

Abstract
When a protein folds or unfolds, it has to pass through many half-folded microstates. Only a few of them can be seen experimentally. In a two-state transition proceeding with no accumulation of metastable intermediates [Fersht, A. R. (1995) Curr. Opin. Struct. Biol. 5, 79–84], only the semifolded microstates corresponding to the transition state can be outlined; they influence the folding/unfolding kinetics. Our aim is to calculate them, provided the three-dimensional protein structure is given. The presented approach follows from the capillarity theory of protein folding and unfolding [Wolynes, P. G. (1997) Proc. Natl. Acad. Sci. USA 94, 6170–6175]. The approach is based on a search for free-energy saddle point(s) on a network of protein unfolding pathways. Under some approximations, this search is rapidly performed by dynamic programming and, despite its relative simplicity, gives a good correlation with experiment. The computed folding nuclei look like ensembles of those compact and closely packed parts of the three-dimensional native folds that contain a small number of disordered protruding loops. Their estimated free energy is consistent with the rapid (within seconds) folding and unfolding of small proteins at the point of thermodynamic equilibrium between the native fold and the coil.
The folding nucleus plays a key role in protein folding (1–4). The nucleus is the only observable, though absolutely unstable, intermediate in the simplest two-state folding. It corresponds to the transition state, i.e., to the free-energy maximum on the folding/unfolding pathway (or, better, to the free-energy saddle point on the network of these pathways).
So far, there is only one very difficult experimental method to identify the folding nuclei in proteins: to find the residues whose mutations affect the folding rate changing the nucleus stability as strongly as that of the native protein (5).
Folding simulations held on simple lattice protein models (6–12) and analytical theories (13–15) also give interesting, intriguing, and sometimes contradictory information on the basics of the nucleation process.
As regards the theoretical search for folding/unfolding nuclei in proteins, several different approaches have been suggested recently. The first is based on the search for a set of highly conserved residues having no obvious functional role (16–18); however, this approach can give no more than a common part of the nuclei existing in homologous proteins. The second, more direct approach is based on all-atom molecular dynamic simulations of protein unfolding (19–22). However, these simulations need extremely denaturing conditions (600 K, etc.) to be completed. Therefore the transition state found for such an extreme unfolding can be rather different from that existing for folding (23). The third approach is based on the investigation of peculiarities of free-energy surfaces of folding proteins (24–27).
Here we present an approach to the search for the folding nucleus (or nuclei). It computes the unfolding of the three-dimensional (3D) protein structure. Unlike results shown in refs. 19–22, it uses a simple free-energy function and dynamic programming (not molecular dynamic simulations) to find the transition states; unlike results shown in refs. 24–27, the network of unfolding pathways is considered explicitly. Simulation of unfolding is much simpler than simulation of folding (because exploring numerous high-energy dead ends can be avoided), whereas according to the physical principle of detailed equilibrium, the pathways and transition states for folding and unfolding must coincide when these processes take place under the same conditions. Therefore this study focuses on conditions close to those of thermodynamic equilibrium between the native and the unfolded coil states. Under these conditions, small proteins demonstrate two-state (i.e., “all-or-none”) transitions both in thermodynamics (28) and kinetics (1, 2). This absence of the other accumulating states allows us to take into account only the pathways going from the native to the unfolded state and to neglect those leading to misfolded globules (14, 29).
Following the earlier developed theory (14, 29), we consider a simplified stepwise unfolding, each step of which is the removal of a chain link (i.e., a chain fragment of one or a few residues) from the native 3D structure. The removed links are assumed to form a random coil; they lose all nonbonded interactions and gain coil entropy (except that spent to close the disordered loops protruding from the remaining globule; Fig. 1). This is our first simplification. The next is the assumption that the links remaining in the globule keep their native positions and that the unfolded regions do not fold into another nonnative globule. Thus, we actually neglect nonnative interactions that make our model similar to that of Gō (30). Further, to facilitate the computations, we now limit the number of loops protruding from the folding intermediates according to the estimates obtained in refs. 14 and 29 and use “chain links” of two (or four for larger proteins) residues. The last and main simplification is that we concentrate on the transition states, i.e., on the stability (actually, the instability) of partly unfolded intermediates rather than on a detailed description of the chain motions.
Figure 1.

A pathway of sequential unfolding (and folding) of the native 3D structure S0. SU is the coil. The U–ν links in the intermediate Sν keep their native positions and conformations (they are shown as a solid line against the background of a dotted cloud denoting the globule), whereas the other ν links (shown in dashed line) are unfolded.
All computations presented in this study refer to the point of thermodynamic equilibrium between the native and coil states.
Free-Energy Estimate.
Semifolded proteins with some given residues fixed in their native positions and the other disordered can be described as being in a “microstate,” because a disordered link corresponds to an ensemble of many conformations. The free energy of microstate S (of nS disordered residues and N − nS residues keeping their native positions and conformations) is taken as
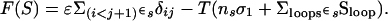 |
1 |
The first sum is taken over all the nonneighbor residues i, j, keeping their native positions in S. The second sum is taken over all the closed disordered loops protruding from the globular native-like part of S (in the scheme given in Fig. 1, the left intermediate contains two closed loops and no tails, and the next one has both the N- and the C-terminal tails and one closed loop). δij is the number of atom–atom contacts (the contact distance is <5 Å) between residues i and j in the native 3D structure; ɛ is the energy of one atom–atom contact; T is the temperature; σ1 is the entropy difference between the coil and the native state of a residue (according to ref. 28, we take σ1 = 2.3R, R being the gas constant). The native state (with nS = 0) and the coil (with nS = N) have equal free energies at the equilibrium temperature. Because both of these states include no closed disordered loops, ɛ and T are connected by the equation ɛ = −TNσ1/(Σ(i<j+1)∈S_nativeδij). Thus, we can express all the free energies in RT units (where T is the transition temperature). The entropy spent to close a disordered loop between fixed residues k and l is estimated (29) as
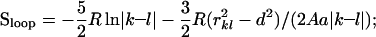 |
2 |
where rkl is the distance between the Cα atoms of the residues k and l, a = 3.8 Å is the distance between the neighbor Cα atoms in the chain, and A is the persistent length for a polypeptide (according to ref. 31, we take A = 20 Å).
Transition States at the Unfolding Pathways.
Let us consider the unfolding of the native structure of the N-residue protein chain. As explained above, each unfolding step is the removal of one “chain link” from the native 3D structure. Let the chain be (mentally) divided into U links (thus, the chain fragment corresponding to the “link” includes N/U residues; the links, rather than separating residues, are used to simplify the computations). Let us consider some unfolding pathway P = (S0→S1→… →SU) corresponding to the above scenario (Fig. 1). Each step of the pathway is reversible; S0 is the native state, SU is the completely unfolded state of the chain of U links, and S1, … , SU-1 are the intermediates at the pathway p. The microstate Sν (ν = 0, 1, .., U) contains ν disordered and U-ν ordered links; the free energy of microstate Sν is F(Sν); then the maximum Fp# = max {F(S0), F(S1), … , F(SU)} points to the free-energy barrier at the pathway p. When this barrier is found, the transition state and the folding nucleus corresponding to the pathway p can be singled out.
However, the possible folding/unfolding pathways are numerous. A large network of these pathways (Fig. 2) forms the “folding funnel” (32): there are many different intermediates Sν for each number ν of disordered links (0 < ν < U), and each sequence of microstates S0, S1, … , SU forms a possible pathway when all the corresponding transitions (S0→S1, S1→S2, … , SU-1→SU) are the above described elementary unfolding steps consisting of the unfolding of one link from the native 3D structure.
Figure 2.

Various unfolding intermediates (only a small number of them are shown) and a network of unfolding pathways. The arrows correspond to elementary unfolding steps, each of which is a transition of one link from the globular native-like part of the intermediate to the coil. Any continuous chain of arrows forms a possible unfolding pathway (see Fig. 1), and a molecule can go up and down along each of them.
The most efficient kinetic pathway has the minimal (over all the pathways) free energy of the transition state, F#min = minpossible p{Fp#}: this pathway passes from S0 (the native state) to SU (the coil) via the lowest barrier, i.e., via the lowest saddle point of the free-energy landscape. When this saddle point is found, the most effective folding nucleus can be singled out.
Each step from S0 leads to some S1; a step from any SU-1 leads to SU; however, when ν = 2,… ,U-1, a step from Sν-1 leads to only some of the Sν microstates. Let Sν-1ε{Sν-1→Sν} mean that Sν-1 can be transformed into Sν in an elementary step (i.e., by removal of one link from the globular part of Sν-1). At any pathway, all the intermediates must obey this condition. Thus, the saddle point free energy can be presented as
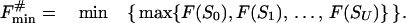 |
3 |
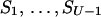 |
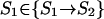 |
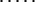 |
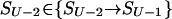 |
Despite the astronomical number of possible pathways, F#min can be calculated by a recursive algorithm similar to that of dynamic programming; the latter is a general approach to optimization of multistage processes (33, 34). The algorithm is as follows.
Let p(Sν) be the altitude of the lowest free-energy barrier at the pathways leading from S0 to Sν inclusively; thus, F#min = p(SU). The p(Sν) values are computed recursively:
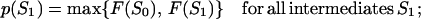 |
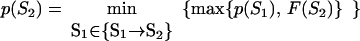 |
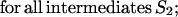 |
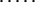 |
4 |
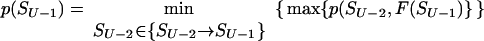 |
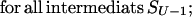 |
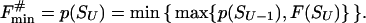 |
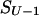 |
This algorithm computes the altitude of the lowest saddle point at the free-energy barrier dividing the native fold and the coil. All the p(S) values are stored for use later to find the saddle point microstate(s) themselves.
To find these microstate(s) [we say “microstate(s),” for there is no guarantee that only one saddle point has the minimal free energy], we perform a similar recursion in the inverse direction (33, 34). The aim of this inverse recursion is to find q(S), the altitude of the lowest free-energy barrier at the pathways following from S (exclusively) to SU, and then to compute
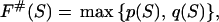 |
5 |
the altitude of the lowest free-energy barrier at the pathways leading from S0 to SU via each intermediate S. The q(S) values are also computed recursively:
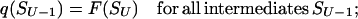 |
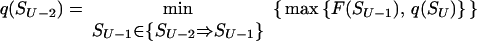 |
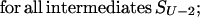 |
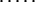 |
6 |
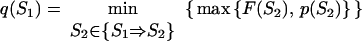 |
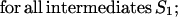 |
here Sv∈{Sv-1⇒Sv} means that microstate Sv can be obtained from Sv-l in one elementary step.
The intermediates S with F#(S)=F#min give an ensemble of transition (micro)states {S#min} with the minimal free energy.
Simplifications.
For all the above described calculations to be feasible, we considered only the intermediates with no more than two closed disordered loops (in the middle of the chain) plus the N- and C-terminal disordered tails (see Fig. 2). These four unfolded regions should be enough to describe the unfolding of a protein of about 100 residues, because the estimated (29) number of coil regions in the folding nucleus is close to L2/3/6 for a protein of L residues. Further, to facilitate the computations, we used “chain links” consisting of a few residues: two for proteins including less than 100 residues and four for larger proteins. These “links” limit the accuracy of our calculations only slightly: the links are still much smaller than the expected size of a nucleus in the vicinity of midtransition between the folded and unfolded phases [where the nucleus should include roughly 1/3 of the protein globule (14, 29)].
With all these limitations, the network of intermediates (Fig. 2) usually includes ≈106 microstates.
Ensemble of Transition States.
There is no guarantee that the protein folds and unfolds only via the transition state(s) having the minimal free energy. It can use also other, not optimal but possibly numerous, passages over the free-energy barrier, and we have to find them to estimate their diversity.
Microstate S is a pass over the barrier if its own free energy F(S) coincides with F#(S), the altitude of the lowest free-energy barrier at the pathways leading from S0 to SU via this S. Therefore we select the microstates with F(S)=F#(S) and thus obtain an ensemble {S#} of all the possible passes over the free-energy barrier dividing S0 from SU. Generally speaking, these passes belong to different pathways (though it is not excluded that some passes belong to one and the same pathway). Thus, the ensemble {S#} gives the utmost estimate of the variety of transition microstates (this set can be redundant because a pathway to a transition state high in free energy can pass via some transition state or states of the lower free energy), while the ensemble {S#min} [which includes only transition microstate(s) with the minimal free energy] gives the lowest estimate of their variety. To outline the nucleus, we studied both these sets.
To investigate the ensemble {S#} (or {S#min}), we compute the Boltzmann weights for all the transition states of the ensemble.
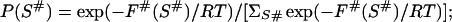 |
7 |
The sum here is taken over all the states forming the ensemble {S#} (or {S#min}, when we are interested only in the lowest-free-energy transition states). All the intermediates forming the ensemble {S#min} have equal free energies and thus equal Boltzmann weights. For the ensemble {S#} members, the free energies and thus the weights P(S#) can be different. The higher the weight P(S#), the more rapid the pathway via this S# [according to the conventional (35) exponential dependence of the reaction rate on the transition state free energy], and therefore the more the chains use this pass S# for folding and unfolding.
Computation of Φ Values.
The experimental data on the transition state structure are expressed in Φf values (1, 2, 5, 36). Φf is close to 1 when a residue has its native conformation and environment in the transition state and to 0 when the residue is unfolded in this state.
In the same way, we can also describe the computed transition state. When this state consists of only one microstate, we can easily say which residues are included in the native part of this microstate and have all their native contacts there (these residues have Φ = 1), which residues are included into the native part but have only some part of their native environment there (these residues have Φ between 1 and 0), and which residues are unfolded and thus have Φ = 0.
However, when the transition state is an ensemble of many microstates, we see only that some residues are often involved in the native-like parts of these microstates and some rarely. Besides, it has to be taken into account that the experimental Φf values are found by point mutations changing the side groups, i.e., Φf values primarily reflect the native environment of a residue side chain. Therefore for each residue r we compute the average fraction of the side chain native contacts preserved in the transition state ensemble {S#}:
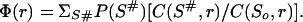 |
8 |
Here the sum is taken over all the transition states of the ensemble {S#}; C(S#,r) is the number of contacts between the side chain atoms of residue r and all the atoms of the other natively positioned residues in microstate S# (except the contacts with the next-neighbor residues: these are ignored because they are mostly present also in the coil); when residue r is not included in the globular part of S#, C(S#,r) = 0; C(S0,r) is this number in the native structure [in the rare cases when C(S0,r) = 0, the value C(S#,r)/C(S0,r) is taken as 1). An analogous value Φmin(r) can be computed also for the case when we consider only the transition states having the minimal free energy.
Thus, the computed Φ values have the same meaning as the Φf values derived from protein engineering experiments. They are compared to estimate the correlation of theory to experiment.
RESULTS AND DISCUSSION
This comparison is done here for five monomeric proteins [barnase (37), CI2 (20, 38), CheY (39), the src SH3 domain (40), and the α-spectrin SH3 domain (41)] where experimental data on the folding nuclei have been obtained for many residues. The 3D coordinates of their native structures have been taken from PDB (42) files 1rnb.ent, 2ci2.ent, 3chy.ent, 1srm.ent, and 1shg.ent, respectively.
The calculations of the unfolding pathways have been done with the above described dynamic programming method and the simple free-energy estimates. Although the presented approach can investigate various folding and unfolding conditions, all the given results below refer to the point of thermodynamic equilibrium between the native and the coil states of the protein where one can expect the simplest kinetic behavior (see refs. 14, 29).
For each of the studied proteins, we see only one transition microstate of the minimal free energy (thus, the ensemble {S#min} consists of only one microstate for these proteins). However, the whole ensemble {S#} of all the transition microstates includes thousands of them, the free energies of tens of these microstates exceeding the minimum by ≈RT only, in spite of significant variations in sizes and positions of the globular parts of these microstates. The statistical properties of the {S#} ensembles are described by Φ values; see Eq. 8.
The level of correlation between theory and experiment is demonstrated in Fig. 3 and Table 1.
Figure 3.

Unfolding nuclei: correlation of theoretical and experimental results for CI2 (a), barnase (b), CheY (c), the src SH3 domain (d), and the α-spectrin SH3 domain (e). Hatched rectangles at the top of each plot show how the nucleus of the minimal free energy is located in the protein chain according to the calculations. The experimental Φf factors are shown with open circles (connected by dotted line for better presentation). The Φ factors calculated for the ensemble of the possible transition states are shown as a solid line with filled circles (the circles correspond to residues with experimentally determined Φf values). The experimental data do not include negative Φf values, because they have no clear structural interpretation (38); Φf values exceeding 1 are taken as 1; when various mutations give different Φf values, we take the highest ones. For barnase, we take Φf as 1-Φu (37) because its unfolding (u) at high denaturant concentration, as well as folding/unfolding at moderate concentration, is a two-state process, whereas its folding in water proceeds via a metastable intermediate (37). The rectangles and lines (at the bottom of each plot) show the native positions of the α-helices and the β-strands in the chain.
Table 1.
Coefficient of correlation between theoretical and experimental Φ values
| Protein | Computed transition state with lowest free energy | Computed ensemble of all transition states | No. of points (Fig. 3) used for Φexp/Φteor comparison |
|---|---|---|---|
| C12 | 0.50 | 0.56 | 37 |
| Barnase | 0.19 | 0.54 | 29 |
| CheY | 0.59 | 0.50 | 17 |
| Ser SH3 | 0.00 | −0.02 | 14 |
| α-sp.SH3 | 0.54 | 0.39 | 6 |
| Average | 0.36 | 0.46 | 21.2 |
Fig. 3a shows the results for CI2 protein. It demonstrates a definite correlation between the chain fragments forming the globular native-like part of the calculated single lowest-free-energy transition state (rectangles) and the regions of high experimental Φf values. The correlation coefficient is 0.50. The correlation is even better when the Φ values are computed from the ensemble {S#} of the possible transition states (see the plot). Now the correlation coefficient is 0.56 (Table 1). These results mean that the theory catches some of the main peculiarities of the protein folding nuclei.
Like the experiment, the theory shows high Φ values for the N-terminal α-helix and the C-terminal β-hairpin of CI2 (this means they are usually involved in the nucleus), but the peaks for theoretical Φs are broader than those for the experimental Φs. This is probably due, at least partially, to the now neglected specificity of atomic contacts and to the rough estimate of loop entropy in our calculations. In principle, these broad computed peaks could also be caused by the limited number of unfolded loops currently allowed in our calculations. However, the calculations done separately by another (“branch-and-bound”) method (which is free of this limitation but which can search for the lowest-free-energy transition microstates rather than for whole ensembles of them) shows that this is not the case: we very rarely see more than two closed loops in the lowest-free-energy transition microstates computed for many proteins (A. V. Skoogarev and A.V.F., unpublished data).
Of course, the found correlation is far from perfect. In principle, this could be attributed to the inherent limitation of the dynamic programming-based approach. Namely, we consider only the folding/unfolding pathways like those shown in Figs. 1 and 2, where a link detached from the native globule is not allowed to return after the next link has been detached. However, the calculations done by a modification of the “branch-and-bound” method (very slow, but free of this limitation) show that at least the transition microstate free energy does not depend on the above-mentioned limitation (A. V. Skoogarev and A.V.F., unpublished data). Besides, although shuttling back and forth orthogonally to the main reaction coordinate ν can affect the transition state (43), the Φ values computed from direct solution of the kinetic equations describing the folding/unfolding network (D. N. Ivankov and A.V.F., unpublished results) are rather close to the Φ values computed here from the ensemble of transition microstates.
The pictures observed for the other investigated proteins are usually similar to those observed here for CI2 [see Fig. 3 b–e and Table 1; see also the recent works of Wolynes and collaborators (20, 24, 27), where the calculation of the transition state and of the Φ values for the CI2 protein and for the λ-repressor are based on quite another technique]. The only exception is src SH3, where the correlation between our theory and experiment is absent. Usually the correlation between the observed and predicted Φ values is about 50% for the Boltzmann ensembles of the transition states, and the correlation is 10% worse for the single-transition states of the minimal free energy.
It should also be mentioned that (according to computations of A. V. Skoogarev, O.V.G. and A.V.F.; unpublished data) the experimental Φs of the amino acid residues in proteins show a very low correlation (≈0.15) with the number of atom–atom or residue–residue contacts; and the correlation of the experimental Φs with inclusion of residues in the secondary structure is also well below 0.2. Thus, these trivial factors cannot account for the theory-to-experiment correlation found in this work by examination of the unfolding pathways.
CONCLUSIONS
An overview of the transition state ensembles calculated for each of the proteins shows that many of the transition states, though of nearly equal free energy, have substantial variations in size and position (see the differences in the calculated Φ plots and the calculated lowest-free-energy nucleus positions in Fig. 3). Our result correlates with the suggestion that a 3D protein structure can fold by using various folding nuclei (7, 8, 12, 27, 41, 44–46). It should be mentioned also that, on the average, the transition state structures found by us are comparatively large: they usually include from 1/3 to 1/2 and sometimes even up to 3/4 of all the chain residues (see Fig. 3). It is noteworthy that the experiment also suggests rather large globular parts of the transition states (3).
Theoretically (14, 29), a large and not-too-specific critical nucleus must be typical of the folding taking place under conditions close to those of thermodynamic equilibrium between the native globule and the coil. These conditions have been used in this work. Klimov and Thirumalai did their simulations (46) under similar conditions. Shakhnovich and colleagues worked at the temperature of fastest folding (6, 16–18), and this temperature was well below the temperature of thermodynamic midtransition. Here, theoretically (14, 29), the critical nuclei must be smaller and must have smaller variations in size. Thus, a discrepancy highlighted in refs. 9 and 10 can be at least partly caused by the difference in the folding conditions used by different authors. “Wet” experiments also show visible modifications of the transition state size and properties with the change in experimental conditions (47).
The semifolded microstates have high free energies in the examined proteins. This is consistent with the two-state all-or-none transition between the native and the unfolded states. Most of these semifolded microstates have a very high free energy of many tens or even hundreds of RT units. However, our calculations find the passages through this high free-energy landscape where the free energy exceeds that of the native and coil states (for the examined proteins) by only 14–21 RT; this is consistent with the estimates obtained in refs. 14 and 29 for proteins of this size (60–120 residues). Such relatively low free-energy barriers allow these protein to fold within milliseconds or seconds, which is in reasonable, though only semiquantitative, concordance with the experiment (3, 37–41). The computed transition states look like those compact and closely packed parts of semifolded native 3D structures that contain the minimal number of protruding loops.
Acknowledgments
We are grateful to O. B. Ptitsyn for discussions and to A. V. Skoogarev and D. N. Ivankov for assistance. This work was supported by the Russian Foundation for Basic Research and by an International Research Scholar’s Award to A.V.F. from the Howard Hughes Medical Institute.
ABBREVIATION
- 3D
three-dimensional
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Fersht A R. Curr Opin Struct Biol. 1995;5:79–84. doi: 10.1016/0959-440x(95)80012-p. [DOI] [PubMed] [Google Scholar]
- 2.Fersht A R. Curr Opin Struct Biol. 1997;7:3–9. doi: 10.1016/s0959-440x(97)80002-4. [DOI] [PubMed] [Google Scholar]
- 3.Jackson S E. Fold Des (London) 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 4.Dobson C M, Karplus M. Curr Opin Struct Biol. 1999;9:92–101. doi: 10.1016/s0959-440x(99)80012-8. [DOI] [PubMed] [Google Scholar]
- 5.Matouscheck A, Kellis J T, Jr, Serrano L, Bycroft M, Fersht A R. Nature (London) 1990;346:440–445. doi: 10.1038/346440a0. [DOI] [PubMed] [Google Scholar]
- 6.Abkevich V I, Gutin A M, Shakhnovich E I. Biochemistry. 1994;33:10026–10036. doi: 10.1021/bi00199a029. [DOI] [PubMed] [Google Scholar]
- 7.Šali A, Shakhnovich E, Karplus M. Nature (London) 1994;369:248–251. doi: 10.1038/369248a0. [DOI] [PubMed] [Google Scholar]
- 8.Onuchic J N, Socci N D, Luthey-Schulten Z, Wolynes P G. Fold Des (London) 1996;1:441–450. doi: 10.1016/S1359-0278(96)00060-0. [DOI] [PubMed] [Google Scholar]
- 9.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 10.Shakhnovich E. Fold Des (London) 1998;3:R108–R111. doi: 10.1016/s1359-0278(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 11.Thirumalai D, Klimov D K. Fold Des (London) 1998;3:R112–R118. doi: 10.1016/S1359-0278(98)00057-1. [DOI] [PubMed] [Google Scholar]
- 12.Pande V S, Rocksar D S. Proc Natl Acad Sci USA. 1999;96:1273–1278. doi: 10.1073/pnas.96.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bryngelson J D, Wolynes P G. Biopolymers. 1990;30:177–188. [Google Scholar]
- 14.Finkelstein A V, Badretdinov A Ya. Fold Des (London) 1997;2:115–121. doi: 10.1016/s1359-0278(97)00016-3. [DOI] [PubMed] [Google Scholar]
- 15.Wolynes P G. Proc Natl Acad Sci USA. 1997;94:6170–6175. doi: 10.1073/pnas.94.12.6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shakhnovich E, Abkevich V, Ptitsyn O. Nature (London) 1996;379:96–98. doi: 10.1038/379096a0. [DOI] [PubMed] [Google Scholar]
- 17.Michnick S W, Shakhnovich E. Fold Des (London) 1998;3:239–251. doi: 10.1016/S1359-0278(98)00035-2. [DOI] [PubMed] [Google Scholar]
- 18.Mirny L A, Abkevich V I, Shakhnovich E I. Proc Natl Acad Sci USA. 1998;95:4976–4981. doi: 10.1073/pnas.95.9.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li A, Daggett V. J Mol Biol. 1996;257:412–429. doi: 10.1006/jmbi.1996.0172. [DOI] [PubMed] [Google Scholar]
- 20.Daggett V, Li A, Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1996;257:430–440. doi: 10.1006/jmbi.1996.0173. [DOI] [PubMed] [Google Scholar]
- 21.Caflisch A, Karplus M. J Mol Biol. 1995;252:672–708. doi: 10.1006/jmbi.1995.0528. [DOI] [PubMed] [Google Scholar]
- 22.Brooks C L, III, Gruebele M, Onuchic J N, Wolynes P G. Proc Natl Acad Sci USA. 1998;95:11037–11038. doi: 10.1073/pnas.95.19.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finkelstein A V. Protein Eng. 1997;10:843–845. doi: 10.1093/protein/10.8.843. [DOI] [PubMed] [Google Scholar]
- 24.Shoemaker B A, Wang J, Wolynes P G. Proc Natl Acad Sci USA. 1997;94:777–782. doi: 10.1073/pnas.94.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Portman J J, Takada S, Wolynes P G. Phys Rev Lett. 1998;81:5237–5240. [Google Scholar]
- 26.Shoemaker B A, Wolynes P G. J Mol Biol. 1999;287:657–674. doi: 10.1006/jmbi.1999.2612. [DOI] [PubMed] [Google Scholar]
- 27.Shoemaker B A, Wang J, Wolynes P G. J Mol Biol. 1999;287:675–694. doi: 10.1006/jmbi.1999.2613. [DOI] [PubMed] [Google Scholar]
- 28.Privalov P L. Adv Protein Chem. 1979;33:167–241. doi: 10.1016/s0065-3233(08)60460-x. [DOI] [PubMed] [Google Scholar]
- 29.Finkelstein A V, Badretdinov A Ya. Mol Biol (Engl Transl) 1997;31:391–398. [Google Scholar]
- 30.Ueda Y, Taketomi H, Gō N. Int J Pept Protein Res. 1975;7:445–459. [PubMed] [Google Scholar]
- 31.Flory P J. Statistical Mechanics of Chain Molecules. New York: Interscience; 1969. [Google Scholar]
- 32.Leopold P E, Montal M, Onuchic J N. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aho A, Hopcroft J, Ullman J. The Design and Analysis of Computer Algorithms. Reading, MA: Addison–Wesley; 1976. [Google Scholar]
- 34.Finkelstein A V, Roytberg M A. BioSystems. 1993;30:1–19. doi: 10.1016/0303-2647(93)90058-k. [DOI] [PubMed] [Google Scholar]
- 35.Moore J W, Pearson R G. Kinetics and Mechanism. New York: Wiley; 1981. [Google Scholar]
- 36.Fersht A R, Matouschek A, Serrano L. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- 37.Serrano L, Matouschek A, Fersht A R. J Mol Biol. 1992;224:805–818. doi: 10.1016/0022-2836(92)90563-y. [DOI] [PubMed] [Google Scholar]
- 38.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 39.Lopez-Hernandez E, Serrano L. Fold Des (London) 1996;1:43–55. [PubMed] [Google Scholar]
- 40.Grantcharova V P, Riddle D S, Santiago J V, Baker D. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 41.Viguera A R, Serrano L, Wilmanns M. Nat Struct Biol. 1996;3:874–880. doi: 10.1038/nsb1096-874. [DOI] [PubMed] [Google Scholar]
- 42.Bernstein F C, Koetzle T F, Williams G J B, Meyer E F, Brice M D, Rogers J R, Kennard O, Shimanouchi T, Tasumi M. Eur J Biochem. 1977;80:319–324. doi: 10.1111/j.1432-1033.1977.tb11885.x. [DOI] [PubMed] [Google Scholar]
- 43.Plotkin S S, Wang J, Wolynes P G. J Chem Phys. 1997;106:2932–2948. [Google Scholar]
- 44.Socci N D, Onuchic J N, Wolynes P G. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 45.Martinez J C, Pisabarro M T, Serrano L. Nat Struct Biol. 1998;5:721–729. doi: 10.1038/1418. [DOI] [PubMed] [Google Scholar]
- 46.Klimov D K, Thirumalai D. J Mol Biol. 1998;282:471–492. doi: 10.1006/jmbi.1998.1997. [DOI] [PubMed] [Google Scholar]
- 47.Oliveberg M. Acc Chem Res. 1998;31:765–772. [Google Scholar]