

Abstract
Single assemblies of the intact light-harvesting complex LH2 from Rhodopseudomonas acidophila were bound to mica surfaces at 300 K and examined by observing their fluorescence after polarized light excitation. The complexes are generally not cylindrically symmetric. They act like elliptic absorbers, indicating that the high symmetry found in crystals of LH2 is not present when the molecules are immobilized on mica. The ellipticity and the principal axes of the ellipses fluctuate on the time scale of seconds, indicating that there is a mobile structural deformation. The B850 ring of cofactors shows significantly less asymmetry than B800. The photobleaching strongly depends on the presence of oxygen.
The light-harvesting complexes are essential to photosynthesis in plants and bacteria. They absorb light from the sun and efficiently transport the photon energy to chemically reactive centers. The spectroscopic properties of the complex LH2 from photosynthetic bacteria, Rhodopseudomonas acidophila strain 10050, whose structure at atomic resolution was determined by Cogdell and coworkers (1), have been reviewed recently (2). The complex is notable for its high symmetry arrangement of the nine αβ-dipeptides that form the scaffold that holds the associated bacteriochlorophyll (Bchl) cofactors in place. These cofactors form into two rings that have approximate 9-fold rotation symmetry. The B800 ring consists of nine monomeric Bchls located, peripherally, between the β-apoproteins and closest to the N-terminal ends, whereas B850 consists of nine pairs of Bchls each associated with one αβ-dipeptide (1). Light absorbed in the B800 ring is transferred to B850 in less than a picosecond. In vivo the energy is transferred from B850 to the complex LH1, which also has high symmetry, and from there to a reaction center where charge separation occurs. Properties of excitations in extended systems depend on the interplay between the nuclear motions that tend to localize excitations, and the delocalizing effect of the interaction between the cofactors (3). Therefore the nature of the excitations present in the LH1 and LH2 complexes must depend not only on the static or average structures but also on the structural fluctuations that occur in solution at ambient temperature.
Single molecule methods are well suited for the examination of the slow structural fluctuations (4), which are representative of the rough energy landscapes of macromolecules. Fluctuations occurring on the microsecond to minutes time scales would be manifest in conventional bulk spectroscopies as a quasistatic inhomogeneous broadening. Single macromolecules can be studied by means of fluorescent probes. Fluctuations of the macromolecular structure and orientation can result in intensity, lifetime and spectral changes of the probe fluorescence. Such methods were used to examine protein dynamics (5–7), enzyme reactions (8, 9) and nucleic acid motions (10, 11). In the case of the light-harvesting complexes a fluorescent probe is unnecessary because the intrinsic fluorescence can be used to monitor the behavior of single assemblies in solutions at room temperature (12) or under cryogenic conditions (13). Usually single-molecule experiments are carried out by repeatedly exciting the molecule until it photobleaches. The trajectory of detected fluorescent photons reflects the fluctuations in orientation and electronic structure of the probes as they sense the different environments reached during the observation period.
Previously it was shown that LH2 could be immobilized on mica in the presence of detergent and that the fluorescence of single assemblies could be detected (12). A number of very interesting and often puzzling photoproperties were observed in the single-molecule experiments that were not seen in bulk measurements. Typically a molecule is excited 107 times during a trajectory. If its fluorescence lifetime is 1 ns, it will spend 10 ms in its electronically excited state. Thus processes may be evident for a single molecule yet have very low yield in the bulk. For example, it was discovered that the B850 fluorescence of single assemblies undergoes frequent switching between states having different radiative properties (12). Under steady illumination the B850 fluorescence suddenly would be extinguished, which was attributed to the photoformation of an excitation trap, proposed to be a radical cation, in the B850 ring. Indeed oxidation of about 1 Bchl molecule per ring in the structurally homologous LH1 ring results in complete quenching of its fluorescence (14). Continued illumination often led to the recovery of the fluorescence. This process of alternating dark and bright states was seen for essentially all the molecules that were studied. Although the recovered bright states did not always have the same fluorescence intensity as the original state, their fluorescence lifetimes were usually quite similar, suggesting light induced changes in the radiative rates of the excitations. Furthermore it was shown that the appearance of the first dark state was linear in the light intensity, as expected for electron or proton transfer. A similar alternation of dark and bright states, the so-called blinking of fluorescent signals, has been reported for single, green fluorescent proteins (15).
Here we report measurements of the anisotropic properties of the absorption of light-harvesting complexes. Based on the structure of LH2 (1) the anisotropy should have predictable characteristics. The symmetry is sufficiently high in the ideal structure of LH2 for experiments in polarized light to characterize the angular motions and orientation of the single molecules on the mica surface. Because LH2 is electronically cylindrical or uniaxial with the transition moments of the B800 and B850 cofactors confined to the basal plane of the cylinder (16), the absorption tensor of both B800 and B850 should contain only two, equal elements. Thus the orientation of the cylinder axis in the laboratory frame should be easily deduced from experiments in polarized light. We will show here that these predictions are not upheld, that the complex immobilized on mica is not cylindrical, and that the dielectric tensor is undergoing dynamics.
MATERIALS AND METHODS
Fluorescence images and trajectories were recorded on a confocal microscope based on a commercial inverted microscope (Nikon, Diaphot 300, objective Nikon FLUOR ×40, numerical aperture = 1.3, oil immersion). The samples were excited by linearly or circularly polarized light through the back-port of the microscope. A diode laser (Philips, Eindhoven, The Netherlands, CQL7825, 794 nm), diode laser driver (Melles Griot, Irvine, CA, 06DLD201), and dichroic mirror (Chroma Technology, Brattleboro, VT, DRLP825) were used for B800 excitation. In the fluorescence path the excitation light was blocked by a holographic notch plus filter (Kaiser Optical Systems, Ann Arbor, MI, HNPF-800) and two color glass filters (Schott, Mainz, Germany, RG 830, 3 mm). A diode laser (Thorlabs, Newton, NJ, RLD-85PC, λ = 850 nm) and dichroic mirror (Chroma Technology DRLP860) were used for B850 excitation. The excitation light was blocked by a holographic notch plus filter (Kaiser Optical Systems, HNPF-850).
The polarization of the excitation light was under control of an electro-optic modulator (Conoptics, Danbury, CT, 350–105). The electro-optic modulator was configured to sweep the polarization of the excitation light over 155o or switch it between two orthogonal polarization states at a repetition rate of 16 s−1. To switch and to sweep the polarization a synthesized function generator (Stanford Research, Sunnyvale, CA, model DS335) was used. In the polarization sweep experiments the birefringence of mica was compensated by a second, identical mica plate, prepared from the same freshly cleaved sheet. In the switching experiments the fast and slow axes of the mica plates were aligned parallel to the light polarization axes. The fluorescence of an IR 144 solution (A800 = 0.1) was recorded with a fixed polarizer in front of the detector, yielding the expected cos2 dependence during the sweep. Without the polarizer the modulation depth was less than 0.5%, indicating that the detection system is isotropic.
The sample was mounted on a closed loop scan unit (Queensgate, Ascot, U.K., S222, scanning area 30 × 30 μm2) controlled by a modified Nanoscope E controller (Digital Instruments, Santa Barbara, CA), and the emitted fluorescence was collected by a single photon-counting module (EG&G, Salem, MA, SPCM-AQ-161) for fluorescence images and trajectories or by a combination of a monochromator (Acton Research, Acton, MA, model 150) and a backilluminated nitrogen cooled charge-coupled device camera (Princeton Instruments, Trenton, NJ) for spectra. Fluorescence images and trajectories were obtained in the same way as described in an earlier study (12).
Single LH2 complexes from R. acidophila strain 10050 were immobilized on a mica substrate with a thickness of less than 120 μm (12), which was glued to a nylon ring. The buffer was 50 mM Tris⋅HCl, pH 7.8/0.1% lauryldimethylamine oxide. The experiments were conducted on samples deoxygenated by the enzymatic system: glucose oxidase (0.5 μM), glucose (0.3%), and catalase (0.06 μM). The solutions of enzymes and glucose were added to the LH2 samples directly before the experiments. Then the mica-nylon ring cell was sealed with parafilm.
RESULTS AND DISCUSSION
Optical Properties of LH2 Complexes.
We define x, y, z as the molecule fixed coordinate system with z parallel to the cylinder axis of LH2. The transition dipoles for B850 and B800 both lie in the xy-plane of the assembly (16). Thus both B850 and B800 should behave as essentially circular oscillators, having the same absorption cross section for light polarized in any direction in the xy-plane. Equivalently, a circular absorber has a 2-fold degenerate excited state to which transitions from the ground state occur with equal cross section but with perpendicular polarization. All pairs of orthogonal linear combinations of these component states are suitable representations of the excited states. The emission will occur from each component state with equal probability regardless of which component initially was excited (17).
In what follows we use X, Y, Z for the laboratory fixed coordinates with X, Y as the focal plane of the microscope (see Fig. 1). If the LH2 retains the ideal circular characteristics given above, then the absorption cross section of a single molecule in circularly polarized light would depend on only the tilt angle θ between the z-axis and the direction of light propagation, Z:
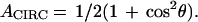 |
1 |
For linear polarization the ideal absorption cross section also depends on the direction in the XY-plane about which tilt occurs:
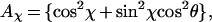 |
2 |
where (φ − α) = χ is the angle in the XY-plane between the linear polarization axis α and the axis of tilt φ both measured with respect to X. The fluorescence detection in our experiment is not polarization sensitive, so the total fluorescence signals for circular and linearly polarized excitation, in the ideal case, are given by Eq. 1 and Eq. 2 multiplied by (1 + cos2θ). The high sensitivity of these signals to the tilt angle is evident. In the experiments either the B800 or the B850 absorbs the light but only B850 emits. For completeness we consider the signal expected from an elliptical absorber having two transitions that have different strength but are polarized perpendicular to one another. In this case the polarization directions x and y are fixed in the molecular frame, the 2-fold degeneracy is split, and the absorption cross sections form the major and minor axes of an ellipse. The quantity ɛ = 〈Imin〉/〈Imax〉 is defined as the ellipticity so that 0 ≤ ɛ ≤ 1, where the average 〈…〉 is over some period of the trajectory. We also use the asymmetry parameter γ = (1 − ɛ)/(1 + ɛ), which is zero when the system is circular. Clearly a tilted circular molecule and an elliptic absorber lying flat cannot be distinguished by measurements with polarization in the XY-plane.
Figure 1.

Coordinates and angles used in text. The linear polarization vector α̂ is in the XY-plane at angle α to X.
Fluorescence Trajectories at 794-nm Excitation.
The fluorescence trajectories obtained with a continuous wave (CW) laser reveal the same types of emitting and photobleaching behavior as was found previously for picosecond pulsed laser excitation (12). All of the complexes show the initial brightly fluorescent state and the final dark state that was reached in one bleaching step 34% of the time. The mean survival time (tmean) in the initial bright state for an emission rate of 1,000 counts/s was 2.2 s for picosecond pulsed excitation and 1.4 s for CW excitation at the same average power. Therefore the dominant mechanism of photodestruction involves only one photon, in agreement with previous conclusions (12). Photobleaching of the whole assembly is caused by the rapid trapping of the B850 excitations at a photobleached Bchl. The B800 molecules have subpicosecond lifetimes so they are probably not photobleached during the experiment.
We found that O2 accounts for some, but not all, of the photobleaching. Single LH2 complexes in the deoxygenated environment survive the CW laser irradiation approximately 10 times longer than at ambient O2 conditions. In the absence of oxygen tmean = 10 s compared with 1.4 s without deoxygenation. Because the triplet state of LH2 is protected by carotenoids (18), photobleaching also might occur by a well-known photphysical process (19) of electron transfer to O2 from the singlet state.
Excitation of B800 with Circularly Polarized Light at 794 nm.
If the surface immobilized LH2 is excited with circularly polarized light, the signal should not depend on the azimuthal angles (see Eq. 1). For excitation at 794 nm, fluorescence images of single LH2 complexes (e.g., Fig. 2A) gave a Gaussian distribution of count rates (Fig. 2B) with a mean of 833, and σ = 140 counts. The σ contains a contribution of ca. 20% from the blinking, which is different for each molecule. Thus the variance in the tilt angle is significantly less than σ.
Figure 2.

The fluorescence images of single mica-immobilized LH2 complexes excited with circularly polarized light (A) and the distribution of the fluorescence count rates of the 273 single LH2 assemblies (B). λex is 794 nm. The excitation power is 7.9 μW.
Switching of 794-nm Excitation Polarization.
The excitation light at 794 nm was switched between two orthogonal polarization directions X and Y at 16 Hz. The corresponding trajectories for fluorescence from B850 are labeled Ix(t) and Iy(t). The total signal is IT(t) = Ix(t) + Iy(t). A common feature of most of the trajectories was that the time average 〈IT(t)〉 was constant for averaging periods longer than several seconds, so that the total signal usually remained constant. Some trajectories showed 〈Ix〉 = 〈Iy〉 and constant 〈IT〉 as expected for circular oscillators lying flat on the mica surface (see Fig. 3A). Others showed 〈Ix〉 ≠ 〈Iy〉 with constant 〈IT〉 as expected for an elliptical or tilted circular absorber and emitter (Fig. 3B). It was common to observe different signals for X and Y polarization along with fluctuations in γ, as in Fig. 3C. These polarization changes are not light induced. These results imply that either the elliptic principal axes rotate in the XY-plane or the molecular z-axis processes about the laboratory Z-axis, keeping the tilt angle constant. The total emission signal does not change during these motions. For the 78 single LH2 complexes that were examined in the polarization switching experiments 33% have similar (e.g., Fig. 3A) or constantly different (e.g., Fig. 3B) values of Ix and Iyduring the measurement. The remaining 67% of the single LH2 complexes undergo dynamic transformations in Ix and Iy on the time scale of seconds.
Figure 3.

The fluorescence trajectories of single LH2 complexes detected with the excitation polarization switching between 0 and π/2. λex is 794 nm in A–C. (A) LH2 assembly with similar Ix and Iy trajectories. (B) LH2 exhibiting different Ix and Iy signals. (C) LH2 undergoing polarization changes on the subsecond and seconds time scales. λex is 850 nm in D–F. (D) LH2 with similar Ix and Iy trajectories. (E and F) LH2 complexes exhibiting fluctuations in the Ix and Iy signals resulting in changes of γ. Black, Ix; red, Iy; blue, the total fluorescence signal.
For the majority of mica-immobilized assemblies γ changed with time and was most often different from zero at any instant. To further characterize the electronic states of Bchls we determined the average values of Ix and Iy for each single LH2 assembly. From these results we obtained the trajectory averaged polarization ratios, 〈Imin〉T/〈Imax〉T shown in Fig. 4A for 78 LH2 complexes. These data include only the portions of each trajectory before the appearance of the first dark state. The average time interval of the first bright state was 7.45 s for 794-nm excitation. For the majority of single LH2 complexes the ratio 〈Imin〉T/〈Imax〉T is significantly less unity.
Figure 4.

The distributions of polarization ratios of single LH2 complexes determined in the polarization switching experiments. (A) λex is 794 nm. (B) λex is 850 nm.
Switching of 850-nm Excitation Polarization.
Fig. 3 D–F shows typical Ix and Iy trajectories of single LH2 complexes excited via the B850 ring. Fig. 3D shows overlapping Ix and Iy trajectories similar to the trajectories shown on Fig. 3A for B800 excitation. The Ix and Iy trajectories on Fig. 3 E and F exhibit significant changes in γ. Often, as in Fig. 3F, different values of Ix and Iy show up immediately after the dark state. Of the 72 LH2 complexes investigated by polarization switching at 850 nm, 67% had similar or slightly different Ix and Iy values during the measurement, and 33% of them underwent fluctuations of γ in the range of 0.1 to 0.2. Fig. 4B shows the 〈Imin〉T/〈Imax〉T distribution of single LH2 complexes for the 850-nm excitation. The average time interval of the first bright state for single LH2 complexes at the 850-nm excitation is 7.73 s, similar to what is obtained with 794-nm excitation, confirming that the bleaching occurs in B850. As Fig. 4 indicates, the 〈Imin〉T/〈Imax〉T distribution at 850 nm is much narrower than at 794 nm. The majority of LH2 complexes excited via B850 had 〈Imin〉T/〈Imax〉T values close to unity.
Sweeping of 794-nm Excitation Polarization.
When the linear polarization is swept in the XY-plane, the detected fluorescence of a single LH2 complex (S) should have the general form:
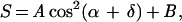 |
3 |
where A is an amplitude, δ is a relative phase, B is an offset, and α = 0 to 155o is the sweep angle. We cannot distinguish δ from π-δ. This relation is a generalization of Eq. 2 according to which only B would contribute to the total fluorescence if the complex were lying flat and B800 and B850 were optically ideal. Fig. 5 shows the phase and count rate trajectories of three typical single LH2 complexes, determined from the detected fluorescence trajectories by using Eq. 3 (e.g., Fig. 6 B and C). Fig. 5A shows an example with a relatively constant δ during the whole measurement. This assembly is unusual in that there is a gradual decrease in the fluorescence count rate, but there is no significant change in δ. The δ value of another LH2 complex (Fig. 5B) is relatively stable during the first 8 s, then it undergoes several consecutive changes by ca. 40o before entering a dark state. Two changes in δ, each of ca. 40o, are observed during the last brightly emitting state of this complex. After a long dark state it re-emits with the phase changed by 40o. These changes in δ are occurring when the total count rate is relatively stable. The δ trajectory of the one shown on Figs. 5C and 6 undergoes numerous transitions. Again the changes in δ take place while the total count rate is constant (e.g., Fig. 6A). Fig. 5 D–F also shows the distributions of δ for these three single LH2 complexes. The data of Figs. 5 and 6 are typical of what was seen for a large number of single LH2 complexes undergoing the frequent changes in δ: the δ value often appears to change in jumps of ca. 40o.
Figure 5.

The polarization asymmetry of single LH2 complexes determined via the sweeping of the excitation polarization over 155o. (A–C) The δ and count rate trajectories. (A) LH2 complex with a stable δ value during the whole fluorescence trajectory. (B) LH2 complex having several sudden changes in δ by ca. 40o. (C) LH2 complex undergoing numerous changes in δ. (D–F) The distributions of the δ values determined from the δ trajectories shown on A–C.
Figure 6.

The dynamics of a typical LH2 assembly. (A) A portion of the δ (blue) and count rate (red) trajectories of the LH2 complex shown in Fig. 5C. (B and C) The two segments of the detected fluorescence trajectory and their fits by Eq. 3 resulting in the δ values of 20° (B) and 70° (C). (D) The time-resolved probability histogram of the phase. The number of occurrences varies from 0 (black) to 15 (white).
Correlation Between Intensity and Ellipticity.
If the polarization and its dynamics were caused by tilting of cylindrical LH2 there should be a correlation between the apparent ellipticity and the total signal. A statistical analysis for 91 molecules revealed a very small correlation coefficient of ca. 0.28, suggesting that tilting is not the main contribution to the polarization anisotropy magnitudes or changes.
Fluorescence Spectra of Single LH2 Complexes.
The average fluorescence spectra of free and mica-bound LH2 complexes, obtained by shifting the focus from the solution to the mica surface, were identical. When detected with an integration time of 0.5 or 0.7 s single assemblies show fluctuations in spectral peak position and width (Fig. 7). The peak position fluctuates by more than 100 cm−1 during the measurement, and the width by more than 200 cm−1. The bright state appearing after a dark period often had an unchanged peak position (e.g., Fig. 7A). The peak distribution for the 311 spectra of the 35 single LH2 complexes measured with integration times of 0.5 and 0.7 s (Fig. 8) is peaked at 11,540 cm−1 with a shoulder near 11,460 cm−1. The shoulder might arise from molecules that emit from the lowest exciton level of the B850 band of states (2, 20): this transition is forbidden for the ideal structure but allowed for elliptically distorted structures. In the experiments represented in Fig. 8 the effect of homogeneous broadening of B850 is removed, which is why the distribution is different from the linear spectrum.
Figure 7.

Fluorescence spectra of two LH2 complexes detected with an integration time of 0.7 s (A) and 0.5 s (B). λex is 794 nm. The excitation power is 333 μW.
Figure 8.

Distribution of the fluorescence peak positions, νmax, of single LH2 complexes and least-square fit (solid curve) to two Gaussians (mean1 = 11,540 cm−1, σ1 = 30 cm−1, amplitude = 30.0; mean2 = 11,460 cm−1, σ 2 = 30 cm−1, amplitude = 6.7).
Structural Implications.
The immobilization of LH2 complexes on the surface of mica is intended to model the protein-membrane interactions occurring in bacterial cells. The mica has some negative charges on its atomically flat surface but it also may have hydrophobic interactions with the protein. The most positively charged part of LH2 corresponds to the C-terminal ends where there are 18 positively charged lysines, two associated with each α-peptide. The N-terminal regions of LH2 are largely negatively charged because of the presence of 27 glutamates and should avoid the surface of mica. Therefore we expect the LH2 to bind on the mica surface with the N termini facing the solution and C termini facing the mica. This configuration has the B850 ring closest to the surface. The B800 Bchls are more exposed to the solution and perhaps more sensitive to structural fluctuations of the complex.
The main results of this paper come from the polarization switching and sweeping experiments. The data cannot be reasonably interpreted by the ideal oscillator model. The polarization changes seldom are accompanied by changes in total signal, suggesting they are not caused by variations in tilt angle. In principle they could be caused by variations in the φ angle of a tilted molecule. However, significant changes in δ are seen in the sweeping experiments (see Figs. 5 and 6). To explain the data with the ideal model would require all molecules to be tilted within a narrow distribution of angles (see Fig. 2). In addition, a given molecule would need to undergo significant (0–140o) precession in φ angle (see Figs. 5 and 6) without any significant changes in the tilt angle θ.
The results are consistent with the immobilized assembly being an elliptic absorber and emitter. The changes seen in the experiments then are attributed to fluctuations of the ellipticity and of the directions of the principal axes of the absorption ellipse. An elliptical absorber is defined here as having two nearly degenerate excitations with unequal transition dipoles whose vectors are fixed in the molecular frame. Any structural distortion that destroys the 3-fold rotational symmetry of the B800 and/or B850 rings would, in principle, yield an electronically elliptical absorber and emitter. The results show that the distortion has a greater effect on B800 than on B850 (Fig. 4). The phase measurement records the changes in δ, which defines the location of the principal axes of the ellipse in the XY-plane. Very large changes in δ are most reasonably explained by distortions occurring at different locations around the ring structures rather than by a precession around the z axis of the whole molecule. It is interesting that phase fluctuations of ca. 40o (i.e., 2π/9) are very commonly seen in the data. This angle is needed to rotate one αβ-dipeptide into the neighboring one, suggesting that a distortion at one location often can shift around the structure by 2π/9 steps. This explanation is reasonable because a distortion near the interface between two αβ-dipeptides might more easily transfer to a neighboring interface than to some random location around the ring. It is important that in a 155o scan we do not observe eight phase peaks separated by π/9, which is expected if successive distortions were to occur in a random stochastic manner around the circle of cofactors.
The different polarization behavior observed between B800 and B850 excitation is also significant (see Fig. 4). It suggests either a larger effect of the distortion on the electronic properties, or a larger distortion of B800 compared with B850. The B850 spectrum may be less sensitive to distortions because of its larger exciton bandwidth, which implies more effective motional averaging of the distortion (21). Also, the B800 Bchls are held in the LH2 more peripherally than are those of B850. The B850 Bchls each are constrained by histidine residues of α-, β-peptides, whereas the B800 Bchls are located between β-peptides and coordinated by the N-terminal formylmethionines of α-peptides (1). The distortion might involve a small rotation of one or two of the B800 cofactors, or it might involve a partial dissociation of the assembly at the interface between two dipeptides. The attachment to the mica surface may perturb the structure of LH2 but the B800 cofactors, which are furthest from the surface, are the most perturbed. It would be challenging to observe these effects in the bulk, where an ellipticity of 0.75 would result in only a 3% change in bulk fluorescence polarization. However, the energy transfer between LH2 units in a concentrated solution as found in vivo would be rendered anisotropic by the elliptical distortions.
Acknowledgments
We thank Professor G. J. Small for helpful comments. This research was supported by the National Institutes of Health and the National Science Foundation, with instrumentation developed under National Institutes of Health Grant RR03148 (R.M.H.); and by the Biotechnology and Biological Sciences Research Council, United Kingdom (R.J.C.).
ABBREVIATION
- Bchl
bacteriochlorophyll
References
- 1.McDermott G, Prince S M, Freer A A, Hawthornthwaite-Lawless A M, Papiz M Z, Cogdell R J, Isaacs N W. Nature (London) 1995;374:517–521. doi: 10.1016/s0969-2126(96)00050-0. [DOI] [PubMed] [Google Scholar]
- 2.Sundström V, Pullerits T, van Grondelle R. J Phys Chem B. 1999;103:2327–2346. [Google Scholar]
- 3.Davydov A S. Theory of Molecular Excitons. New York: Plenum; 1971. [Google Scholar]
- 4.Xie X S, Trautman J K. Annu Rev Phys Chem. 1998;49:441–480. doi: 10.1146/annurev.physchem.49.1.441. [DOI] [PubMed] [Google Scholar]
- 5.Kinosita K, Jr, Yasuda R, Noji H, Ishiwata S, Yoshida M. Cell. 1998;93:21–24. doi: 10.1016/s0092-8674(00)81142-3. [DOI] [PubMed] [Google Scholar]
- 6.Kitamura K, Tokunaga M, Iwane A H, Yanagida T. Nature (London) 1999;397:129–134. doi: 10.1038/16403. [DOI] [PubMed] [Google Scholar]
- 7.Jia Y, Talaga D S, Lau W L, Lu H S M, DeGrado W F, Hochstrasser R M. Chem Phys. 1999;247:69–83. [Google Scholar]
- 8.Lu H P, Xun L Y, Xie X S. Science. 1998;282:1877–1882. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- 9.Ha T, Ting A Y, Liang J, Caldwell W B, Deniz A A, Chelma D S, Schultz P G, Weiss S. Proc Natl Acad Sci USA. 1999;96:893–898. doi: 10.1073/pnas.96.3.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edman L, Mets U, Rigler R. Proc Natl Acad Sci USA. 1996;93:6710–6715. doi: 10.1073/pnas.93.13.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia Y, Sytnik A, Li L, Vladimirov S, Cooperman B S, Hochstrasser R M. Proc Natl Acad Sci USA. 1997;94:7932–7936. doi: 10.1073/pnas.94.15.7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bopp M A, Jia Y, Li L, Cogdell R J, Hochstrasser R M. Proc Natl Acad Sci USA. 1997;94:10630–10635. doi: 10.1073/pnas.94.20.10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Oijen A M, Ketelaars M, Kohler J, Aartsma T J, Schmidt J. J Phys Chem B. 1998;102:9363–9366. [Google Scholar]
- 14.Law C J, Cogdell R J. FEBS Lett. 1998;432:27–30. doi: 10.1016/s0014-5793(98)00826-6. [DOI] [PubMed] [Google Scholar]
- 15.Dickson R M, Cubitt A B, Tsien R Y, Moerner W E. Nature (London) 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 16.Freer A A, Prince S M, Sauer K, Papiz M, Hawthomthwaite-Lawless A, McDermott G, Cogdell R J, Isaacs N W. Structure (London) 1996;4:449–462. doi: 10.1016/s0969-2126(96)00050-0. [DOI] [PubMed] [Google Scholar]
- 17.Wynne K, Hochstrasser R M. Chem Phys. 1993;171:179–188. [Google Scholar]
- 18.Cogdell R J, Frank H A. Biochim Biophys Acta. 1987;895:63–79. doi: 10.1016/s0304-4173(87)80008-3. [DOI] [PubMed] [Google Scholar]
- 19.Turro N J. Modern Molecular Photochemistry. Mill Valley, CA: University Science Books; 1991. [Google Scholar]
- 20.Wu H M, Ratsep M, Lee I J, Cogdell R J, Small G J. J Phys Chem B. 1997;101:7654–7663. [Google Scholar]
- 21.Kumble R, Hochstrasser R M. J Chem Phys. 1998;109:855–865. [Google Scholar]