

Abstract
The evolutionary implications of transposable element (TE) influences on gene regulation are explored here. An historical perspective is presented to underscore the importance of TE influences on gene regulation with respect to both the discovery of TEs and the early conceptualization of their potential impact on host genome evolution. Evidence that points to a role for TEs in host gene regulation is reviewed, and comparisons between genome sequences are used to demonstrate the fact that TEs are particularly lineage-specific components of their host genomes. Consistent with these two properties of TEs, regulatory effects and evolutionary specificity, human-mouse genome wide sequence comparisons reveal that the regulatory sequences that are contributed by TEs are exceptionally lineage specific. This suggests a particular mechanism by which TEs may drive the diversification of gene regulation between evolutionary lineages.
Historical perspective
Controlling elements
The influence of transposable elements (TEs) on gene regulation has been apparent for as long as these genetic elements have been known to exist. In fact, the discovery of TEs was predicated upon their ability to regulate the expression patterns of the genes of the host organisms in which they reside. Barbara McClintock (1984) originally referred to the mobile genetic elements that she discovered in maize as “controlling elements” based on their ability to control the expression of genes involved in pigmentation. Beginning in 1944, McClintock observed many cases of variegation, in other words differences in the pattern of expression, for the distribution of chlorophyll among maize seedling leaves. Importantly, McClintock noticed that distinct chlorophyll patterns were localized to discrete sectors and that these sectors occurred in adjacent pairs where each member of the pair was the reciprocal of the other with respect to their pigmentation. Similar observations were made for patterns of gain and loss of genetic markers on maize kernels (Fig. 1A). These observations led her to conclude that differences in the regulation of genes involved in pigmentation between sectors were due to an event that occurred at mitosis where one daughter cell gained some genetic element and the other daughter cell lost it (McClintock, 1946). Shortly thereafter she was able to demonstrate that these controlling elements could move from one location in the genome to another and thus conclusively postulated the existence of TEs (McClintock, 1948).
Fig. 1.
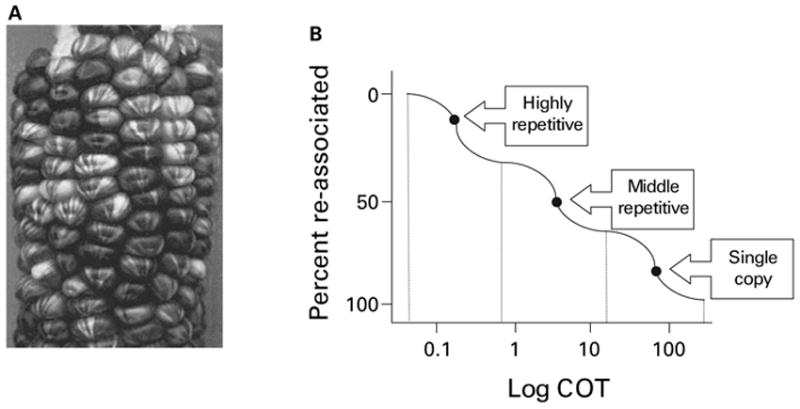
Historically relevant implications of TEs for gene regulation. (A) Variegated pigmentation of maize kernels resulting from TE activity. (B) COT curve showing the dynamics of DNA reassociation used to infer the presence of repetitive DNA in eukaryotic genomes. Repetitive DNA reassociates more rapidly than single copy DNA.
Despite McClintock’s standing as a highly respected geneticist and the volume of evidence that she presented, the implications of these findings were not widely appreciated or even accepted until much later. In her recollections of this period, McClintock has attributed the initial reluctance of the scientific community to embrace her conclusions to two aspects of her discovery, both of which were particularly difficult to reconcile with the understanding of genetics that existed at that time (McClintock, 1987). First and foremost, the notion of mobile genetic elements implied a dynamic genome that was radically at odds with the prevailing notion of a static genome based on the “beads on a string” model of chromosomal organization. Somewhat less obviously, at that time even the basic concept that the expression of genes was developmentally regulated was generally not conceived of and would not become widely appreciated until more than a decade later with the publication of the classic work of Jacob and Monod (1961). Of course, the significance of McClintock’s work would come to be fully appreciated in time, and a reflection on the path to her discovery may be taken colloquially to suggest that the very essence of TEs is tied to their ability to influence patterns of host gene regulation.
COT curves
Another critical early line of research that underscored the potential influence of repetitive DNA on gene regulation was founded on studies of the kinetics of DNA reassociation pioneered by Roy Britten and colleagues (Britten and Kohne, 1968). In short, they observed that the rate of DNA reassociation for relatively large eukaryotic genomes was much more rapid than would be expected if all or even most of the genomic DNA was single copy. Reassociation kinetics were visualized on so-called COT curves where the fraction of reassociated DNA was plotted against COT, a parameter that is equal to the product of the DNA concentration in the solution times the time of incubation (moles of DNA times seconds per liter). Careful examinations of these plots revealed distinct fractions of genomic DNA that reassociate at different rates, and these fractions were inferred to represent different classes of genomic DNA consisting of more (relatively rapidly reassociating) or less (more slowly reassociating) repetitive DNA (Fig. 1B). Mammalian genomes, like those of the mouse and human, were estimated to contain as much as 20–35% repetitive DNA. While these figures are now known to be underestimates (Lander et al., 2001; Waterston et al., 2002), at the time they represented a far greater fraction of repetitive DNA than had previously been imagined.
The significance of this experimental work was of course the novel demonstration of the prevalence of repetitive DNA in eukaryotic genomes. Fortunately however, Britten and colleagues did not stop there. They considered the preponderance of repetitive DNA with respect to their interest in both evolutionary theory and gene regulation and hypothesized at length on the significance of repetitive DNA to the evolution of regulatory differences. In fact, their theoretical work of that era represented one of the strongest assertions to date of the importance of regulatory changes driving evolutionary diversification. Britten and Davidson (1969) articulated a detailed model on the genomic architecture of regulatory networks and suggested that repetitive DNA may influence gene expression patterns by providing binding sites for regulatory factors in the 5′ regions of genes. Further elaboration of this model placed even more of an emphasis on the role of repetitive DNA in gene regulation and demonstrated how repetitive sequences could move in the genome and serve as source of evolutionary variation in regulatory patterns. In their model, repetitive sequences were considered to move via chromosomal rearrangement and not transposition per se (Britten and Davidson, 1971). Of course, the precise nature of repetitive DNA was unknown at the time as was the preponderance of TE sequences among this fraction of genomic DNA. However, the predictions of Britten and colleagues were subsequently born out in a number of cases where TEs were demonstrated to alter expression patterns by providing cis-regulatory sequences after insertion into the vicinity of a host gene (Britten, 1996a).
Examples of TE influences on gene regulation
Molecular evidence
Over the last 15 years, an abundance of experimental evidence has accumulated that directly points to the contribution of repetitive DNA to gene regulation. This evidence consists largely of examples where TEs have been shown to contribute to the regulation of a host gene by providing cis-regulatory sequences that interact with host trans factors. Interestingly, the vast majority of these cases were uncovered fortuitously in the sense that the investigators were not out to assess the role of TEs in gene regulation but rather were seeking to understand the molecular basis of the regulatory properties of the particular system that they were working on. The first example of this kind came from the study of the sex-limited protein (Slp) encoding gene in mouse (Stavenhagen and Robins, 1988). Slp is one of two tandem genes and is closely related to the adjacent C4 gene that encodes the fourth component of complement. Apparently, after the duplication of these two genes an endogenous retrovirus (ERV) inserted upstream of the Slp gene and this insertion resulted in an altered expression pattern for Slp which in turn drove the functional divergence of the protein (van den Berg et al., 1992). Unlike the C4 gene, Slp is expressed only in males due to androgen dependence conferred by androgen response elements found in the long terminal repeat of the ERV (Adler et al., 1992, 1993).
Pursuant to his interest in the relationship between repetitive DNA and gene regulation, Britten reviewed a number of such cases where insertions of TEs have resulted in fixed novel regulatory patterns and established four criteria for the identification of convincing examples: 1 – the presence of a known TE sequence in the gene region, 2 – evidence that the insertion has been present long enough to be fixed, 3 – evidence that part of the TE sequence participates in the regulation of the nearby gene and 4 – evidence that the gene encodes some function (Britten, 1996a, b; 1997). By 1997, Britten was able to find more than 20 examples that conformed to all four of these criteria and many more similar examples have been uncovered since that time. For instance, a number of cases where human TEs can be shown to serve as promoters for adjacent genes have recently been identified (Landry et al., 2001, 2002; Medstrand et al., 2001; Dunn et al., 2003). The most extensive literature survey to date of TE contributions to host gene regulation identified almost 80 cases where regulatory elements of vertebrate genes are derived from TEs (Brosius, 1999).
In addition to serving as promoter and enhancer sequences for nearby genes, TE insertions have also been shown to influence host gene expression by providing alternative splice sites (Varagona et al., 1992; Feuchter-Murthy et al., 1993; Baban et al., 1996; Davis et al., 1998) and polyadenylation sites (Goodchild et al., 1992; Sugiura et al., 1992; Mager et al., 1999). Alu elements may be particularly prone to providing alternative splice sites to host genes and being incorporated into mRNA sequences as a result (Makalowski et al., 1994; Sorek et al., 2002; Lev-Maor et al., 2003).
Genomic evidence
The accumulation of genomic sequence data has led to a number of efforts to systematically assess the contribution of TEs to gene regulation. These studies have consisted of computer-based inquiries that rely on large scale analyses of sequence data. The earliest examples of these types of studies were conducted on plant genome sequences; investigators interested in the relationship between TEs and plant genes took the novel approach of computationally searching plant gene sequences for the presence of TEs. An initial survey of maize and barley gene sequences revealed that quite a few members of one specific TE family – Tourist, a miniature inverted repeat element (MITE) – were inserted in the regions just flanking genes or in intron sequences (Bureau and Wessler, 1992). This observation suggested that these elements may be often associated with genes, and this was confirmed with more extensive analyses that revealed the frequent association of Tourist elements with genes from a number of different cereal grass genomes (Bureau and Wessler, 1994a; Bureau et al., 1996). Evidence that these TE-gene associations may include functionally significant cases was supplied by studies that revealed that MITEs had contributed regulatory sequences such as cis-binding sites and polyadenylation signals to host genes (Bureau and Wessler, 1994b; Wessler et al., 1995).
The recent availability of complete eukaryotic genome sequences provided increased opportunities to systematically evaluate the contribution of TEs to host gene regulatory sequences. For example, the majority of retrotransposons discovered in the complete genome sequence of Caenorhabditis elegans were found to be located in close proximity to host gene sequences suggesting that they may contribute to the regulation of these genes (Ganko et al., 2003). In addition, a survey of the retrotransposons of the fission yeast Schizosaccharomyces pombe revealed that these elements were disproportionately associated with pol II promoters in complete genome sequence (Bowen et al., 2003).
TE sequences make up a much greater fraction of vertebrate genomes and studies of the human genome in particular have underscored the substantial contribution of TEs to regulatory sequences. For example, the initial analysis of the human genome sequence revealed that hundreds of transcriptional terminator sequences were donated by one class of retrotransposon alone (Lander et al., 2001). Subsequently more detailed analyses revealed the extent to which human regulatory sequences are derived from TEs. For instance, a survey of human genome sequences found that almost 25% of proximal promoter sequences (i.e. 500 bp upstream of the transcription start site) as well as numerous 5′ and 3′ untranslated regions (UTRs) contained TE derived sequences (Jordan et al., 2003). Clearly, as was the case with the plant sequences studied earlier, there is a strong association between TE sequences and gene sequences in the human genome. However, this fact alone does not necessarily imply functionally relevant relationships where TEs provide working regulatory sequences to host genes; the association of TEs with promoters may simply be due to the prevalence of TE-derived sequences in the human genome at large. To address this issue, experimentally characterized human regulatory sequences were mapped to their gene sequences to examine whether they may have been donated by TEs. When experimentally characterized regulatory sequences were evaluated, it was shown that TEs have donated sequences to both cis-binding regulatory elements that act in a gene-specific manner as well as scaffold/matrix attachment regions (S/MARs) and locus control regions that exert their regulatory effects in a more global manner (Jordan et al., 2003). A subsequent genomic scale analysis of human and mouse sequences confirmed the abundance of TE-derived sequences in regulatory regions and the donation of experimentally characterized regulatory elements by TEs (van de Lagemaat et al., 2003). Interestingly, this study also found that TEs were found more often in the regulatory regions of genes that are rapidly evolving and those with relatively narrow phylogenetic distributions (i.e. those that are mammalian specific). For example, genes involved in immune suppression and those involved in the response to external stimuli were particularly enriched for TE sequences. These observations were taken to suggest that TEs may have contributed substantially to the evolutionary diversification of mammalian genomes presumably by generating lineage-specific patterns of gene regulation.
Lineage-specific regulatory sequences contributed by TEs
TEs are lineage-specific
TEs may be the most lineage-specific elements of eukaryotic genomes. For instance, a recent comparison of a single 12-megabase (Mb) genomic region among 12 vertebrate species indicated that the distribution of different TE types differed greatly within and between vertebrate lineages (Thomas et al., 2003). Among the nine mammalian species examined in this study, species-specific TE insertions account for the majority of size differences seen between lineages. In addition, when the complete mouse genome sequence was compared to that of the human, it was shown that mouse-specific TEs made up 87.0% of all mouse TEs (32.4% of the mouse genome) and human-specific TEs accounted for 51.9% of all human TEs (24.4% of the human genome) (Waterston et al., 2002). In other words, mouse lineage-specific TEs have contributed well over 800 Mb of DNA to the mouse genome and human lineage-specific TEs make up over 700 Mb of the human genome. On the other hand, the same comparison revealed that only 1% of mouse protein coding genes do not have any human homolog and only 20% of mouse genes do not have a direct 1:1 human ortholog (i.e. are not descended from precisely the same ancestral gene). TEs are clearly far more lineage-specific than the host genes of these two mammals. Even more remarkably, TE insertions can generate substantial genomic fractions over much shorter periods of evolutionary time than have elapsed since the human-mouse divergence. Comparison of several primate genomic sequences suggests that transposition rates vary widely across lineages and that the human lineage has experienced a particularly high rate of retrotransposition (Liu et al., 2003). This has led to a TE generated expansion of over 500 Mb in the human lineage over the last 50 million years and an increase of 30 Mb in the human lineage just since the divergence from chimpanzee ~6 million years ago.
When these findings are considered with respect to the influence of TEs on gene regulation, it suggests that TEs may exert regulatory effects in a way that is most likely to cause differences between evolutionary lineages. The implications for this aspect of TE influence on gene regulation with respect to methods used to identify regulatory sequences are explored below. Also, in support of the notion that TEs may contribute to lineage-specific regulatory differences, data on the lineage-specific contributions that TEs make to human regulatory sequences are presented.
Phylogenetic footprinting may overlook TEs
Recently, a sustained effort based on the comparative analysis of genomic sequence data has been made to improve methods for the prediction of cis-regulatory sites in genomic DNA. This approach is known as “phylogenetic footprinting” and it rests on the plausible assumption that functionally important regions of genomic DNA will evolve more slowly than non-important regions due to the effects of purifying selection (Gumucio et al., 1992; Zhang and Gerstein, 2003). From this it follows that when non-coding sequences are compared between species, functionally important regulatory sequences (e.g. cis-binding sites) will be characterized by anomalously low levels of sequence divergence. This method has been employed to identify putative regulatory sequences in a number of different systems (McCue et al., 2002; Boffelli et al., 2003; Kellis et al., 2003; Lenhard et al., 2003).
It is worth noting that, at this time, relatively little is known about the pattern of non-coding sequence evolution. The notion that functionally important regulatory sites will be more conserved than neighboring nonfunctional sequences is entirely reasonable and consistent with what is known about molecular evolution (Li, 1997), but it is still mostly an assumption. Only recently have investigators begun to study the pattern of noncoding sequence evolution with respect to the location of known regulatory sites and the results do not entirely support the phylogenetic footprinting rationale. There is evidence that the rate of evolution for noncoding DNA at regulatory sites is lower than the rate of evolution for the surrounding, presumably nonfunctional, noncoding sequence (Dermitzakis and Clark, 2002; Moses et al., 2003). However, several recent studies indicate that there is a rapid evolutionary turnover of regulatory sites, which suggests that the phylogenetic footprinting approach may yield numerous false negatives. For example, when Drosophila pseudoobscura genomic sequences were compared to D. virilis sequences, only 50% of the known regulatory regions were found to be located in sequences that are conserved between the two species (Alkema and Wasserman, 2003). A comparative analysis of genomic sequence from 12 vertebrates fared only slightly better with respect to the identification of functionally validated regulatory elements; in this case, 63% of the known regulatory elements were shown to be located in conserved sequence regions (Thomas et al., 2003). Another analysis of regulatory sequence evolution compared experimentally characterized transcription factor binding sites between human and rodent genomes and found extensive sequence variation at these sites (Dermitzakis and Clark, 2002). Based on this survey, 32–40% of human functional sites were estimated to be nonfunctional in rodents. It appears that the assumption that functionally important regulatory sites will be highly conserved is not always true, and one can expect that phylogenetic footprinting will yield numerous false negatives as a result.
An approach that employs the same rationale as that of phylogenetic footprinting has recently been used to evaluate the potential contribution of TEs to functionally important non-coding sequences in mammalian genomes (Silva et al., 2003). In this study, orthologous intergenic regions were compared between the human and mouse genomes. Consistent with the fact that they are very lineage specific, TEs were shown to make up 40–60% of the regions with low similarity between species and only 20% of the regions of high similarity. However, certain families of elements, namely MIR a family of DNA-type elements and L2 a family of LINE-like elements, were found to be common within the conserved segments. From this observation, it was inferred that these ancient conserved TE sequences have been under purifying selection based on some functional utility that they provide to their hosts. Remarkably, the recruitment of these TEs to perform some function that benefits their hosts was shown to be quite common having occurred two times or more for each host gene examined.
As demonstrated by the study of Silva et al. (2003), TEs clearly make up an important component of functionally important noncoding DNA. However, the problem of false positives discussed above with respect to phylogenetic footprinting would seem to be even more exacerbated for TEs. Because TEs are so lineage specific, they should be expected to rarely show up as conserved regions in sequence alignments between species. Below, cross-species comparisons of experimentally characterized regulatory sites are shown to suggest that TE-contributed regulatory sequences are far more lineage specific and much less conserved than regulatory sites that are not derived from TEs. Thus, TEs appear to be particularly likely to contribute to the generation of lineage-specific regulatory elements and as such may play a role in driving the diversification between evolutionary lineages.
Evidence of TE contributions to lineage-specific regulatory sequences
One way to assess the contributions of TEs to regulatory sequences is to search for their presence among promoter sequence regions proximal to host genes. Indeed, a number of studies have inferred a possible role for TEs in gene regulation based on their proximity to host genes (Bureau and Wessler, 1992, 1994b; Bureau et al., 1996; Ganko et al., 2003; Jordan et al., 2003; van de Lagemaat et al., 2003). To this end, we have surveyed the proximal promoter sequences of 4,737 human transcripts for the presence of TE sequences as well as for low complexity repetitive sequences. Full-length human transcript sequences were taken from the database of transcriptional start sites (Suzuki et al., 2002) and proximal promoter regions from the transcripts that mapped unambiguously to the human genome sequence (National Center for Biotechnology, build 33, ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/) were used for analysis. Each of these proximal promoter sequences consists of nucleotides from −2,000 bp to +1,000 bp with respect to the transcriptional start site. Promoter sequences were analyzed with the RepeatMasker program (http://ftp.genome.washington.edu/RM/RepeatMasker.html) to determine the location of TEs and low complexity repetitive sequences (Fig. 2). Consistent with previous observations, there are numerous TE-derived sequences in the proximal promoter regions of human genes. Low complexity sequences are also present but they are not nearly as abundant as TE sequences.
Fig. 2.
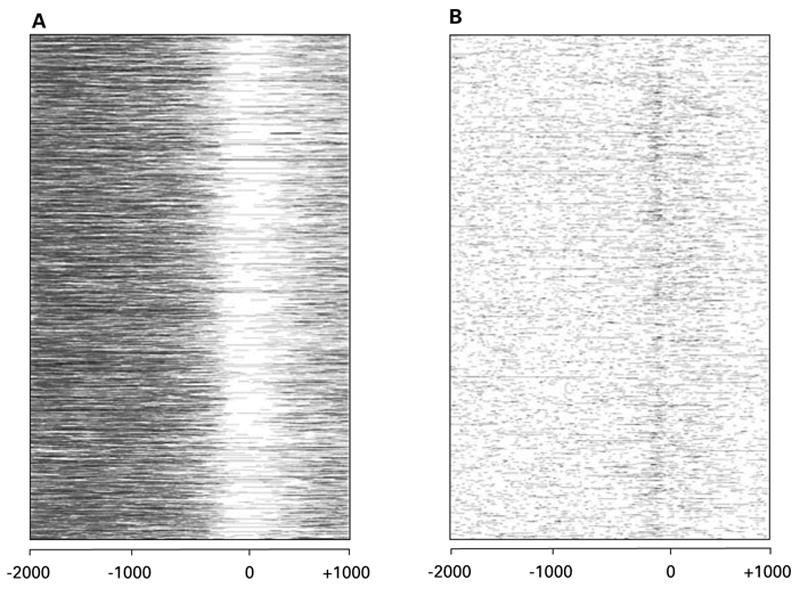
Density of repetitive DNA in human genome promoter regions. 4,737 human promoter sequences, beginning at position −2,000 bp and ending at position +1,000 bp with respect to the transcriptional start sites, were scanned for the presence of TE derived sequences (A) and low complexity repetitive sequences (B). In each promoter sequence, residues that overlap with TE (A) and low complexity sequences are colored black (B).
One problem with the approach described above is that it is difficult if not impossible to definitively claim a role for TEs in host gene regulation based simply on their presence in sequence regions that are involved in regulation. In the case of the human genome for instance, the presence of TEs in host gene regulatory regions may simply reflect their abundance in the genome. Indeed, the pattern of TE insertions in the human promoter regions is consistent with this possibility, and even suggests that most TE insertions in promoter regions are actually deleterious and selected against. This is because the density of TE sequences in human promoters is greatest in the most distal regions and steadily declines closer to the start site of transcription (Fig. 2A). TE density is lowest just adjacent to the transcription start site and increases slightly after the start sites in the 5′ UTRs. Interestingly, the density of low-complexity DNA in human promoter regions shows virtually the opposite pattern (Fig. 2B). The density of low-complexity DNA is fairly uniformly low in distal promoter regions and 5′ UTRs but increases markedly in the core promoter regions that contain the transcription start sites. This may reflect the prevalence of certain transcription factor binding sites that have low complexity recognition sequences. For example, Sp1 sites are particularly prevalent around transcription start sites and are often found in tandem arrays of multiple sites. The Sp1 recognition sequence is GC rich, and a tandem array of such sites would certainly result in a low complexity sequence region. In addition, core promoter sequences where the transcriptional start sites are located are known to be enriched for CpG islands and this too is probably reflected in the abundance of low complexity sequences detected in this region. The prevalence of low complexity sequences in core promoter regions suggests that error-prone mechanisms such as DNA replication may play an important role in generating regulatory sequence variation.
One way to make definitive inferences about the contribution of TEs to regulatory sequences is to start with experimentally characterized sites that are known to contribute to the regulation of host genes and then search for cases where such sites can be shown to have been donated by TEs. This approach has been employed successfully to identify TE-derived cis-regulatory sequences as well as TE-derived S/MARs that regulate gene expression in a more global manner (Jordan et al., 2003; van de Lagemaat et al., 2003). We combine a similar approach here, employing the identification of experimentally characterized cis-regulatory sites that overlap with TE sequences, with human-mouse sequence comparisons to evaluate the level of evolutionary conservation of regulatory sites that have been derived from TEs.
The TRANSFAC database (Matys et al., 2003) was used to identify experimentally characterized human regulatory sequences. The data that were taken from TRANSFAC (professional version 7.1) are cis-binding sites that have been identified with a number of different experimental procedures including footprinting, gel-shift assays, promoter deletion experiments and mutagenesis. A total of 1,145 of these cis-regulatory sites were mapped to the complete human genome sequence (National Center for Biotechnology, build 33, ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/). The locations of the regulatory sites in the human genome sequence were compared to the location of TE sequences detected using the program Repeat-Masker (http://ftp.genome.washington.edu/RM/RepeatMasker.html). A total of 38 cases where experimentally characterized regulatory sites overlapped with TE-derived sequences were identified in this way (Table 1 and Fig. 3). Next, the locations of experimentally characterized regulatory sites mapped to the human genome were compared with the sequence alignments between orthologous aligned regions of the human and mouse genomes (Schwartz et al., 2003) found at the UCSC genome browser (Karolchik et al., 2003). The alignments used were made between the April 2003 assembly of the human genome (build 33) and the February 2003 assembly of the mouse genome (MGSCv4 or mm3, http://genome.cse.ucsc.edu/goldenPath/10april2003/vsMm3/). These alignments cover only ~40% of the human genome sequence, but almost 90% (1,026 out of 1,145) of the experimentally characterized regulatory sites mapped to the human genome can be found in the regions that align to the mouse genome. To a great extent, this may reflect the fact that the characterized regulatory sites are more conserved than most noncoding DNA, but it is also likely to be partially due to the fact that the mapped sites are located in the close proximity of genes that are well studied experimentally and such regions are generally evolutionarily conserved (i.e. it may be largely due to an experimental sampling bias). In any case, less than 25% (9 out of 38) of TE-derived cis-regulatory sites are found in the regions of the human genome that align to the mouse genome (Table 1). Thus, TE-derived sites are far less conserved than the non-TE-derived cis-regulatory sites analyzed (P = 8.2 × 10−5, Fisher’s exact test). In addition, all but two of the nine cis-regulatory sites that map to aligned human-mouse segments were donated by the relatively ancient MIR and L2 TE families (Table 1 and Fig. 3). Thus, ancient TE families are disproportionately represented among the conserved set of TE-donated regulatory sites (P = 2.1 × 10−4, Fisher’s exact test). This finding is consistent with the previous analysis that showed many MIR and L2 element sequences have been conserved to serve some function in the human and mouse genomes (Silva et al., 2003). However, the majority of TE-derived cis-regulatory sequences in the human genome come from the relatively younger Alu and L1 TE families (Table 1 and Fig. 3). None of these TE derived cis-regulatory sequences are conserved between the human and mouse genome, and this can be attributed to the fact that the TE insertions that generated the regulatory sequences occurred after the human and mouse evolutionary lineages diverged. This is one important route by which TEs may contribute to lineage-specific regulatory sequence divergence. However, there is also a case where an ancient TE insertion donated regulatory sequences but was only conserved in one evolutionary lineage. The long terminal repeat (LTR) of a mammalian apparent LTR retrotransposon (MaLR) has donated four cis-binding sites to the γA-globin gene enhancer (Fig. 3). Despite the fact that sequence comparison between the MaLR insertion at this locus and an MaLR consensus sequence indicates that this insertion occurred before human and mouse diverged from a common ancestor, this particular TE insertion and the regulatory sequences that it provides can be found only in the human genome (Jordan et al., 2003). Thus, lineage-specific regulatory sequences donated by TEs may also result from asymmetric use by the host of TE sequences, after their insertion, along different evolutionary lineages.
Table 1.
TE-derived cis-regulatory binding sites in the human genome
| Human genea | Acc. nob | Coord1c | Coord2c | Elementd | Coord1e | Coord2e | Conservationf | TRANSFAC-IDg | PMIDh |
|---|---|---|---|---|---|---|---|---|---|
| pS2 | NT_030188 | 536479 | 536500 | SINE/Alu | 536469 | 536755 | − | HS$PS2_02
HS$PS2_03 |
9166778 |
| CETP | NT_010498 | 5716332 | 5716347 | DNA/MER1 | 5716004 | 5716385 | − | HS$CETP_01 | 1429586 |
| CETP | NT_010498 | 5716175 | 5716198 | DNA/MER1 | 5716004 | 5716385 | − | HS$CETP_02 | 11331284
0683381 |
| LPL | NT_030737 | 3517296 | 3517320 | LINE/L1 | 3517023 | 3517312 | + | HS$LPL_01 | 1406652 |
| HFH-4 | NT_009237 | 38132217 | 38132229 | SINE/Alu | 38131899 | 38132229 | − | HS$PTHRO_03 | 9096351 |
| c-myb | NT_025741 | 39609496 | 39609501 | SINE/MIR | 39609412 | 39609581 | + | HS$CMYBI1T_01 | 10739671 |
| c-Ha-ras | NT_029289 | 2446412 | 2446429 | SINE/Alu | 2446156 | 2446467 | − | HS$RAS1_11 | 9415707 |
| HFH-8 | NT_010799 | 865284 | 865296 | SINE/Alu | 865272 | 865585 | − | HS$INOS_01 | 9486531 |
| hPTH | NT_009237 | 4921983 | 4922000 | LINE/L1 | 4920816 | 4921991 | − | HS$PTH_03 | 7961715
532588 939213 |
| hPTH | NT_009237 | 4922826 | 4922840 | SINE/Alu | 4922559 | 4922858 | − | HS$PTH_04 | 7961715
532588 939213 |
| cyclin A | NT_016354 | 47240654 | 47240673 | SINE/MIR | 47240564 | 47240708 | + | HS$CYCA_09 | 7843287 |
| MSH2 | NT_022184 | 26445211 | 26445222 | SINE/Alu | 26445051 | 26445477 | − | HS$MSH2_01 | 7761476 |
| IFN-B | NT_011512 | 20371677 | 20371682 | SINE/Alu | 20371435 | 20371742 | − | HS$IFNB_02 | 2475256
409321 850164 |
| A gamma globin | NT_028310 | 4028828 | 4028840 | LTR/MaLR | 4028565 | 4028901 | − | HS$GG_29 | 2259631 |
| A gamma globin | NT_028310 | 4028799 | 4028813 | LTR/MaLR | 4028565 | 4028901 | − | HS$GG_30 | 2259631 |
| A gamma globin | NT_028310 | 4028744 | 4028766 | LTR/MaLR | 4028565 | 4028901 | − | HS$GG_31 | 2259631 |
| A gamma globin | NT_028310 | 4028677 | 4028696 | LTR/MaLR | 4028565 | 4028901 | − | HS$GG_32 | 2259631 |
| CD8 alpha | NT_022184 | 65830044 | 65830074 | SINE/Alu | 65829796 | 65830106 | − | HS$CD8A_01 | 8413295 |
| CD8 alpha | NT_022184 | 65829957 | 65829985 | SINE/Alu | 65829796 | 65830106 | − | HS$CD8A_02 | 8413295 |
| CD8 alpha | NT_022184 | 65829823 | 65829851 | SINE/Alu | 65829796 | 65830106 | − | HS$CD8A_03 | 8413295 |
| cdc2 kinase | NT_008583 | 11088405 | 11088410 | SINE/Alu | 11088245 | 11088434 | − | HS$CDC2_01 | 7867724 |
| StAR | NT_008251 | 56686 | 56712 | SINE/MIR | 56623 | 56754 | ++ | HS$STAR_01 | 8703908 |
| beta globin | NT_028310 | 4009002 | 4009022 | LTR/ERVL | 4008990 | 4009043 | − | HS$BG_08 | 2587218 |
| beta globin | NT_028310 | 4007552 | 4007583 | LINE/L1 | 4007493 | 4007588 | − | HS$BG_26 | 7499351 |
| E-Cadherin | NT_010498 | 17490712 | 17490744 | SINE/Alu | 17490692 | 17490983 | − | HS$CDH1_01 | 11278651
543680 |
| GPIIb | NT_010748 | 1121030 | 1121042 | LINE/L2 | 1120863 | 1121056 | ++ | HS$GP2B_12 | 2026605 |
| GPIIb | NT_010748 | 1120906 | 1120926 | LINE/L2 | 1120863 | 1121056 | ++ | HS$GP2B_15, HS$GP2B_13 | 8408012
026605 |
| GPIIb | NT_010748 | 1121048 | 1121057 | LINE/L2 | 1120863 | 1121056 | ++ | HS$GP2B_14 | 8408012 |
| PLOD | NT_021937 | 2505360 | 2505379 | SINE/Alu | 2505092 | 2505394 | − | HS$PLOD1_02 | 11157981 |
| PLOD | NT_021937 | 2505724 | 2505743 | SINE/Alu | 2505580 | 2505864 | − | HS$PLOD1_03 | 11157981 |
| alpha fetoprotein | NT_006216 | 2805460 | 2805474 | DNA/AcHobo | 2805441 | 2805517 | − | HS$AFP_01 | 9204933
468995 |
| CD2 | NT_004754 | 1228369 | 1228379 | SINE/Alu | 1228321 | 1228620 | − | HS$CD2_05 | 2209539 |
| BAX | NT_011109 | 21725891 | 21725928 | SINE/Alu | 21725821 | 21725957 | − | HS$BAX_01
HS$BAX_02 |
11278953 |
| delta globin | NT_028310 | 4017070 | 4017077 | LINE/L1 | 4017076 | 4017508 | + | HS$DG_02 | 1717993 |
| fra-1 | NT_033903 | 10769015 | 10769034 | SINE/Alu | 10768926 | 10769145 | − | HS$FRA1_01 | 9990071 |
| alpha globin | NT_037887 | 168818 | 168827 | SINE/Alu | 168673 | 168930 | − | HS$AG_07 | 1642094 |
| PLTP | NT_011362 | 9594014 | 9594043 | SINE/MIR | 9593958 | 9594084 | ++ | HS$PLTP_01 | 11867625
0744760 0998425 |
| talin | NT_008413 | 35713354 | 35713375 | SINE/Alu | 35713102 | 35713400 | − | HS$TLN_01 | 11278651 |
Names of the human genes regulated by the TE-derived cis-binding sites. Gene name conventions follow the listed publications (see PMID).
GenBank accession numbers for the human genome contigs (build 33) where the TE-derived cis-binding sites are located.
Contig coordinates of the TE-derived cis-binding sites.
Class and family names of the TEs from which the cis-binding sites are derived.
Contig coordinates of the TEs from which the cis-binding site are derived.
Human-mouse conservation for the TE-derived cis-regulatory sequences (see text for description). ++ Means that the cis-element maps to a region of the human genome that aligns to the mouse genome and shows visible sequence similarity between human and mouse. + Means that the cis-element maps to a region of the human genome that aligns to the mouse genome but does not show visible sequence similarity between human and mouse. – Indicates no conservation between human and mouse for the cis-element (i.e. it is lineage-specific).
Identification numbers for the TRANSFAC (professional version 7.1) entries that contain the descriptions of the cis-binding sites.
PubMed identification numbers for the publications that describe the characterization of the cis-binding sites.
Fig. 3.

Sequence alignments that show the relationship of TE-derived sequences, host promoter sequences and experimentally characterized cis-binding sites. TE family consensus sequences are aligned with host genome sequences. Cis-binding sites are characterized for human sequences and their locations in the alignments are boxed. (A) An Alu element that inserted after the diversification of the human and mouse lineages donated three cis-binding sites to human (Hs) CD8α gene regulatory sequences. (B) A L2 element that inserted prior to the diversification of the human (Hs) and mouse (Mm) lineages, and was then conserved, donated three cis-binding sites to the GPIIb gene regulatory region. (C) A MaLR element that inserted prior to the diversification of the human and mouse lineages but was only conserved in the human (Hs) lineage donated four cis-binding sites to the γA-globin enhancer region.
Conclusion
TEs are perhaps the most lineage-specific elements of eukaryotic genomes and they are known to contribute regulatory sequences that control the expression of host genes. Taken together, these facts suggest that TE-derived regulatory sequences may be particularly lineage specific. A comparison of human and mouse genome sequences with respect to the location of TE-derived regulatory sequences suggests that this is indeed the case. This result is consistent with a recent survey that showed TEs to be more prevalent in the UTRs of relatively unconserved human genes (van de Lagemaat et al., 2003). Thus, the activity of TEs may provide one specific mechanism that drives the regulatory diversification of host genome evolutionary lineages.
References
- Adler AJ, Danielsen M, Robins DM. Androgen-specific gene activation via a consensus glucocorticoid response element is determined by interaction with nonreceptor factors. Proc Natl Acad Sci USA. 1992;89:11660–11663. doi: 10.1073/pnas.89.24.11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler AJ, Scheller A, Robins DM. The stringency and magnitude of androgen-specific gene activation are combinatorial functions of receptor and nonrecep-tor binding site sequences. Mol Cell Biol. 1993;13:6326–6335. doi: 10.1128/mcb.13.10.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkema W, Wasserman WW. Understanding the language of gene regulation. Genome Biol. 2003;4:327. doi: 10.1186/gb-2003-4-7-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baban S, Freeman JD, Mager DL. Transcripts from a novel human KRAB zinc finger gene contain spliced Alu and endogenous retroviral segments. Genomics. 1996;33:463–472. doi: 10.1006/geno.1996.0221. [DOI] [PubMed] [Google Scholar]
- Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, et al. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–1394. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- Bowen NJ, Jordan IK, Epstein JA, Wood V, Levin HL. Retrotransposons and their recognition of pol II promoters: a comprehensive survey of the transposable elements from the complete genome sequence of Schizosaccharomyces pombe. Genome Res. 2003;13:1984–1997. doi: 10.1101/gr.1191603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ. Cases of ancient mobile element DNA insertions that now affect gene regulation. Mol Phylogenet Evol. 1996a;5:13–17. doi: 10.1006/mpev.1996.0003. [DOI] [PubMed] [Google Scholar]
- Britten RJ. DNA sequence insertion and evolutionary variation in gene regulation. Proc Natl Acad Sci USA. 1996b;93:9374–9377. doi: 10.1073/pnas.93.18.9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ. Mobile elements inserted in the distant past have taken on important functions. Gene. 1997;205:177–182. doi: 10.1016/s0378-1119(97)00399-5. [DOI] [PubMed] [Google Scholar]
- Britten RJ, Davidson EH. Gene regulation for higher cells: a theory. Science. 1969;165:349–357. doi: 10.1126/science.165.3891.349. [DOI] [PubMed] [Google Scholar]
- Britten RJ, Davidson EH. Repetitive and non-repetitive DNA sequences and a speculation on the origins of evolutionary novelty. Q Rev Biol. 1971;46:111–138. doi: 10.1086/406830. [DOI] [PubMed] [Google Scholar]
- Britten RJ, Kohne DE. Repeated sequences in DNA. Hundreds of thousands of copies of DNA sequences have been incorporated into the genomes of higher organisms. Science. 1968;161:529–540. doi: 10.1126/science.161.3841.529. [DOI] [PubMed] [Google Scholar]
- Brosius J. Genomes were forged by massive bombardments with retroelements and retrosequences. Genetica. 1999;107:209–238. [PubMed] [Google Scholar]
- Bureau TE, Wessler SR. Tourist: a large family of small inverted repeat elements frequently associated with maize genes. Plant Cell. 1992;4:1283–1294. doi: 10.1105/tpc.4.10.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau TE, Wessler SR. Mobile inverted-repeat elements of the Tourist family are associated with the genes of many cereal grasses. Proc Natl Acad Sci USA. 1994a;91:1411–1415. doi: 10.1073/pnas.91.4.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau TE, Wessler SR. Stowaway: a new family of inverted repeat elements associated with the genes of both monocotyledonous and dicotyledonous plants. Plant Cell. 1994b;6:907–916. doi: 10.1105/tpc.6.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau TE, Ronald PC, Wessler SR. A computer-based systematic survey reveals the predominance of small inverted-repeat elements in wild-type rice genes. Proc Natl Acad Sci USA. 1996;93:8524–8529. doi: 10.1073/pnas.93.16.8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MB, Dietz J, Standiford DM, Emerson CP., Jr Transposable element insertions respecify alternative exon splicing in three Drosophila myosin heavy chain mutants. Genetics. 1998;150:1105–1114. doi: 10.1093/genetics/150.3.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Clark AG. Evolution of transcription factor binding sites in mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol. 2002;19:1114–1121. doi: 10.1093/oxfordjournals.molbev.a004169. [DOI] [PubMed] [Google Scholar]
- Dunn CA, Medstrand P, Mager DL. An endogenous retroviral long terminal repeat is the dominant promoter for human (beta)1,3-galactosyltransferase 5 in the colon. Proc Natl Acad Sci USA. 2003;100:12841–12846. doi: 10.1073/pnas.2134464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuchter-Murthy AE, Freeman JD, Mager DL. Splicing of a human endogenous retrovirus to a novel phospholipase A2 related gene. Nucleic Acids Res. 1993;21:135–143. doi: 10.1093/nar/21.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganko EW, Bhattacharjee V, Schliekelman P, McDonald JF. Evidence for the contribution of LTR retro-transposons to C. elegans gene evolution. Mol Biol Evol. 2003;20:1925–1931. doi: 10.1093/molbev/msg200. [DOI] [PubMed] [Google Scholar]
- Goodchild NL, Wilkinson DA, Mager DL. A human endogenous long terminal repeat provides a polyadenylation signal to a novel, alternatively spliced transcript in normal placenta. Gene. 1992;121:287–294. doi: 10.1016/0378-1119(92)90133-a. [DOI] [PubMed] [Google Scholar]
- Gumucio DL, Heilstedt-Williamson H, Gray TA, Tarle SA, Shelton DA, et al. Phylogenetic footprinting reveals a nuclear protein which binds to silencer sequences in the human gamma and epsilon globin genes. Mol Cell Biol. 1992;12:4919–4929. doi: 10.1128/mcb.12.11.4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003;19:68–72. doi: 10.1016/s0168-9525(02)00006-9. [DOI] [PubMed] [Google Scholar]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, et al. The UCSC genome browser database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Landry JR, Medstrand P, Mager DL. Repetitive elements in the 5′ untranslated region of a human zinc-finger gene modulate transcription and translation efficiency. Genomics. 2001;76:110–116. doi: 10.1006/geno.2001.6604. [DOI] [PubMed] [Google Scholar]
- Landry JR, Rouhi A, Medstrand P, Mager DL. The Opitz syndrome gene MID1 is transcribed from a human endogenous retroviral promoter. Mol Biol Evol. 2002;19:1934–1942. doi: 10.1093/oxfordjournals.molbev.a004017. [DOI] [PubMed] [Google Scholar]
- Lenhard B, Sandelin A, Mendoza L, Engstrom P, Jareborg N, et al. Identification of conserved regulatory elements by comparative genome analysis. J Biol. 2003;2:13. doi: 10.1186/1475-4924-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Maor G, Sorek R, Shomron N, Ast G. The birth of an alternatively spliced exon: 3′ splice-site selection in Alu exons. Science. 2003;300:1288–1291. doi: 10.1126/science.1082588. [DOI] [PubMed] [Google Scholar]
- Li WH. Molecular Evolution. Sinauer Associates; Sunderland: 1997. [Google Scholar]
- Liu G, Zhao S, Bailey JA, Sahinalp SC, Alkan C, et al. Analysis of primate genomic variation reveals a repeat-driven expansion of the human genome. Genome Res. 2003;13:358–368. doi: 10.1101/gr.923303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager DL, Hunter DG, Schertzer M, Freeman JD. Endogenous retroviruses provide the primary polyadenylation signal for two new human genes (HHLA2 and HHLA3) Genomics. 1999;59:255–263. doi: 10.1006/geno.1999.5877. [DOI] [PubMed] [Google Scholar]
- Makalowski W, Mitchell GA, Labuda D. Alu sequences in the coding regions of mRNA: a source of protein variability. Trends Genet. 1994;10:188–193. doi: 10.1016/0168-9525(94)90254-2. [DOI] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. Maize genetics. Carnegie Inst Washington Year Book. 1946;45:176–186. [PubMed] [Google Scholar]
- McClintock B. Mutable loci in maize. Carnegie Inst Washington Year Book. 1948;47:155–169. [PubMed] [Google Scholar]
- McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- McClintock B. The Discovery and Characterization of Transposable Elements: The Collected Papers of Barbara McClintock. Garland; New York: 1987. [Google Scholar]
- McCue LA, Thompson W, Carmack CS, Lawrence CE. Factors influencing the identification of transcription factor binding sites by cross-species comparison. Genome Res. 2002;12:1523–1532. doi: 10.1101/gr.323602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medstrand P, Landry JR, Mager DL. Long terminal repeats are used as alternative promoters for the endothelin B receptor and apolipoprotein C-I genes in humans. J Biol Chem. 2001;276:1896–1903. doi: 10.1074/jbc.M006557200. [DOI] [PubMed] [Google Scholar]
- Moses AM, Chiang DY, Kellis M, Lander ES, Eisen MB. Position specific variation in the rate of evolution in transcription factor binding sites. BMC Evol Biol. 2003;3:19. doi: 10.1186/1471-2148-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, et al. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JC, Shabalina SA, Harris DG, Spouge JL, Kondrashov AS. Conserved fragments of transposable elements in intergenic regions: evidence for widespread recruitment of MIT- and L2-derived sequences within the mouse and human genomes. Genet Res. 2003;82:1–18. doi: 10.1017/s0016672303006268. [DOI] [PubMed] [Google Scholar]
- Sorek R, Ast G, Graur D. Alu-containing exons are alternatively spliced. Genome Res. 2002;12:1060–1067. doi: 10.1101/gr.229302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavenhagen JB, Robins DM. An ancient provirus has imposed androgen regulation on the adjacent mouse sex-limited protein gene. Cell. 1988;55:247–254. doi: 10.1016/0092-8674(88)90047-5. [DOI] [PubMed] [Google Scholar]
- Sugiura N, Hagiwara H, Hirose S. Molecular cloning of porcine soluble angiotensin-binding protein. J Biol Chem. 1992;267:18067–18072. [PubMed] [Google Scholar]
- Suzuki Y, Yamashita R, Nakai K, Sugano S. DBTSS: DataBase of human Transcriptional Start Sites and full-length cDNAs. Nucleic Acids Res. 2002;30:328–331. doi: 10.1093/nar/30.1.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JW, Touchman JW, Blakesley RW, Bouffard GG, Beckstrom-Sternberg SM, et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature. 2003;424:788–793. doi: 10.1038/nature01858. [DOI] [PubMed] [Google Scholar]
- van de Lagemaat LN, Landry JR, Mager DL, Med-strand P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 2003;19:530–536. doi: 10.1016/j.tig.2003.08.004. [DOI] [PubMed] [Google Scholar]
- van den Berg CW, Demant P, Aerts PC, Van Dijk H. Slp is an essential component of an EDTA-resistant activation pathway of mouse complement. Proc Natl Acad Sci USA. 1992;89:10711–10715. doi: 10.1073/pnas.89.22.10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varagona MJ, Purugganan M, Wessler SR. Alternative splicing induced by insertion of retrotransposons into the maize waxy gene. Plant Cell. 1992;4:811–820. doi: 10.1105/tpc.4.7.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Wessler SR, Bureau TE, White SE. LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev. 1995;5:814–821. doi: 10.1016/0959-437x(95)80016-x. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gerstein M. Of mice and men: phylogenetic footprinting aids the discovery of regulatory elements. J Biol. 2003;2:11. doi: 10.1186/1475-4924-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]