP shares no significant amino acid sequence homology with IL-18R. This knowledge may be beneficial for the development of reagents to block varvIL-18BP as part of a therapy for smallpox.
The revelation of the mechanism by which varvIL-18BP neutralizes IL-18 may also help the development of small molecule inhibitors of IL-18. IL-18 activity have been implicated in autoimmune diseases, and human IL-18BP is currently in clinical trials for the treatment of rheumatoid arthritis and severe psoriasis (Dinarello, 2000; Dinarello, 2006). Our study on IL-18’s binding site for varvIL-18BP represents the first such study for any IL-18BP. The role of Lys53 in binding with human IL-18BP was previously suggested by molecular modeling and by hIL-18BP neutralization assay (Kim et al., 2001; Kim et al., 2000), but the direct evidence for such a role was not available. Leu5 and Ser55 were identified in this study by analyzing 17 mutations of hIL-18 surface residues, the majority of which did not affect the affinity with varvIL-18BP. Interestingly, Leu5, Lys53 and Ser55 form a contiguous surface patch (Fig. 7A), consistent with their being a single binding site for IL-18BP. These residues are conserved among mammalian IL-18 proteins and possibly serve as a binding site for other viral and mammalian IL-18BPs, which bind to hIL-18 via a conserved set of residues (Fig. 7B) (Xiang and Moss, 2001a; Xiang and Moss, 2001b).
The finding that K53A substitution reduces the affinity of hIL-18 with varvIL-18BP by 100-fold indicates that Lys53 is a binding free energy “hot spot”. Previously, three aromatic residues of human IL-18BP were identified to be the hot spot for the interaction with hIL-18 (Fig. 7B), as these three residues contribute 68% of the total binding free energy of hIL-18 and human IL-18BP (Xiang and Moss, 2001b). Subsequently, the corresponding residues in molluscum contagiosum virus and ectromelia virus IL-18BPs were also found to be critical for the interaction with both human and mouse IL-18 (Esteban and Buller, 2004; Xiang and Moss, 2001a). Amino acids with aromatic side chains are often found at the interface of protein complex to form high energy π-cation pair with side chains bearing positive charge (Crowley and Golovin, 2005; Zacharias and Dougherty, 2002). This π-cation interaction is electrostatic in nature and calculated to be stronger than a typical salt-bridge (Zacharias and Dougherty, 2002). Half of the protein complexes that were examined contain at least one π-cation pair (Crowley and Golovin, 2005). It is possible that Lys53 of hIL-18 interacts with the critical aromatic residues of IL-18BP to form the high energy π-cation pair.
The finding that Lys53 and Ser55 contribute to the binding with hIL-18Rα expands the known boundary of the binding site II for hIL-18Rα. This finding on Lys53 was initially surprising for us, as K53A substitution was previously shown to enhance hIL-18 activity (Kim et al., 2001), a finding that was confirmed by our current study. The fact that K53A enhances IL-18 activity also suggests that the substitution does not have any deleterious effect on hIL-18 structure. K53A reduces the affinities with varvIL-18BP and IL-18Rα by 100- and 4-fold, respectively, so Lys53 is clearly more critical for the binding with varvIL-18BP than with IL-18Rα. Lys53 may similarly be more critical for the binding with human IL-18BP than with IL-18Rα, which is consistent with the previous report that K53A is more resistant to neutralization by human IL-18BP (Kim et al., 2001). K53A enhances IL-18 activity probably by greatly reducing the ability of human IL-18BP to bind and neutralize the mutated hIL-18. Although human IL-18BP expression has not been shown in the human macrophagic KG-1 cells that were used in this study, its expression was shown to be induced by IFN-γ in primary monocytes/macrophages (Corbaz et al., 2002; Veenstra et al., 2002) as well as in nonleukocytic cell lines such as keratinocytes, epithelial cells and renal mesangial cells (Muhl et al., 2000; Paulukat et al., 2001). Our results are consistent with the idea that hIL-18 activity is regulated by its relative affinities with hIL-18R and human IL-18BP. S55A reduces the affinities with varvIL-18BP and IL-18Rα by 6- and 2-fold, respectively, so Ser55 is more important for the binding with varvIL-18BP than with hIL-18Rα. Ser55 may similarly be more important for binding with human IL-18BP than with hIL-18Rα, which could explain why S55A enhances IL-18 activity at low IL-18 concentration. In contrast to Lys53 and Ser55, Leu5 is more important for the interaction with hIL-18Rα than with varvIL-18BP, as L5A decreases the affinity with hIL-18Rα and varvIL-18BP by 10- and 4-fold, respectively. Leu5 may similarly be more important for the binding with hIL-18Rα than with human IL-18BP, so L5A reduces IL-18 activity. The fact that each substitution caused different effects on IL-18 activity and on IL-18’s affinities with varvIL-18BP and hIL-18Rα also demonstrate that the substitution does not cause the effect by a general, indirect effect on hIL-18 structure.
Materials and Methods
Construction of expression vectors for hIL-18, varvIL-18BP and hIL-18Rα ectodomain
The coding sequence for hIL-18 with its caspase-1 cleavage site mutated to the Factor Xa site was generated by recombinant PCR as follows. Two separate PCR reactions were performed with a hIL-18 cDNA clone (MGC-12320; ATCC) as the template and the primers sets (5-GGGAATTCCATATGGCTGCTGAACCAGTAG-3' and 5'-GCCAAAGTAACGTCCTTCGATGTTTTCATC-3') or (5'-GATGAAAACATCGAAGGACGTTACTTTGGC-3' and 5'- CGCGGATCCTAGTCTTCGTTTTGAACAGTG-3'). The sequences in the primers that change the coding sequence of the caspase-1 cleavage site are underlined and the appended Nde I and the BamH I sites are shown in bold italics. The two PCR products were gel-purified and used together as the template in a recombinant PCR reaction. The product was cloned into the Nde I and BamH I sites of a vector that was constructed by removing the coding sequence for the thrombin cleavage site from pET15b (Novagen). The resulting plasmid, designated as pYW1, encodes proIL-18 with the Factor Xa cleavage site and an N-terminal 6-His tag (Fig. 1B). Recombinant PCR reaction was similarly performed to introduce single amino acid substitutions into hIL-18 coding sequence in pYW1.
The coding sequence for variola virus IL-18BP (varvIL-18BP) was constructed by site-directed mutagenesis of ectromelia virus IL-18BP (ectvIL-18BP) as follows. The coding sequence for amino acid 71 to 121 of ectvIL-18BP was amplified by PCR from genomic DNA of ectromelia virus naval strain (a gift from Dr. Antonio Alcami), with primers 5’-CTGGGCGATGGCATCAAAGAAGATGAAACCATTCGTACCA- 3’ and 5’-TTTTGAAACGCCATTTAGCGTAGTGAGGAC-3’. The underlined sequence in the primers represents changes from the wild type sequence to simultaneously introduce N76K, V81I and D116N substitutions. The PCR product was then further amplified by PCR to extend the coding sequence to amino acid 63 to 127, with primers 5’-CGAGTATACCAAGTTTATAGAACATCTGGGCGATGGCATC-3’ and 5’-CTTCAGCCAAATATTCTTTTTTGAAACGCCATTT-3’. The underlined sequence in the primers denotes changes from the wild type sequence to introduce the D64Y substitution. The PCR product, which encodes the C-terminal half of ectvIL-18BP with four amino acids substituted, was cloned into pCR2.1 vector using a TA cloning system (Invitrogen), yielding pTS1. Separately, the coding sequence for N-terminal half of ectvIL-18BP containing the H38Y and E48K substitutions were constructed by recombinant PCR with ectromelia virus genomic DNA as the template and the primer sets (5’- ATATAGGATCCATGAGAATCCTA-3’ and 5’-GTTCCACACATCCTGAACAATAAAATTCTCCAG-3’) and (5’- CAGGATGTGTGGAACATATGCCTAAGTTTAGCTA-3’ and 5’- AAACTTGGTATACTCGTCCGATTTC-3’). The underlined sequence in the primers denotes changes from the wild type sequence to introduce the H38Y and E48K substitutions. The BamH I and the Acc I sites in the primers are shown in bold italics. These sites were used to clone the recombinant PCR product into the corresponding sites of pTS1 to generate the entire coding sequence for varvIL-18BP.
The mammalian expression vector for varvIL-18BP with a C-terminal biotinylation site and a 6-His tag was constructed as previously described for the construction of other IL-18BPs expression vectors (Xiang and Moss, 2001b). Briefly, the coding sequence for varvIL-18BP was amplified by PCR using primers 5'-CTATCAGCTAGCATGAGAATCCTATTTCT-3' and 5'-TAATGGATCCACCACCTCCACCCTTCAGCCAAATATT-3'. The Nhe I and BamH I sites in the primers are shown in bold italics, and they were used to clone the PCR product into the corresponding sites in pYX45 (Xiang and Moss, 2001b), resulting in the fusion of the varvIL-18BP coding sequence in frame with the coding sequence for the biotinylation site and a 6-His tag that are present in pYX45. Similarly, the coding sequence for ectvIL-18BP was cloned into pYX45 for making the mammalian expression vector for ectvIL-18BP.
The mammalian expression vector for hIL-18Rα ectodomain with a C-terminal biotinylation site and a 6-His tag was constructed as follows. The coding sequence for hIL-18Rα ectodomain (amino acid 1-229) was amplified by PCR from reverse-transcripted cDNA of human KG-1 cells using primers 5'-CTATCAGCTAGCGCCACCATGAATTGTAGAGAATTACCCTTGACCC-3’ and 5'-GAAGATCTACCACCTCCACCTCTTGTGAAGACGTG-3’. The Nhe I and Bgl II sites in the primers are shown in bold italics. The PCR product was digested with Nhe I and Bgl II and ligated with pYX45 vector that was digested with Nhe I and BamH I.
The fidelity of each construct was confirmed by sequencing with an ABI 3100 Genetic Analyzer.
Expression and purification of hIL-18 proteins
The expression construct for wild type or mutated hIL-18 was transformed into E. coli strain BL21(DE3). A 5 ml overnight culture from a single colony was inoculated to 500 ml of LB medium containing 100 μg/ml ampicillin. When the culture grew to a density of 0.7–0.8 A600, protein expression was induced by adding 1 mM of isopropylthiogalactoside (IPTG) and incubated at 25 °C with shaking for 4–5 h. Bacteria were harvested by centrifugation (5,000 × g for 15 min at 4 °C). The pellet was suspended with 10 ml of B-PER buffer (Pierce), and further lysed by sonication on ice for 2 min. The soluble protein was clarified by centrifugation (27,000 × g, 15 min, 4 °C) and bound to a 1 ml Ni-nitrilotriacetic acid resin (Ni-NTA; Qiagen) overnight in the presence of 10 mM imidazole. The resin was washed three times with 10 ml of 10mM imidazole in phosphate-buffered saline (PBS) and once with Factor Xa buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl and 2 mM CaCl2). The proteins bound to the resin were digested with Factor Xa at 4°C for 8 h by suspending the resin in 1.5 ml of Factor Xa buffer with 5 μg of Factor Xa (New England Biolabs). The clarified supernatant from the resin was applied to a sephacryl S-100HR column controlled by an Amersham ACTA Purifier, and 1 ml fraction was collected. Purity of the products was determined by 10% SDS-PAGE under reducing conditions followed by Coomassie blue and silver staining. The protein concentrations were determined by Bradford protein assay (Biorad).
IL-18BP and IL-18Rα Protein purification and in vitro biotinylation
The varvIL-18BP and IL-18Rα were expressed in HEK293T cells, purified from the culture medium and biotinylated essentially as described previously (Xiang and Moss, 2001b). Briefly, 293T cells were transfected with the expression plasmid using Lipofectamine (Invitrogen). After overnight incubation, the medium was replaced with serum free DMEM (Invitrogen). After another three days of incubation, the medium was harvested, clarified by centrifugation and incubated with Ni-NTA resin. The resin was then washed and added with E. coli biotin holoenzyme synthetase (BirA; Avidity). The resin was incubated with shaking at 37°C for 1 to 4 h, washed with 15 mM imidazole in PBS containing 150 mM NaCl. The recombinant protein was eluted with 2 ml of 300 mM imidazole in PBS and was used directly for analysis with a BIAcore 3000 sensor (BIACORE, Piscataway, N.J.).
Surface Plasmon Resonance (SPR)
The SPR analysis was done essentially as described previously (Xiang and Moss, 2001a; Xiang and Moss, 2001b). Briefly, biotinylated varvIL-18BP or hIL-18Rα was captured onto a BIAcore SA chip coated with streptavidin. Various concentrations of hIL-18 were injected at a flow rate of 20 μl/min. The chip coated with varvIL-18BP was regenerated with a 10-μl injection of 10 mM glycine (pH 1.5), while the chip coated with hIL18Rα was regenerated with a 10-μl injection of 10 mM NaOH. The sensorgrams were analyzed with BIAEVALUATION software (BIACORE). The binding data from the injection of at least four different concentrations of analyte were globally fitted to a 1:1 binding model. Analyses with the same concentration series were done twice.
IL-18 Bioassay
The IL-18 bioassay was done similarly as previously described (Xiang and Moss, 1999). Briefly, KG-1 cells (0.25 ml at 1 × 106 per ml) in RPMI medium containing 10% FBS were seeded in wells of a 96-well plate and stimulated with different concentrations of hIL-18. After stimulation at 37°C for 24 h, 100 μl of clarified supernatants were assayed for human IFN-γ by ELISA (BD Biosciences). The IL-18 assay was done in duplicates.
Acknowledgments
We thank Dr. Antonio Alcami for providing ectromelia virus genomic DNA, Jie Chao and Yimin Wu for providing technical assistance. Y. Xiang was supported by a Research Scholar Development Award from NIAID (K22 AI053230).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizawa Y, Akita K, Taniai M, Torigoe K, Mori T, Nishida Y, Ushio S, Nukada Y, Tanimoto T, Ikegami H, Ikeda M, Kurimoto M. Cloning and expression of interleukin-18 binding protein. FEBS Lett. 1999;445(2–3):338–42. doi: 10.1016/s0014-5793(99)00148-9. [DOI] [PubMed] [Google Scholar]
- Born TL, Morrison LA, Esteban DJ, VandenBos T, Thebeau LG, Chen N, Spriggs MK, Sims JE, Buller RM. A poxvirus protein that binds to and inactivates IL-18, and inhibits NK cell response. J Immunol. 2000;164(6):3246–54. doi: 10.4049/jimmunol.164.6.3246. [DOI] [PubMed] [Google Scholar]
- Calderara S, Xiang Y, Moss B. Orthopoxvirus IL-18 binding proteins: affinities and antagonist activities. Virology. 2001;279(1):22–6. doi: 10.1006/viro.2000.0689. [DOI] [PubMed] [Google Scholar]
- Corbaz A, ten Hove T, Herren S, Graber P, Schwartsburd B, Belzer I, Harrison J, Plitz T, Kosco-Vilbois MH, Kim SH, Dinarello CA, Novick D, van Deventer S, Chvatchko Y. IL-18-Binding Protein Expression by Endothelial Cells and Macrophages Is Up-Regulated During Active Crohn's Disease. J Immunol. 2002;168(7):3608–3616. doi: 10.4049/jimmunol.168.7.3608. [DOI] [PubMed] [Google Scholar]
- Crowley PB, Golovin A. Cation-pi interactions in protein-protein interfaces. Proteins. 2005;59(2):231–9. doi: 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Targeting interleukin 18 with interleukin 18 binding protein. Ann Rheum Dis 59 Suppl. 2000;1:i17–20. doi: 10.1136/ard.59.suppl_1.i17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr. 2006;83(2):447S–455. doi: 10.1093/ajcn/83.2.447S. [DOI] [PubMed] [Google Scholar]
- Esteban DJ, Buller RM. Identification of residues in an orthopoxvirus interleukin-18 binding protein involved in ligand binding and species specificity. Virology. 2004;323(2):197–207. doi: 10.1016/j.virol.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Esteban DJ, Nuara AA, Buller RM. Interleukin-18 and glycosaminoglycan binding by a protein encoded by Variola virus. J Gen Virol. 2004;85(Pt 5):1291–9. doi: 10.1099/vir.0.79902-0. [DOI] [PubMed] [Google Scholar]
- Fujioka N, Akazawa R, Ohashi K, Fujii M, Ikeda M, Kurimoto M. Interleukin-18 protects mice against acute herpes simplex virus type 1 infection. J Virol. 1999;73(3):2401–9. doi: 10.1128/jvi.73.3.2401-2409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb SL, Myskowski PL. Molluscum contagiosum. Int J Dermatol. 1994;33(7):453–61. doi: 10.1111/j.1365-4362.1994.tb02853.x. [DOI] [PubMed] [Google Scholar]
- Gracie JA, Robertson SE, McInnes IB. Interleukin-18. J Leukoc Biol. 2003;73(2):213–24. doi: 10.1189/jlb.0602313. [DOI] [PubMed] [Google Scholar]
- Kato Z, Jee J, Shikano H, Mishima M, Ohki I, Ohnishi H, Li A, Hashimoto K, Matsukuma E, Omoya K, Yamamoto Y, Yoneda T, Hara T, Kondo N, Shirakawa M. The structure and binding mode of interleukin-18. Nat Struct Biol. 2003 doi: 10.1038/nsb993. [DOI] [PubMed] [Google Scholar]
- Kim SH, Azam T, Yoon DY, Reznikov LL, Novick D, Rubinstein M, Dinarello CA. Site-specific mutations in the mature form of human IL-18 with enhanced biological activity and decreased neutralization by IL-18 binding protein. Proc Natl Acad Sci U S A. 2001;98(6):3304–9. doi: 10.1073/pnas.051634098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Eisenstein M, Reznikov L, Fantuzzi G, Novick D, Rubinstein M, Dinarello CA. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc Natl Acad Sci U S A. 2000;97(3):1190–5. doi: 10.1073/pnas.97.3.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Takeuchi M, Ohta T, Nishida Y, Arai N, Ikeda M, Ikegami H, Kurimoto M. Interleukin-18 activates the IRAK-TRAF6 pathway in mouse EL-4 cells. Biochem Biophys Res Commun. 1998;244(1):183–6. doi: 10.1006/bbrc.1998.8236. [DOI] [PubMed] [Google Scholar]
- Liu B, Novick D, Kim SH, Rubinstein M. Production of a biologically active human interleukin 18 requires its prior synthesis as PRO-IL-18. Cytokine. 2000;12(10):1519–25. doi: 10.1006/cyto.2000.0749. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 2849–2883. 2 vols. [Google Scholar]
- Muhl H, Kampfer H, Bosmann M, Frank S, Radeke H, Pfeilschifter J. Interferon-gamma mediates gene expression of IL-18 binding protein in nonleukocytic cells. Biochem Biophys Res Commun. 2000;267(3):960–3. doi: 10.1006/bbrc.1999.2064. [DOI] [PubMed] [Google Scholar]
- Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10(1):127–36. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- Paulukat J, Bosmann M, Nold M, Garkisch S, Kampfer H, Frank S, Raedle J, Zeuzem S, Pfeilschifter J, Muhl H. Expression and release of IL-18 binding protein in response to IFN-gamma. J Immunol. 2001;167(12):7038–43. doi: 10.4049/jimmunol.167.12.7038. [DOI] [PubMed] [Google Scholar]
- Pirhonen J. Regulation of IL-18 expression in virus infection. Scand J Immunol. 2001;53(6):533–9. doi: 10.1046/j.1365-3083.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- Reading PC, Smith GL. Vaccinia Virus Interleukin-18-Binding Protein Promotes Virulence by Reducing Gamma Interferon Production and Natural Killer and T-Cell Activity. J Virol. 2003;77(18):9960–9968. doi: 10.1128/JVI.77.18.9960-9968.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Shibuya K, Mui A, Zonin F, Murphy E, Sana T, Hartley SB, Menon S, Kastelein R, Bazan F, O'Garra A. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity. 1997;7(4):571–81. doi: 10.1016/s1074-7613(00)80378-7. [DOI] [PubMed] [Google Scholar]
- Smith VP, Bryant NA, Alcami A. Ectromelia, vaccinia and cowpox viruses encode secreted interleukin-18-binding proteins. J Gen Virol 81 Pt. 2000;5:1223–30. doi: 10.1099/0022-1317-81-5-1223. [DOI] [PubMed] [Google Scholar]
- Symons JA, Adams E, Tscharke DC, Reading PC, Waldmann H, Smith GL. The vaccinia virus C12L protein inhibits mouse IL-18 and promotes virus virulence in the murine intranasal model. J Gen Virol. 2002;83(Pt 11):2833–44. doi: 10.1099/0022-1317-83-11-2833. [DOI] [PubMed] [Google Scholar]
- Tanaka-Kataoka M, Kunikata T, Takayama S, Iwaki K, Ohashi K, Ikeda M, Kurimoto M. In vivo antiviral effect of interleukin 18 in a mouse model of vaccinia virus infection. Cytokine. 1999;11(8):593–9. doi: 10.1006/cyto.1998.0453. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Matsui K, Okamura H, Nakanishi K. Pathophysiological roles of interleukin-18 in inflammatory liver diseases. Immunol Rev. 2000;174:192–209. doi: 10.1034/j.1600-0528.2002.017418.x. [DOI] [PubMed] [Google Scholar]
- Veenstra KG, Jonak ZL, Trulli S, Gollob JA. IL-12 Induces Monocyte IL-18 Binding Protein Expression Via IFN-{gamma} J Immunol. 2002;168(5):2282–2287. doi: 10.4049/jimmunol.168.5.2282. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Moss B. IL-18 binding and inhibition of interferon gamma induction by human poxvirus-encoded proteins. Proc Natl Acad Sci U S A. 1999;96(20):11537–42. doi: 10.1073/pnas.96.20.11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Moss B. Correspondence of the functional epitopes of poxvirus and human interleukin-18-binding proteins. J Virol. 2001a;75(20):9947–54. doi: 10.1128/JVI.75.20.9947-9954.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Moss B. Determination of the functional epitopes of human interleukin-18-binding protein by site-directed mutagenesis. J Biol Chem. 2001b;276(20):17380–6. doi: 10.1074/jbc.M009581200. [DOI] [PubMed] [Google Scholar]
- Zacharias N, Dougherty DA. Cation-[pi] interactions in ligand recognition and catalysis. Trends in Pharmacological Sciences. 2002;23(6):281–287. doi: 10.1016/s0165-6147(02)02027-8. [DOI] [PubMed] [Google Scholar]