

Abstract
To explore the analytic relevance of unbinding force measurements between complementary DNA strands with an atomic force microscope, we measured the forces to mechanically separate a single DNA duplex under physiological conditions by pulling at the opposite 5′-ends as a function of the loading rate (dynamic force spectroscopy). We investigated DNA duplexes with 10, 20, and 30 base pairs with loading rates in the range of 16–4,000 pN/s. Depending on the loading rate and sequence length, the unbinding forces of single duplexes varied from 20 to 50 pN. These unbinding forces are found to scale with the logarithm of the loading rate, which is interpreted in terms of a single energy barrier along the mechanical separation path. The parameters describing the energy landscape, i.e., the distance of the energy barrier to the minimum energy along the separation path and the logarithm of the thermal dissociation rate, are found to be proportional to the number of base pairs of the DNA duplex. These single molecule results allow a quantitative comparison with data from thermodynamic ensemble measurements and a discussion of the analytic applications of unbinding force measurements for DNA.
The direct measurement of forces between individual biological ligand receptor pairs is an emerging field. With the atomic force microscope (AFM), it has been possible to measure unbinding forces between single ligand receptor pairs, which are typically in the pico-Newton range (1–5). These sensitive force measurements can be performed with a high spatial resolution (5). However, the relevance of the force measurements as an (local) analytical tool depends on the understanding of the relationship between the measured forces and thermodynamic data characterizing the complex (6).
The measured unbinding forces are not a fundamental property of a ligand-receptor pair but depend on the loading rate that is applied to the complex: If the load on the complex increases sufficiently slowly, there is time for thermal fluctuations to drive the system over the energy barrier, and the unbinding force will be small (7). A scaling of the force with the logarithm of the loading rate is expected for a single energy barrier along the unbinding path (8) and was found in AFM experiments on unfolding of protein domains (9, 10) and on rupture force measurements of the P-selectin/ligand complex (11). With a different force probe technique, it has been possible to measure the loading rate dependence of the unbinding forces of biotin/(strept)avidin complexes (12). In this system the loading rate dependence of the force is more complicated, indicating that more than one energy barrier is present along the separation path.
The aim of our paper is to present a deeper understanding of the forces that arise if one separates the two strands of a DNA duplex by pulling at both 5′-ends. For this purpose, we have measured the loading rate dependence of the unbinding forces between complementary DNA strands to get information about the energy profile of the separation path. The knowledge of this dependence will allow us to discuss the possibility of analytic applications of unbinding force measurements between complementary strands of DNA. Our aim is to study whether the unbinding force can be predicted from knowledge of the base pair content of the duplex. Therefore, we determined as a first step how the unbinding forces depend on the number of base pairs (bp) of the DNA duplex.
Following the work of Lee et al. (4), we covalently immobilized complementary oligonucleotides with a 5′-SH modification via a cross-linker on the tip of an AFM cantilever and a substrate (schematically shown in Fig. 1A), both modified with aminosilane, respectively (13). We used a 30-nm-long poly(ethylene glycol) (PEG) cross-linker between the oligonucleotides and the surfaces to reduce unspecific forces between the tip and the substrate, which had made the interpretation of previous AFM experiments difficult (4, 14).
Figure 1.

Measurement of unbinding forces. (A) The complementary single strands of a DNA are immobilized on an AFM tip and a surface via their 5′-ends by PEG linker molecules. On approach of the surface to the tip, a duplex may form that is loaded on retract until an unbinding occurs. (B) A typical force-versus-piezo displacement for the DNA duplex a⋅b during the retract of the sample with a velocity of 100 nm/s shows that, after initial contact of the tip with the surface (positive force values), the DNA duplex is loaded and the 30 nm long PEG-linker molecules are stretched (negative force values). At a displacement of ≈50 nm, the duplex unbinds at a loading of 50 pN. (C) A force displacement curve in which two molecules unbind one after the other, the last unbinding event also being at ≈50 pN loading.
The most probable unbinding force can be determined from a histogram of the rupture forces. To facilitate the interpretation of the data, we used oligonucleotides that contain no self-complementary regions and where an “all-or-none” type of binding of the complementary strands is favored. By comparing the forces that arise between the oligonucleotide and itself and the oligonucleotide and its complement, we were able to demonstrate that the measured forces are specific. To investigate the dependence of the unbinding forces on the duplex length, we probed the same single-stranded DNA molecule immobilized on the cantilever tip against complementary DNA strands of different length immobilized on different surfaces. A comparison of the unbinding forces is possible without an exact knowledge of the spring constant of the cantilever. For each duplex, the unbinding force was measured as a function of the loading rate by measuring at different retract velocities of the piezo element where the sample is mounted. The results were interpreted in terms of a single energy barrier along the unbinding path. The rate of dissociation over the barrier (thermal off-rate) was found to decrease exponentially with the number of base pairs in the DNA duplex, allowing a quantitative comparison with thermodynamic data from the literature (15). The separation of the energy minimum from the barrier was found to increase linearly with the number of base pairs. These scaling relations lead to a nonlinear dependence of the unbinding forces on the number of base pairs, which has to be considered when discussing possible analytic applications of force measurements.
METHODS
Surface and Tip Modification.
All chemicals were purchased from Fluka unless noted otherwise. The immobilization follows essentially the protocol in ref. 13. Glass slides were cleaned in ethanol for 20 min in an ultrasonic bath and were dried under a stream of argon. From now on, both surfaces, the glass slides and the AFM-tips (Si3Ni4-Microlever, Park Scientific, Sunnyvale, CA), were treated in parallel. The surfaces were silanized immediately after a 60-min treatment with a strong UV light source (UV-Clean, Boekel Scientific, Feasterville, PA) in a 2% solution of aminopropyltriethoxysilane in dry toluene for 2 h. After rinsing with toluene and drying under a stream of argon, the surfaces were immersed in a 1 mM solution of poly(ethylene glycol)-α-maleimide-ω-N-hydroxysuccinimide-ester (molecular weight 3,400; Shearwater Polymers, Huntsville, AL) in DMSO for 2–3 h. The surfaces again were rinsed with DMSO and were dried in a stream of argon. The oligonucleotides with a 5′-SH modification were synthesized by Microsynth (Balgach, Switzerland) and were stored in a pH 6.5 phosphate buffer containing 10 mM DTT at −18° until use. Immediately before use, the oligonucleotides were diluted to a final concentration of 25 μM with a pH 6.5 phosphate buffer, and DTT was extracted from an aliquot of typically 200 μl by washing three times with 1 ml of ethylacetate. A 50-μl drop of the oligonucleotide solution then was incubated on the poly(ethylene glycol)-α-maleimide-modified surfaces overnight at room temperature in a humid chamber. After rinsing with PBS buffer (pH 7.3; Life Technologies), the tips and surfaces were ready for use in the force experiments.
Dynamic Force Spectroscopy.
Force-displacement measurements were performed with a commercial AFM (Nanoscope III, Digital Instruments, Santa Barbara, CA) in which the acquisition of the cantilever deflection data and the control of the piezo displacement was improved by using a 16-Bit AD/DA card (National Instruments, Austin, TX) in connection with an additional personal computer and a home-build high voltage amplifier.
We implemented a dynamic force spectroscopy mode in the software-controlled (labview, National Instruments) piezo displacement: To acquire the distribution of the rupture forces at different retract velocities, the value of the velocity was changed after every approach/retract cycle (keeping the approach velocity constant) to the next faster value. When the largest retract velocity was reached, the cycle started again at the smallest velocity. In this way, the forces at different velocities were acquired quasi in parallel to reduce the influence of drift in the sample versus tip position. All measurements were performed at 25°C in PBS buffer (pH 7.3; Life Technologies)
The spring constants of all cantilevers were calibrated by the thermal fluctuation method (16) with an absolute uncertainty of 20%. The measurements presented here were performed with eight different cantilevers with spring constants ranging from 12 to 17 pN/nm.
RESULTS AND DISCUSSION
Unbinding Force Measurement.
The base sequence of the oligonucleotides we have investigated were designed to favor the binding to its complementary oligonucleotides in the ground state with respect to intermediate duplexes in which the strand is shifted relative to its complement. Therefore, we have chosen the DNA oligomer a = 5′-G-G-C-T-C-C-C-T-T-C-T-A-C-C-A-C-T-G-A-C-A-T-C-G-C-A-A-C-G-G-3′, which contains 30 bases and in which every three base motive, e.g., G-G-C, occurs only once in the sequence. To avoid self-complementarity, the complement of each three-base motive is not contained in the sequence. The sequence a is tested against its complement b = 3′-C-C-G-A-G-G-G-A-A-G-A-T-G-G-T-G-A-C-T-G-T-A-G-C-G-T-T-G-C-C-5′ and the truncated complements c = 3′-A-T-G-G-T-G-A-C-T-G-T-A-G-C-G-T-T-G-C-C-5′ and d = 3′-T-A-G-C-G-T-T-G-C-C-5′ of length 20 and 10, respectively. The duplexes a⋅b, a⋅c, and a⋅d have approximately the same CG content of 60, 55, and 60%, respectively.
The force spectra between the different oligonucleotides are acquired by approaching the surface to the tip until a repulsive force is sensed. To prevent unspecific binding, the data presented here were recorded under conditions in which this repulsive force on approach never exceeded 150 pN. On retraction of the sample from the tip, the force-displacement curve may show a typical unbinding event like in Fig. 1B. Here, a tip modified with the sequence a is tested against a surface with the complement b. The force-displacement curve shows the typical nonlinear elastic behavior of the linker polymer on stretching before the DNA duplex unbinds.
To test the specificity of the measured unbinding forces between the a-tip and a complementary b-surface, we measured the forces between the a-tip and an a-surface. The activity of the a-surface was afterward proven by testing it against a b-tip and the b-tip against the previously used b-surface. The result of such a measurement is shown in the force histograms in Fig. 2, where the distribution of the last unbinding forces for all four tip-sample combinations is shown. This figure demonstrates that the observed unbinding forces are attributable to the specific interaction of the complementary base pairs.
Figure 2.

The probability distribution of the unbinding forces of the last rupture events for ≈500 approach/retract cycles for a retract velocity of 100 nm/s. (A) The histograms show the specific a-tip/b-surface interaction (gray) and the unspecific a-tip/a-surface interaction (black). (B) Histograms of the b-tip versus a- (gray) and b-surface (black) unbinding force distribution. This tip shows an increase in multiple unbinding forces and unspecific binding. The comparison of all four tip/surface combinations demonstrates the specificity of the unbinding force experiment. In both histograms, the most probable unbinding force is ≈50 pN. The maximum of the distribution is found by a Gaussian fit to be, for example, 48 ± 2 pN in A.
The forces observed in the measurements of the sequence against itself are very likely caused by unspecific tip-surface interactions, which is indicated by the fact that the unspecific events occur closer to the surface than the specific interaction where both polymeric linkers of 30-nm length have to be stretched before the unbinding of the complex arises. From the histograms in Fig. 2A, we conclude that not more than 10% of the unbinding forces in the complementary a-tip/b-surface interaction are unspecific.
To quantify the most probable value of the unbinding force for a single complex, one has to work under conditions in which the probability that two or more duplexes unbind at the same time can be neglected. The immobilization of the oligonucleotides via the 30-nm-long PEG linker favors this conditions: When two complexes form, it is very unlikely that the two linker molecules, being attached to different points on the tip with a radius of curvature of ≈50 nm, are extended to the same length on stretching. Because of the nonlinear elastic behavior of the linker, the complex with the most extended linker molecule will hold most of the loading force and rupture first. This leads to a situation in which multiple complexes rupture sequentially (Fig. 1C). In the a-tip/b-surface interaction of Fig. 2A, where the total binding probability is ≈75%, 33% of the curves show one rupture event, 20% show two, 15% three, and 7% more than three subsequent rupture events. If the binding obeys Poisson-statistics, one complex would form in 35%, two complexes in 24%, three in 11%, and more than three in 5% of all trials. This excellent agreement with the observed number of rupture events indicates that all multiple binding events have been detected and that it is very unlikely that the last rupture event is attributable to the unbinding of more than one DNA duplex. It is, therefore, reasonable to assume that the observed unbinding forces are attributable to the unbinding of a single DNA duplex. We found the most probable unbinding force by fitting a Gaussian to the observed rupture force distribution, giving a value of 48 ± 2 pN for the unbinding of the 30 base pairs (with a retract velocity of 50 nm/s) in Fig. 2A. We estimate the statistical error of 2 pN by 2σ/√N (96% confidence level), where σ is the width of the distribution of the N rupture events in the histogram.
Dynamic Force Spectroscopy.
We measured the most probable unbinding force for the three duplexes a⋅b, a⋅c, and a⋅d in dependence on the retract velocity. For each duplex, typically, 200–400 unbinding events (from 300–600 approach/retract cycles) were recorded at five or six different values of the retract velocity in the range between 8 nm/s and 2,000 nm/s, keeping the approach velocity constant to 100 nm/s. Fig. 3A shows the shift in the unbinding force distribution for the a⋅d duplex by increasing the retract velocity from 8 to 1,600 nm/s. The maximum of the distribution shifts by 20 pN in this case.
Figure 3.

Velocity dependence of the unbinding forces. (A) The maximum of the unbinding force distribution for the a⋅d duplex (10 bp) is shifted from 21 to 41 pN (from Gaussian fits) when increasing the retract velocity from 8 to 1,600 nm/s. The broadening of the distribution is partially attributable to an increase of noise in the force displacement curves at higher retract velocities (because the rate of data acquisition is constant, fewer deflection values are acquired to determine the force at higher velocities). (B) The dependence of the most probable unbinding force on the retract velocity for an a-tip against a b-surface (30 bp), c-surface (20 bp), and d-surface (10 bp) and linear fits to the respective data sets. The extrapolation to zero unbinding force gives an estimate for the thermal off-rate.
The results of a measurement, where it was possible to acquire the data for the three duplexes with the same a-tip, is shown in Fig. 3B. Over the range of the measured retract velocities, the data could be interpreted in terms of a cooperative unbinding of the base pairs in the duplex. The cooperative thermal dissociation of short oligonucleotides (17) is described by the thermal off-rate ν = ν0exp(−E/kBT), where the frequency ν0 is a prefactor, E is the activation energy for dissociation, and kBT (4.1 pNnm at room temperature) is the thermal energy. In a model with a single energy barrier of height E along the separation path, the application of an external force f leads to an exponential increase of the off-rate ν(f) = νexp(fx/kBT), where the separation x is a length scale describing the separation of the energy minimum from the barrier. If the load on the complex increases with a constant rate uc, which is given by the retract velocity u times the elasticity c, the most probable unbinding force F depends logarithmically on the loading rate (7):
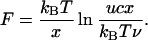 |
1 |
By a linear fit of the data in Fig. 3B, one is able to determine the parameters in Eq. 1 for each DNA duplex: namely, the separation x from the slope of the linear fit and the thermal off-rate ν by extrapolating to zero force F. To calculate the values for the thermal off-rates ν, the elasticity c had to be determined from the force-displacement curves: Although the elasticity (force increase per piezo displacement) is a nonlinear function of the force, attributable to the nonlinear entropic elasticity of the PEG linker, the relation in Eq. 1 is still a good approximation if the elasticity c is chosen to be the elasticity at the most probable unbinding force (18). This elasticity is the slope of the force-displacement curve before the unbinding event occurs, and values from 1.5 to 3 pN/nm for unbinding forces from 20 to 50 pN have been found. The moderate increase of the elasticity with the unbinding forces preserves the linear scaling of the unbinding forces with the logarithm of the retract velocity to a good approximation. We have adopted a constant value of 2 pN/nm for the elasticity to calculate a numeric value for the thermal off-rate. The values for the data in Fig. 3B and an additional data set (obtained with different tips and surfaces) are compiled in Fig. 4, where the errors reported are the standard deviations from the linear regression.
Figure 4.

Measured parameters of the single energy barrier model (Eq. 1). The thermal off-rate ν (A) and the separation x (B) were determined from the linear fits of Fig. 3B (filled squares) and an additional data set [obtained from b-tip/a-surface, a-tip/c-surface, and a-tip/d-surface measurements (open squares)]. Error bars are the statistical errors of the fits. The separation and the logarithm of the thermal off-rate scale linearly with the number of base pairs.
The data in Fig. 4 indicate clearly that the values for the separation x and the thermal off-rate ν are well reproduced within the statistical error for different data sets. The most important fact to understand the unbinding forces that arise between the complementary oligonucleotides is that both the separation x and the logarithm of the thermal off-rate ν scale in a first approximation linearly with the number of base pairs in the duplex.
Thermal Off-Rates.
Measurements of the thermal dissociation rates (thermal off-rates) of the investigated duplexes with other methods are not available, but a similar scaling of the thermal off-rate with the number of base pairs n has been found for oligo(A)⋅oligo(U) duplexes of 8–18 bp with a temperature jump method (18, 19). An exponential decrease of the thermal off-rate with the number of base pairs is expected because of the increase of the activation energy E for dissociation (≈10 kT per A⋅U base pair). However, the frequency prefactor ν0 also strongly increases with the number of base pairs because of the increasing number of degrees of freedom of the system, so that the thermal off-rate was found to decrease approximately as 108−0.5n⋅s−1 at room temperature (17). Our measured off-rates also can be described by
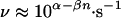 |
2 |
where we found α = 3 ± 1 and β = 0.5 ± 0.1 from a linear regression of the data in Fig. 4A. The fact that the off-rates are lower for duplexes containing CG pairs is known (17), but literature values for CG-rich systems are rare (15). For instance, the thermal off-rate for the duplex (5′-G-G-G-C-3′)⋅(3′-C-C-C-G-5′) was found to be 40 s−1 (15) at room temperature compared with ≈10 s−1 (calculated on the basis of Eq. 2 with n = 4). This suggests a reasonable agreement of our measured off-rates with thermodynamic data. To determine the height of the energy barrier, temperature-dependent off-rate measurements would be necessary.
Forced Unbinding Pathway.
The length scale x describes the difference in 5′-end to 5′-end separation between the bound state representing the energy minimum on the one hand and the state representing the energy barrier, i.e., the transition state, on the other hand. To estimate the order of magnitude of the separation x, it is reasonable to assume that the transition states along the mechanical unbinding path are similar to the transition states of the thermal dissociation path: Without applied load, the thermal fluctuations will open the duplex at the helix ends (17), and dissociation occurs if all base pairs are zipped open. Thus, the transition states can be described by a situation in which only a few base pairs are formed. An upper limit for the separation x can be estimated if we assume that the bound state is a B-DNA helix with an axial rise of 3.4 Å per base pair (20). If the transition state consists only of one base pair and totally stretched single-stranded DNA ends with a distance of ≈7 Å between adjacent phosphate groups (20), the separation x is proportional to the number of base pairs (Fig. 5A), increasing by ≈3.6 Å per base pair. A linear regression of the data in Fig. 4B gives
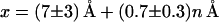 |
3 |
for the increase in separation. The increase in 5′-end to 5′-end separation of ≈1 Å per base pair is significantly smaller than the maximal expected separation increase of 3.6 Å. This may originate from the fact that the extension of the duplex in the bound (double-stranded DNA) and at the transition state (single-stranded DNA) is not described correctly in the above argument. For instance, the duplex deforms under an applied load, and, for a large (>1,000 bp) duplex, a cooperative transition at 60–70 pN from a B-DNA to a so-called overstretched S-DNA (B-S transition) is found, where the difference in extension between single- and double-stranded DNA almost vanishes (21, 22). However, the elasticity of the duplex itself cannot be extracted from the force displacement curves directly because the elasticity is dominated by the PEG linker. Certainly, more sophisticated models are needed to understand the 5′-end to 5′-end separation of a DNA duplex at a transition state and to clarify the significance of the offset length of 7 Å in the linear regression (Eq. 3) that accounts for a length scale in the unbinding pathway that is independent of the number of base pairs.
Figure 5.

Geometrical considerations. (A) The mechanical unbinding path is suggested to be similar to the thermal dissociation path: Starting from a B-DNA helix, representing the energy minimum, thermal fluctuations will open the helix at the ends and drive the system over the reduced energy barrier E-Fx. Here, the transition state has one base pair and stretched single-stranded ends that would lead (as an upper limit) to a separation increase x of 3.6 Å per base pair. (B and C) A nick (B) (located far away form the helix ends) experiences a force that should be independent of the geometry of the force at the helix ends—i.e., if the force is applied via the 3′- or 5′-ends. Therefore, the melting on stretching of a DNA (B) corresponds to the unbinding of the DNA duplex on loading via both 5′-ends (C). (C and D) The difference in the unbinding geometry of the 5′-end to 3′-end (D) and the 5′-end to 5′-end (C) separation of a DNA duplex leads to different unbinding forces because the length gained on opening one base pair is ≈2× bigger in the first compared with the second case.
Comparison with Mechanical Properties of DNA.
We first note that, because of the above relations (Eqs. 1, 2, and 3), the unbinding force itself is a nonlinear function of the number of base pairs. Interestingly, the rupture forces for very large DNA duplexes saturate at a finite value of 1.2 kBT/0.7 Å ≈ 70 pN, independent of the loading rate if the scaling found for oligonucleotides with up to 30 bp extends to duplexes of arbitrary length.
There is evidence from experiments on the stretching of long double-stranded DNA (>1,000 bp) (15, 17) that such an upper limit for the 5′-end to 5′-end force of a DNA molecule exists. Although the exact geometry of the force at the helix ends is not known in the stretching experiments of long DNA, a nick in a DNA duplex behaves similar to the free 3′-end in our separation experiment (Fig. 5 B and C). In fact, it was argued (21, 23) that the stretching of DNA can lead to melting because of the separation of one strand starting at the nick (or a free 3′end). This melting was observed as a hysteresis in the force-versus-extension curves at or above the B-S transition for DNA duplexes at forces ranging from 35 pN for poly(dA-dT) (23), 70 pN for λ-DNA (21), 70–200 pN for a λBstEII digest DNA, to 250 pN for poly(dG-dC) (23). However, in the case of the λBstEII digest DNA, the melting force, in contrast to the B-S transition, has been shown to depend on the pulling speed. The melting force is the maximal force a large DNA duplex can hold when stretched at both 5′-ends because, in this case melting, would result in a mechanical separation.
Although the unbinding of the investigated DNA duplexes occurs at forces below the B-S transition, i.e., the bound state is assumed to be a B-DNA, a few unbinding events occur at forces >70 pN because of the statistical nature of the unbinding. A B-S transition is indicated as a region of constant force in a force-displacement curve that should be located around 70 pN. This transition is difficult to resolve unambiguously (because of the experimental noise) for the 30-bp duplex. The fact that the average slope of the force-displacement curves is smaller for forces in the range of 60–70 pN compared with forces in the range of 50–60 and 70–80 pN for the data of Fig. 2 may indicate the B-S transition of the 30-bp duplex.
The 5′-end to 5′end unbinding forces measured with our setup also can be compared with experiments in which λ-phage DNA (24) or poly(dG-dC) and ploy(dA-dT) hairpins (23) were mechanically separated with a different geometry of the acting forces, namely by pulling apart the 3′- and 5′-end of the two DNA strands. Forces of 11–14 pN for λ-phage DNA, 20 pN for a poly(dG-dC), and 9 pN for a poly(dA-dT) hairpin are needed to separate the two strands, independent of the loading rate and number of base pairs. In this case, the forces are interpreted as the average energy (over ≈50 bp) per base pair divided by the 5′-end to 3′-end separation increase per open base pair. One can apply this argument to our 5′-end to 5′-end geometry of the applied force by noting that the 5′-end to 3′-end separation increase is ≈2× the 5′-end to 5′-end separation increase per opened base pair (Fig. 5 C and D) if the stretching of the DNA duplex on pulling at the opposite 5′-ends is neglected. This would yield forces 2× bigger: e.g., 18–40 pN when separating a long duplex.
Analytic Application of Force Measurements.
If one thinks of the detection of hybridization by AFM force measurements (25), e.g., in a DNA sequencing application, the accuracy of the information about the thermal off-rate and the number of base pairs in a duplex is determined by the range of loading rates and the quality of the statistics of unbinding forces at each loading rate. Neglecting a systematic error in the spring constant of 20%, the error in the separation x is already low enough to conclude that one should be able to determine a length difference of a single base pair in a 10-bp duplex. For example, the difference in the most probable unbinding force between a 9- and 10-bp duplex at a loading rate of 300 pN/s should be 3 pN, which is still measurable. Because one mismatched base pair can increase the thermal off-rate by one order of magnitude (26), this should lead to a difference of 7 pN in the unbinding force of a 10-bp duplex at 300 pN/s loading. With the same argument, variations in the CG content should be detectable if they lead to changes in the thermal off-rate by one order of magnitude. For larger duplex sizes, the resolution decreases so that the best contrast in the force is reached for small duplex sizes. Note, however, that, for small duplexes (<5 bp), the thermal off-rate gets too fast to detect an unbinding event in the time scale of the AFM experiment; i.e., the measurement between an a-tip and a surface with the oligonucleotide 3′-T-T-G-C-C-5′ showed no unbinding events.
Conclusion.
The cooperative unbinding of the base pairs in the DNA duplex leads to a scaling of the unbinding forces with the logarithm of the loading rate. This behavior is described with one length scale (the separation x) and one time scale (given by the thermal off-rate). The separation x was found to increase with the number of base pairs, again indicating the cooperativity of the unbinding event. Especially the exponential decrease of the thermal off-rate with the number of base pairs is in good agreement with thermodynamic data. We are thus able to predict values of the unbinding forces from the knowledge of the thermal off-rate of a DNA duplex.
On a molecular scale, the forces between complementary DNA strands can be considered as a model system for ligand-receptor systems in general. Because length scales for separation and thermal off-rates are of a comparable order of magnitude in these systems, our considerations about the analytic application of force measurements can be generalized to these systems.
Acknowledgments
We thank E. C. Constable and J. Engel for a critical reading of the manuscript. This work was supported by the Swiss National Science Foundation.
ABBREVIATIONS
- AFM
atomic force microscope/microscopy
- PEG
poly(ethylene glycol)
References
- 1.Lee G U, Kidwell D A, Colton R J. Langmuir. 1994;10:354–357. [Google Scholar]
- 2.Florin E L, Moy V T, Gaub H. Science. 1994;264:415–417. doi: 10.1126/science.8153628. [DOI] [PubMed] [Google Scholar]
- 3.Dammer U, Popescu O, Wagner P, Anselmetti D, Güntherodt H-J, Misevic G N. Science. 1995;267:1173–1175. doi: 10.1126/science.7855599. [DOI] [PubMed] [Google Scholar]
- 4.Lee G U, Chrisley L A, Colton R J. Science. 1994;266:771–773. doi: 10.1126/science.7973628. [DOI] [PubMed] [Google Scholar]
- 5.Ros R, Schweisinger F, Anselmetti D, Kubon M, Schäfer R, Plückthun A, Tiefenauer L. Proc Natl Acad Sci USA. 1998;95:7402–7405. doi: 10.1073/pnas.95.13.7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moy V T, Florin E L, Gaub H E. Science. 1994;266:257–259. doi: 10.1126/science.7939660. [DOI] [PubMed] [Google Scholar]
- 7.Evans E, Ritchie K. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bell G I. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 9.Rief M, Gautel M, Oesterhelt F, Fernandez J M, Gaub H E. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 10.Carrion-Vazquez M, Oberhauser A F, Fowler S B, Marszalek P E, Broedel S E, Clarke J, Fernandez J M. Proc Natl Acad Sci USA. 1999;96:3694–3699. doi: 10.1073/pnas.96.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fritz J, Katopodis A G, Kolbinger F, Anselmetti D. Proc Natl Acad Sci USA. 1998;95:12283–12288. doi: 10.1073/pnas.95.21.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. Nature (London) 1999;397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]
- 13.Chrisely L A, Lee G U, O’Ferral E. Nucleic Acids Res. 1996;24:3031–3039. doi: 10.1093/nar/24.15.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noy A, Vezenov D V, Kayyem J F, Meade T J, Lieber C M. Chem Biol. 1997;4:519–527. doi: 10.1016/s1074-5521(97)90324-0. [DOI] [PubMed] [Google Scholar]
- 15.Turner D H, Sugimoto N, Freier S M. In: Landolt-Börnstein, New Series VII/1c. Saenger W, editor. Berlin: Springer; 1990. pp. 201–227. [Google Scholar]
- 16.Hutter J L, Bechhoefer J. Rev Sci Instrum. 1993;64:1868–1873. [Google Scholar]
- 17.Pörschke D. Mol Biol Biochem Biophys. 1977;24:191–218. doi: 10.1007/978-3-642-81117-3_5. [DOI] [PubMed] [Google Scholar]
- 18.Evans E, Ritchie K. Biophys J. 1999;76:2439–2447. doi: 10.1016/S0006-3495(99)77399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pörschke D, Eigen M. J Mol Biol. 1971;62:361–381. doi: 10.1016/0022-2836(71)90433-5. [DOI] [PubMed] [Google Scholar]
- 20.Saenger W. Principles of Nucleic Acid Structure. Berlin: Springer; 1984. p. 261. [Google Scholar]
- 21.Smith S B, Cui Y, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 22.Cluzel P, Lebrun A, Heller C, Lavery R, Viovy J-L, Chatenay D, Caron F. Science. 1996;271:792–794. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- 23.Rief M, Clausen-Schaumann H, Gaub H E. Nat Struct Biol. 1999;6:346–349. doi: 10.1038/7582. [DOI] [PubMed] [Google Scholar]
- 24.Essevaz-Roulet B, Bockelmann U, Heslot F. Proc Natl Acad Sci USA. 1997;94:11935–11940. doi: 10.1073/pnas.94.22.11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazzola L T, Frank C W, Fodor S P A, Mosher C, Lartius R, Henderson E. Biophys J. 1999;76:2922–2933. doi: 10.1016/S0006-3495(99)77447-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tibanyenda N, de Bruin S H, Haasnoot C A G, Van der Marel G A, Van Boom J H, Hilbers C W. Eur J Biochem. 1984;139:19–27. doi: 10.1111/j.1432-1033.1984.tb07970.x. [DOI] [PubMed] [Google Scholar]