

Abstract
Most diseases that cause catastrophic loss of vision do so as a result of abnormal angiogenesis and wound healing, often in response to tissue ischemia or inflammation. Disruption of the highly ordered tissue architecture in the eye caused by vascular leakage, hemorrhage, and concomitant fibrosis can lead to mechanical disruption of the visual axis and/or biological malfunctioning. An increased understanding of inflammation, wound healing, and angiogenesis has led to the development of drugs effective in modulating these biological processes and, in certain circumstances, the preservation of vision. Unfortunately, such pharmacological interventions often are too little, too late, and progression of vision loss frequently occurs. The recent development of progenitor and/or stem cell technologies holds promise for the treatment of currently incurable ocular diseases.
Introduction
To see well, we must maintain a clear visual axis and normally functioning cellular phototransduction. Light entering the eye passes through the cornea (the major refractive surface), the lens, the vitreous (gel in the posterior chamber of the eye), the inner retina, and, finally, into the photoreceptors of the outer retina (Figure 1). These photoreceptors are the site at which photons of light are converted into electrical signals that are transmitted to the visual cortex of the brain by a complex series of synaptic transmissions (Figure 1). To maintain a visual axis through which light can pass undisturbed, a highly ordered tissue structure is required. Any disturbance in normal cell-cell relationships can lead to biological malfunctioning and/or diffraction, absorbance, or reflection of photons, resulting in disturbed or diminished vision.
Figure 1. Schematic representation of the eye and principal types of retinal neovascularization and fibrosis/gliosis.
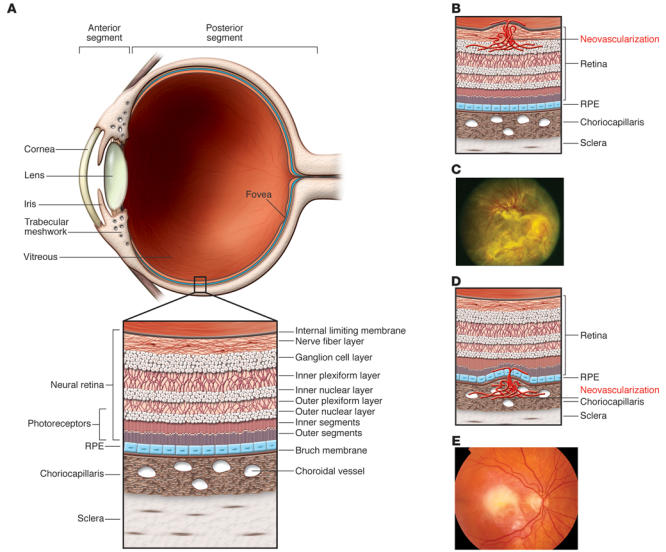
(A) The anterior segment of the eye, consisting primarily of the cornea and iris, is separated from the posterior segment by the lens. The posterior segment consists primarily of the vitreous and the retina. (B) The retina is a highly ordered, multilayered structure that is richly vascularized. Ischemic retinopathies, such as DR, can lead to ischemia and neovascularization on the surface of the retina. (C) In extreme cases, associated gliosis can lead to tractional retinal detachments. Reproduced with permission from the American Academy of Ophthalmology (122). (D) ARMD can be associated with subretinal neovascularization originating from the choriocapillaris, and this can lead to subretinal hemorrhage and fibrosis (E).
Homeostasis of the eye, as in tissues elsewhere in the body, depends on the presence of normal vasculature, ECM, and various cell types. If homeostasis is disturbed by infection, inflammation, or metabolic disease, visual function becomes impaired. The end result of these conditions is often fibrosis. In the CNS, of which the retina is a part, such wound-healing responses and associated fibrosis are mediated by glial cells, which perform functions in the CNS similar to those performed by fibroblasts in the rest of the body. Therefore, gliosis is frequently used to describe the glial cell–mediated wound-healing response observed in the CNS, much as fibrosis (which is fibroblast mediated) is used to describe similar processes in non-CNS tissues. In the skin, fibrosis can lead to a cosmetic blemish in the form of a scar; in the eye this can have disastrous consequences for vision — mechanically disrupting the visual axis or sufficiently disturbing the tissue microenvironment such that proper cellular functioning is no longer possible. For example, fibrosis of the cornea can occur after a viral infection, leading to corneal opacification and thereby loss of vision. In the posterior segment of the eye (Figure 1), uncontrolled retinal vascular proliferation, as a result of diabetes-associated retinal hypoxia, can lead to fibrosis and traction retinal detachment, a dreaded complication of advanced diabetic retinopathy (DR). Under the retina, similar fibrosis can occur subsequent to subretinal hemorrhage associated with neovascular age-related macular degeneration (ARMD).
Collectively, these conditions of fibrosis in the eye lead to vision loss in millions of individuals worldwide. In this Review, I discuss the cellular pathophysiology associated with fibrosis in the anterior and posterior segments of the eye (Figure 1), with a focus on the latter. Therapeutic approaches for treating these disorders, based on advances in our understanding of the biological mechanisms underlying these conditions, are reviewed and then discussed in the context of recent novel advances in the area of cell-based therapies.
Fibrosis in the eye: general considerations
Fibrosis commonly refers to the response of a tissue to injury. The injury can occur as a result of a mechanical wound or various metabolic malfunctions, including responses to inflammation, ischemia, and degenerative disease. The local response to such injuries includes infiltration by inflammatory cells, neovascularization, altered vascular permeability, proliferation of fibroblasts and fibroblast-like cells, modification of the ECM, and, ultimately, some sort of resolution of the damaged tissue. The CNS is highly specialized in many ways, including the types of inflammatory and wound-healing cells present. Since the retina is part of the CNS, its response to injury utilizes mechanisms very similar to those observed in the rest of the brain; this is true not only for the wound-healing response but also for utilization of migratory cues functional during development of the neuronal and vascular components of this highly organized tissue (1, 2). As discussed below, the response of the anterior segment of the eye to wound healing more closely resembles the response of non-CNS tissues than do such events in the posterior segment or the eye. Therefore, I refer to such wound-healing events in the anterior segment as fibrosis, whereas comparable events in the retina are referred to as gliosis. Although such distinction is somewhat artificial, it does serve to differentiate between the fibroblasts and glial cells that effect the wound-healing and scar-formation events.
Anterior segment fibrotic diseases of the eye
Two major diseases of the anterior segment of the eye leading to visual loss are corneal opacification and glaucoma. In glaucoma, there is progressive loss of ganglion cells of the nerve fiber layer; this results in degeneration of the neuronal tracts through which efferent signals travel from the retina to the visual cortex (3). Typically associated with increased intraocular pressure, this disease can lead to progressive constriction of the visual fields and, eventually, complete loss of vision. Although increased intraocular pressure can occur from either increased production of intraocular fluid or increased resistance to outflow, it is more commonly believed that progressive fibrosis of the tracts through which the intraocular fluid leaves the eye (called the trabecular meshwork) accounts for most of the damage that causes glaucoma. Increased understanding of the molecular basis for malfunctioning of the trabecular meshwork (4) (in particular, the aberrant production of ECM components) and of the fibrosis associated with increased resistance to outflow, holds promise for developing therapeutics for this relentlessly progressive disease (5).
Although there are those who consider the cornea simply a “dustcover for the retina,” it in fact is a highly organized tissue through which light must pass before entering the rest of the eye. The cornea is covered externally by a stratified nonkeratinizing epithelium and internally by a single layer of transporting endothelium with multiple orthogonal arrays of collagen in between. It is normally avascular due to the high concentration of soluble VEGFR-1 (6) and is surrounded by a transitional margin, the corneal limbus, within which resides nascent endothelium and corneal epithelial stem cells (7), which have high potential for therapeutic value (8). Diseases of the cornea can be genetic (e.g., inherited dystrophies) or acquired secondary to infection (e.g., herpetic keratitis) or inflammation (e.g., pterygia). Elastoid degeneration of the conjunctiva, resulting in pingueculae and pterygia (fibrovascular growths on the surface of the cornea), can lead to visual loss secondary to induced astigmatism and/or obstruction of the visual axis and would be amenable to topically applied inhibitors of fibrosis and/or angiogenesis (9). The final common events in all of these diseases are often inflammatory changes associated with neovascularization, tissue edema, and, ultimately, fibrosis of the corneal stroma, which leads to opacification and decreased vision (10). Nearly 20 years ago, penetrating keratoplasty (or corneal transplants) changed the uniformly dismal prognosis for patients with opacified or failed corneas; in a substantial percentage of patients undergoing this procedure, if there are no other associated abnormalities, the visual axis is cleared and vision is restored. Despite advances in the use of antiinflammatory drugs, antibiotics, and hypertonic solutions to reduce corneal edema associated with the immune response to the transplant, there is a substantial failure rate, typically due to recurrent opacification. Recent advances in corneal limbal stem cell biology hold the promise of reducing the failure rate for this procedure (11).
Posterior segment fibrotic diseases of the eye
General comments.
The posterior segment of the eye consists of structures behind the lens; the interior of the back of the eye is filled with vitreous, a viscoelastic material consisting largely of water, collagen, and hyaluronic acid (12). The vitreous serves as a shock absorber, among other things, for the retina, the most posterior tissue in the eye. In addition, the vitreous can provide scaffolding over which glial and endothelial cells migrate from their normal intraretinal position anteriorly over the retinal surface and/or into the vitreous in certain disease states (e.g., diabetes, proliferative vitreoretinopathy, retinopathy of prematurity [ROP]). The retina consists of multiple layers of neurons, blood vessels, ECM, and various resident and transient cells such as glial cells and monocytes. The vascular supply of the retina consists of the retinal blood vessels (found in three layers on the innermost portion of the retina) and the choriocapillaris (a rich vascular plexus found in the outermost portion of the retina). The photoreceptors are in the outermost portion of the neurosensory retina and rest on a monolayer of cells, the retinal pigmented epithelium (RPE), discussed further below. The RPE rests on a collagenous basement membrane (Bruch membrane), and directly beneath this structure flows the choriocapillaris, providing blood supply for the outer third of the retina. Although there is a blood-retina barrier and relative immune privilege in this part of the eye, normal inflammatory responses to irritation and hypoxia can be quite robust and can lead to much of the pathology observed in diseases that decrease vision (Figure 1).
Most diseases that lead to vision loss in industrialized nations do so as a result of abnormalities in the retinal or choroidal vasculature. These diseases, characterized by macula edema, retinal and vitreous hemorrhage, and fibrovascular scarring, include ARMD, DR, ROP, and neovascular glaucoma. The final common pathophysiological denominator in all of these diseases is the retinal response to injury, with chronic wound healing leading to fibrosis. Although the underlying principles of wound healing in other tissues apply to this process in the eye, it is the uniqueness of the cellular composition and anatomical structure of the retina that makes this normal biological process so potentially devastating to vision. The photoreceptors are located in the outermost portion of the neurosensory retina, just anterior to the RPE and choriocapillaris. Overlying these cells are the vitreous, nerve fiber layer, inner nuclear layer, numerous capillary plexuses, and Mueller-glial cells and their processes (Figure 1). For light to hit the photoreceptors in an undisturbed manner such that visual images can be formed, it is important that the highly organized architecture of the retina is preserved. When abnormal blood vessels form in response to inflammatory or hypoxic stimuli, they can leak fluid, causing retinal thickening and edema, and/or bleed, leading to fibrovascular proliferation and tractional retinal detachment. The following discussion focuses on the unique aspects of wound healing, fibrosis, and scar formation as it occurs in the posterior segment of the eye.
Fibrovascular scarring and gliosis in the retina.
In simplest terms, fibrovascular scarring is a consequence of the underlying inflammatory or hypoxia-driven neovascularization and its associated fibrosis. Therefore, prevention of the primary vascular abnormality is the most appropriate therapeutic target to preserve retinal structure and function. To understand fibrosis and its consequences in the back of the eye, understanding the unique aspects of retinal fibrosis is necessary. Glial cells are the CNS counterparts of peripheral fibroblasts, with several key distinctions, and are therefore the primary participants in the formation of fibrotic scars in response to retinal injury and disease. In addition to their fibrotic tendencies, glial cells also perform a myriad of supportive functions for the neurons with which they are intimately associated. In the retina, this trophic relationship to neurons is extended to the vascular endothelium, with which certain glia are intimately associated in both developing and mature tissue. For example, activated astrocytes form the template over which retinal vascular endothelial cells migrate during formation of the superficial vascular plexus (1, 13); disturbances in the number or distribution of these cells disrupts the normal development of the retinal vasculature (14). Glial cells of the retina include the resident immune cells, microglial cells, and two types of macroglial cell, the astrocyte and the retina-specific Mueller-glial cell (15). Two broad categories of disease account for most of the conditions that lead to fibrovascular scarring in the retina and its associated vision loss — inflammatory diseases (e.g., ARMD) and ischemic diseases (e.g., DR).
Subretinal fibrosis: ARMD.
The leading cause of vision loss in Americans over the age of 65 is ARMD; 12–15 million Americans over the age of 65 have this disease and 10%–15% of them will lose central vision as a direct effect of choroidal (subretinal) neovascularization and fibrosis. Clinically, most of these individuals develop atrophic changes in the RPE, which performs a myriad of functions associated with normal photoreceptor functioning (16) and is the cellular interface between the underlying choriocapillaris and the outermost portion of the neurosensory retina, the photoreceptors. As the RPE ages or becomes diseased, it can function improperly, and a build-up of subretinal deposits, called drusen, accumulate. These drusen contain, among other things, angiogenic lipids and damaged proteins (17). RPE dysfunction and the accumulation of drusen can lead to thickening of Bruch membrane, and the accumulation of angiogenic drusen associated with this fibrosis can lead to decreased diffusion of oxygen from the choriocapillaris to the photoreceptors, further exacerbating conditions that can lead to choroidal neovascularization. Once these new abnormal blood vessels begin to grow in the subretinal space, they often hemorrhage, leading to further wound-healing responses and, ultimately, to subretinal fibrosis (Figure 1, D and E). Needless to say, local destruction of photoreceptors, the RPE, and choroidal blood vessels leads to permanent reduction in macular function and vision. Efforts to develop animal models to study this process have been hampered by the fact that rodents do not seem to faithfully mimic the human disease, although transgenic mice have provided some use in this regard (18).
Advances in therapeutic options available to treat neovascular ARMD have provided some benefit to small subsets of patients with this disease (19, 20). Most drugs currently in clinical trials or approved for treating ARMD-associated choroidal neovascularization are directed at inhibiting promoters of angiogenesis, such as VEGF. There is extensive literature covering these approaches, and I refer the reader to several excellent recent reviews (refs. 16, 19). Unfortunately, inhibiting angiogenic cytokines does not address the underlying pathophysiology — ischemia and inflammatory stimuli. Efforts to minimize sub- and epiretinal fibrosis have met with limited success and, in any event, would represent a therapeutic intervention occurring too late to rescue vision, since such scarring would have already led to photoreceptor death.
Epiretinal fibrosis: DR.
The leading cause of visual loss for Americans under the age of 65 is diabetes; 6%–8% of the American population is diabetic, and 40,000 patients each year suffer visual loss from complications of the disease, often as a result of retinal edema or neovascularization (21). Virtually every diabetic has some form of DR after 20 years of the disease (21). Ischemia occurs as a result of the diabetic microvasculopathy that includes pericyte cell death, microaneurysms, intraretinal microvascular abnormalities, altered vascular permeability, and macular edema (22). As the hypoxia increases, neovascularization can occur, leading to intraretinal, subhyaloid (between the retinal surface and posterior vitreous base) and vitreous hemorrhage (Figure 1B). These proliferating blood vessels are accompanied by fibrosis that occurs as a consequence of glial cell activation and proliferation (gliosis) (Figure 1C). As abnormal vessels continue to proliferate on the retinal surface, they can extend into the vitreous and contract, causing traction on the retinal surface and leading to retinal detachment, a dreaded complication of proliferative DR. Retinal neovascularization and associated gliosis and fibrosis are also observed in ROP (23) and as a complication of surgery to treat retinal detachment (24, 25). Surgical intervention and laser obliteration of the peripheral retina (to decrease the metabolic demand and thereby match up supply and demand) are the current treatments and are of limited benefit. Although animal models of ischemic retinopathy have been very useful in helping to develop a better understanding of factors that control retinal vascular proliferation (24, 26), the rodent does not develop the associated preretinal fibrosis, limiting its utility in studying the gliosis observed in the human condition. Given that abnormal vascular proliferation serves as the stimulus for pathological fibrotic responses in these diseases, the following discussion focuses on known basic molecular and/or biological pathways of angiogenesis and rational approaches to therapeutic interventions based on this knowledge.
Neovascularization of the retina leads to gliosis and fibrous scarring
General considerations.
The ocular response to hypoxia and inflammatory insults typically leads to retinal or choroidal neovascularization. During development, this process is highly regulated and leads to the establishment of a well organized, mature vasculature (1). In the adult eye, this is often not the case, and associated glial cells (e.g., astrocytes, microglia, and Mueller-glial cells) proliferate with the endothelial cells, leading to fibrosis and scar formation. To understand this gliotic response, it is important to understand angiogenesis.
In the normal adult, angiogenesis (defined as the growth of new blood vessels from preexisting ones) is tightly regulated and limited to wound healing, pregnancy, and uterine cycling. Our understanding of the molecular events involved in the angiogenic process has advanced substantially since the purification of the first angiogenic molecules nearly two decades ago (27). This process, under physiologic conditions, can be activated by specific angiogenic molecules (Table 1), such as basic and acidic FGF (28), VEGF (29), angiogenin (30), TGF-β (31), IFN-β (32), TNF-α (33), and PDGF (34). Angiogenesis can also be suppressed by inhibitory molecules (Table 2), such as IFN-α (35), thalidomide (36), thrombospondin-1 (37), angiostatin (38), endostatin (39), a naturally occurring form of the carboxyterminal, noncatalytic domain of MMP-2 (PEX) (40), transfer RNA (tRNA) synthetases (41, 42), and pigment epithelium–derived factor (43). It is the balance of these naturally occurring stimulators and inhibitors of angiogenesis that is thought to tightly control the normally quiescent capillary vasculature (44). When this balance is upset, as in certain disease states (e.g. DR), capillary endothelial cells are induced to proliferate, migrate, and differentiate.
Table 1 .
Molecules with angiogenic activity in the eye

Table 2 .
Molecules with antiangiogenic activity in the eye

The role of cell adhesion molecules, such as integrins, in regulating the relationship between proliferating vascular cells and their environment, has been the focus of many studies (45). At least three cytokine-dependent pathways of angiogenesis have been described and defined by their dependency on distinct vascular cell integrins, αvβ3 (46), αvβ5 (47), and αvβ1 (48). Cell migration through the ECM also depends on proteolysis of the matrix. Integrins (32, 49, 50), MMPs (51–53), and tissue inhibitors of metalloproteinases (TIMPs) (54) are found throughout the eye, where they interact with each other (40) to maintain a quiescent vasculature until the balance is upset, resulting in pathological angiogenesis. Signaling molecules, including SRC tyrosine kinases (55), modify endothelial cell behavioral responses to changes in the microenvironment, and similar pathways are operational in migrating neurons, differentiating progenitor cells, and glial cells (56). Vascular endothelial cells are protected from apoptotic stimuli by αv integrin subunit interaction with RAF kinase (57). This response is differentially regulated by two distinct pathways, one involving FGF and the other involving VEGF-stimulated endothelial cell apoptosis (47).
Ocular angiogenesis.
Several reports suggest that VEGF is the dominant angiogenic stimulus in experimentally induced iris neovascularization (58, 59) as well as endogenous neovascular retinopathies (60–62). Although there clearly is a direct correlation between intraocular VEGF levels and ischemic retinopathic ocular neovascularization, a role for FGF cannot be ruled out (63–65). Substantial inhibition of retinal vascular proliferation in a mouse model of hypoxic retinopathy is observed with antibody (66) and aptamer (67) antagonists of VEGF as well as with selective PKC antagonists (68).
Although normal human ocular blood vessels do not ordinarily display αvβ3 or αvβ5 integrins, only αvβ3 has been consistently observed in ocular tissue from patients with ARMD, whereas both αvβ3 and αvβ5 were present in tissues from patients with proliferative DR (49). Systemically administered peptide antagonists of both integrins blocked new blood vessel formation in a mouse model of retinal angiogenesis (49, 69, 70), reinforcing the concept that both integrins might have a role in active fibrovascular proliferation of the type seen in proliferative DR. Consistent with the concept that integrins such as αvβ3 and αvβ5 are required for proliferating endothelial cells to successfully navigate the extracellular milieu is the observation that these integrins can selectively bind MMPs, including PEX (40). Small-molecule PEX mimetics also bind αvβ3, preventing binding of MMP2, and thereby mimicking the action of PEX (71, 72). Clearly, interactions between developing vasculature and the ECM are critical during normal and abnormal angiogenesis (73).
The antiangiogenic activity of several compounds exhibits strain-related differences in various animal models of angiogenesis (74). Steroids (75), in particular an angiostatic steroid devoid of glucocorticoid activity (anecortave acetate) (76, 77), pigment epithelium–derived factor (43), MMP antagonists (78), and somatostatin analogs (79–81) have demonstrated potent antiangiogenic activity in various animal models of ocular neovascularization. Laser photocoagulation has been effective in preventing severe visual loss in subgroups of high-risk diabetic patients but, with few exceptions, has not been effective at preventing visual loss in patients with choroidal neovascularization due to ARMD or inflammatory eye diseases such as ocular histoplasmosis. Photodynamic therapy using nonthermal lasers to activate photoactivateable dyes reduces severe vision loss in a small subset of ARMD patients (82). Very recent substantial advances in the use of antiangiogenic monotherapy, principally VEGF antagonists, have led to the reduction of severe vision loss in a subset of patients with ARMD (83). Although slowing, or even minimally improving, vision loss in some patients, this approach does not offer relief for the underlying condition or the ischemia driving the neovascularization. Thus, a substantial challenge in developing effective treatments for these diseases remains the relief of retinal ischemia. Combination therapy holds great promise in this area (84) but remains relatively unexplored.
Retinal neovascularization leads to gliosis.
Retinal glial cells, and astrocytes in particular, have an important role in establishing and maintaining the highly ordered retinal vasculature (1, 14). Retinal injury due to hypoxia or inflammatory changes, similar to injuries in other tissues, is typically associated with neovascularization and fibrosis (85). Reactive gliosis can be both neuro- and vasculoprotective but can also directly contribute to scar formation and tractional lesions that lead to vision loss. Therefore, if we are to better control the retinal response to injury, a better understanding of the regulation of glial cell proliferation is necessary. A number of studies have examined the control of Mueller-glial cell proliferation and activation after retinal injury, and a role for p27Kip1 has been demonstrated in the regulation of Mueller-glial cell proliferation during injury-associated gliosis (86). These cells not only participate in glial scar formation after injury but can also upregulate the production of trophic factors that facilitate neuronal survival following injury through the production of neurotrophic factors such as ciliary neurotrophic factor (CNTF) (87).
RPE can modulate the retinal response to injury.
The cells of the RPE are highly specialized stationary macrophages, of sorts, that perform many functions relating to maintenance of healthy photoreceptors (16) and often participate in injury response that involves the outer retina and choriocapillaris (e.g., choroidal and intraretinal neovascularization) (88). The cells of the RPE not only phagocytose debris but are known to upregulate the production of a number of cytokines that serve to modulate the inflammatory and neovascular response to injury and stress (89). Although substantial progress has been made in identifying many of the factors that promote and inhibit angiogenesis and gliosis, the complexity of cellular interactions and the number of factors involved have made meaningful therapeutic intervention very difficult.
Cell-based therapies for the treatment of diseases of retinal fibrosis/gliosis
Current therapies for ARMD and DR aim to inhibit cytokines that mediate the vasoproliferative response or to destroy the tissue that is creating the increased metabolic demand. A more rational therapeutic approach would be to relieve the hypoxia or to replace or normalize the cells that underlie the fibrotic response to injury. Our increased understanding of the angiogenic process has led to the identification of effector molecules, their cellular receptors, intracellular signaling cascades, and post-transcriptional regulators; this knowledge has translated into the use of compounds that can inhibit the pathological angiogenesis associated with tumors and neovascular eye diseases (90). Understanding of cell-cell and cell-ECM interactions that affect angiogenesis at the tissue level has also improved, and more recently a role for circulating cells in the regulation of vascular homeostasis in the adult organism has been identified in a number of organs, including the eye (91). In addition to the endothelial cells and their progenitors, it is critical to identify and modulate cells that contribute to the fibrotic response; key cells to target in this regard would be Mueller-glial cells or the cells of the RPE.
More recent work suggests that cell-based therapy, using adult bone marrow–derived progenitor cells, could have substantial clinical utility in the treatment of ischemic retinopathies such as ROP and DR. One advantage of such an approach, in contrast to inhibiting angiogenesis with small molecules or recombinant proteins, lies in the ability of the cell to adapt and respond to a changing environment. Current therapeutic approaches to the treatment of neovascular eye diseases generally involve the application of a single angiostatic or antiproliferative agent to the eye. Cell therapy probably involves numerous factors produced by the cell that can be appropriately modulated in response to changing conditions. This shift, from antiangiogenics and destructive procedures that inhibit or ablate hypoxia-driven neovascularization to cell-based therapies that facilitate repair and stabilization, could be of benefit in the treatment of certain ocular disorders.
Could RPE cell-based therapies modulate the injury response observed in ARMD? The RPE and photoreceptors enjoy an intimate relationship both anatomically and functionally.
This interdependency has historically contributed to the difficulty in determining whether the principal underlying defect in many inherited retinal degenerations lies in the photoreceptor or the cells of the RPE. With the advent of molecular genetics this confusion has decreased, but the interdependency between these two cell types remains, and concomitant degeneration of both cell types is often observed in various inherited and acquired degenerative diseases of the retina. In this regard, RPE cell transplantation has been evaluated for its potential to serve as a cell-based drug-delivery vehicle, to replace diseased RPE, and to provide a source of cells whose phenotypic differentiation might be manipulated by various cytokines and trophic substances. In a number of macula and retinal degenerative disorders, there is atrophy of the RPE and associated malfunctioning in the phototransducing cellular machinery. Damaged RPE cells and associated atrophy are hallmarks of ARMD, and heroic surgical approaches involving retinal translocation and the insertion of RPE sheets have been considered to provide photoreceptors in individuals with healthier, RPE-rich regions of the retina (92). Although retinal translocation has enjoyed a limited clinical effort (93), the insertion of RPE sheets is still in the laboratory stages of development (94, 95). Recent clinical studies of autofluorescence in patients with ARMD have shown a surprisingly high level of functional RPE (96), even in the face of choroidal neovascularization and early subretinal fibrosis, suggesting that interventions aimed at sustaining RPE might be productive means to maintaining photoreceptor function in the face of choroidal neovascularization.
Immortalized human cell lines derived from the RPE have been created to facilitate both the transplantation of cells of the RPE and their use in cell-based therapeutic approaches (97, 98). When transformed RPE-derived cell lines are transfected with a plasmid encoding one therapeutic factor, CNTF, and transplanted directly into the vitreous of dogs with retinal degeneration, photoreceptor degeneration is reduced (97, 98). Encapsulated cell-based delivery devices (99) might be useful in the treatment of various retinal degenerative diseases and, in fact, have recently been used in a human clinical trial evaluating the efficacy of CNTF in the treatment of retinal degeneration. Cells with many of the molecular and functional characteristics of the cells of the RPE have recently been isolated from spontaneously differentiating human embryonic stem cell lines and have been touted as a potential source of transplantable RPE for subretinal transplantation into human retinas (100). If these cell lines can be maintained in cell banks and altered so as to facilitate immune acceptance, they might represent a source of transplantable tissue. Embryonic stem cells might also be a source of transplantable cells of the RPE that retain RPE characteristics or can differentiate into photoreceptor cells (101). Finally, cells of the RPE modified to suppress normal wound-healing/fibrotic responses to subretinal injuries such as choroidal neovascularization might be useful in the treatment of neovascular ARMD.
Adult bone marrow–derived stem cells.
Adult bone marrow is known to contain a population of HSCs that can differentiate into various cell types including myeloid and endothelial lineage cells. Endothelial progenitor cells (EPCs) mobilize from the bone marrow in response to various signaling molecules (102) and can target sites of angiogenesis in ischemic peripheral vasculature, myocardium, and induced ocular injury. The EPC-containing fraction of the HSCs, when injected directly into the eye, can localize to activated astrocytes, a hallmark of many ocular vascular and degenerative diseases, and participate in normal developmental angiogenesis in neonatal mice and injury-induced neovascularization in the adult (103). These intravitreally injected adult bone marrow–derived progenitor cells can completely prevent the retinal vascular degeneration observed in the rd1 and rd10 mouse models of retinal degeneration, and this vascular rescue correlates with neuronal rescue (104). Large-scale genomic analysis of rescued eyes revealed substantial upregulation of antiapoptotic genes, and this effect is also observed when human bone marrow–derived progenitor cells are used to treat SCID mice with retinal degeneration. In addition, this subset of HSCs, if previously transfected with a plasmid encoding a secreted antiangiogenic peptide, profoundly inhibits retinal angiogenesis (103), suggesting that cell-based therapy might be useful in the inhibition of proliferative retinopathies.
The specific identity of the potentially therapeutic, pluripotent EPC-containing fraction of HSCs remains an enigma. HSCs have been reported to differentiate into various cell types other than hematopoietic cells, including neurons, glial cells, and muscle cells, depending on their environment (105), although this is not without controversy (106). The observation that HSCs contain a pool of EPCs able to incorporate into retinal vasculature has been demonstrated, but there is continuing controversy as to the precise identity of these cells (107). Similarly, systemically administered HSCs can function as hemangioblasts during hypoxia-stimulated retinal neovascularization (108) and contribute to wound healing and angiogenesis in a mouse model of laser-stimulated choroidal neovascularization (109–111). Recent studies have demonstrated a role for R-cadherin (112) and the integrin α4β1 (102) in the targeting of HSCs to the retinal and tumor vasculature, respectively. Therefore, inhibiting the targeting of the circulating HSCs that contribute to abnormal ocular neovascularization might, in fact, reduce the incidence of new blood vessel growth. Unfortunately, inhibition of neovascularization under ischemic conditions can serve to promote ongoing ischemia. Would it be better to coax the newly forming vessels to develop in a functional manner that could alleviate hypoxia, thereby avoiding neovascularization and associated complications, including fibrosis?
Retinal microglial cells for use in cell-based therapy of retinal fibrotic disease.
Retinal microglial cells have historically been viewed as immunocompetent cells that respond to inflammation and infection, phagocytosing debris created during normal developmental remodeling or degenerative disease. A role for microglial cells in promoting retinal vascularization has not been clearly described. We have recently demonstrated that adult bone marrow–derived cells expressing surface markers of myeloid progenitors differentiate into microglia and facilitate the enhanced recovery of vasculature after hypoxic injury (113). This process is dependent on hypoxia-inducible factor 1α (HIF-1α) and describes a novel role for myeloid progenitors in modulating angiogenesis. In addition, endogenous microglial cells have an important role in promoting and maintaining retinal vascularization during normal development.
The use of progenitor cell therapy to promote vascularization has been spearheaded by the field of cardiology with the goal of collateralizing ischemic myocardium (114) and extremities (115) or of using the levels of circulating cells to predict clinical outcomes (116, 117). Clinical studies suggest that bone marrow–derived progenitor cells are effective at improving perfusion and cardiac function (118). Numerous studies from other investigators have explored the potential role of bone marrow–derived EPCs and have demonstrated that these cells are present in newly formed myocardial vessels. However, recent data suggest that heterogeneous bone marrow populations, such as mononuclear cells or unfractionated cells that contain very small numbers of stem cells and/or EPCs, can also substantially enhance collateral (e.g., new vessels that facilitate revascularization of poorly perfused regions of tissue) development. This suggests that perivascular cells, as well as those directly incorporating into vessels, are likely to be involved. These perivascular cells include myeloid cells (119) recruited from the circulation in response to local VEGF expression in the adult heart or liver. These cells assume a perivascular location and are retained there by stromal cell–derived factor-1 (SDF-1) promoting vessel growth in these organs in a paracrine fashion. In addition to this paracrine function, myeloid progenitors have recently been shown to differentiate into endothelial cells, further underscoring their role in promoting normal vessel growth and facilitating angiogenesis in the adult (120). Before such concepts can be applied to using these cells to restore normal vasculature in ischemic retinas, it is necessary to better understand the relationships among microenvironmental cues (mediated either through ECM and/or cell-produced signaling molecules), the endothelium, and vascular-associated cells. The signals that determine whether normal or abnormal angiogenesis, with or without associated fibrosis, occurs remain to be clearly defined. Better defining these parameters is the greatest challenge to implementing cell-based therapies for treatment of fibrovascular diseases of the retina.
A cautionary note needs to be added. Although selective targeting of progenitor cells to sites of neovascularization and/or gliosis/fibrosis might be beneficial under certain circumstances, there is data suggesting that these cells can, under appropriate circumstances, contribute to the disease process. Much of these data have come from studies of experimental models in which sublethal irradiation is followed by bone marrow transplantation with fluorescently-labeled cells that are then observed to home to sites of pathological neovascularization following mechanical or laser-induced injury (109–111). Whether an intact blood-retinal barrier (compromised by laser in most of these studies), endogenous glial cells, or the RPE ordinarily modulates the wound-healing response to exclude circulating cells is not clear. Another example of a potentially negative effect of enhancing microglial cells and/or macrophages during an ocular wound-healing response comes from a recent study of fibrovascular proliferation following experimentally induced retinal detachment (121). In this study, local Mueller-glial cells were observed to upregulate expression of monocyte chemoattractant protein 1 (MCP-1), leading to increased accumulation of microglial cells and macrophages in the detached retina. These cells in turn contributed to local oxidative stress on photoreceptors, leading to photoreceptor apoptosis. Clearly, modulation of heterogeneous, complex cell-cell interactions in diseased tissue requires a detailed understanding of the cell types and cellular interactions that comprise target tissues.
Concluding remarks
The unique anatomy and complex intercellular interactions in the retina contribute to the visually devastating complications of the fibrovascular response of the tissue to injury. Angiogenesis is a critical component of wound healing, and in the retina this is observed under conditions of traumatic injury, hypoxia, and inflammation. Unfortunately, neovascularization under these conditions is associated with altered vascular permeability, retinal edema, hemorrhage, and, in extreme cases, fibrovascular traction and fibrosis. Gliosis is a substantial component of the angiogenic response, and although there are neuroprotective functions associated with glial cells, the end result in the retina is often tractional retinal detachment or intra- or subretinal scarring associated with vision loss. Modulation of glial cell reactivity might provide therapeutic approaches to treat these disorders, and increased understanding of glial cell proliferation and migration should provide therapeutic targets. The complexity of cell-cell interactions in modulating the injury response decreases the likelihood of developing successful molecular monotherapies for these conditions. Cell-based therapies, in which cellular responses to microenvironmental cues regulate the injury response, might provide novel and useful approaches to the treatment of these diseases. With the recent demonstration that adult bone marrow–derived progenitor cells selectively localize to activated glial cells (103), it might be possible to use such cells to modulate the pathological responses in the eye or brain by using rescue activities of the cells themselves (Figure 2) or by delivering transgenes expressing static or trophic molecules useful in the treatment of fibroproliferative disorders.
Figure 2. Cell-based therapy for the treatment of ischemic retinopathies.

Bone marrow–derived progenitor cells are injected into the posterior segment of the eye, where the injected cells localize to activated astrocytes. Both endothelial and myeloid progenitor cells localize to forming vasculature, where they can differentiate into endothelial cells or microglial cells, each participating in vasculotrophic rescue of ischemic blood vessels. Top far right, control-treated eye; bottom far right, progenitor cell–treated eye. Images reproduced from Nature Medicine (103) and Journal of Clinical Investigation (113).
Acknowledgments
I am very grateful to the members of my laboratory and many colleagues at Scripps and other institutions, especially Gunter Blobel, who have challenged me to think critically and move in uncharted directions over the years. Matt Ritter, Mike Dorrell, Eyal Banin, Sheila Friedlander, and Alan Bird provided many helpful and critical comments in the preparation of this manuscript; Ruth Jacobson was a great help in researching the literature. The research cited in this work from our laboratory has been generously supported by the National Eye Institute of the NIH, the MacTel Foundation, the V. Kann Rassmusen Foundation, the Scripps Fonseca/Mericos Fund, and Merck KGaA.
Footnotes
Nonstandard abbreviations used: ARMD, age-related macular degeneration; CNTF, ciliary neurotrophic factor; DR, diabetic retinopathy; EPC, endothelial progenitor cell; PEX, carboxyterminal, noncatalytic domain of MMP-2; ROP, retinopathy of prematurity; RPE, retinal pigmented epithelium; TIMP, tissue inhibitor of metalloproteinases.
Conflict of interest: The author has declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 117:576–586 (2007). doi:10.1172/JCI31030.
References
- 1.Dorrell M.I., Aguilar E., Friedlander M. Retinal vascular development is mediated by endothelial filopodia, a preexisting astrocytic template and specific R-cadherin adhesion. Invest. Ophthalmol. Vis. Sci. 2002;43:3500–3510. [PubMed] [Google Scholar]
- 2.Dorrell M.I., Friedlander M. Mechanisms of endothelial cell guidance and vascular patterning in the developing mouse retina. Prog. Retin. Eye Res. 2006;25:277–295. doi: 10.1016/j.preteyeres.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Gilbert C.D., Wiesel T.N. Intrinsic connectivity and receptive field properties in visual cortex. Vision Res. 1985;25:365–374. doi: 10.1016/0042-6989(85)90061-6. [DOI] [PubMed] [Google Scholar]
- 4.Ferrer E. Trabecular meshwork as a new target for the treatment of glaucoma. Drug News Perspect. 2006;19:151–158. doi: 10.1358/dnp.2006.19.3.985929. [DOI] [PubMed] [Google Scholar]
- 5.Fautsch M.P., Johnson D.H. Aqueous humor outflow: what do we know? Where will it lead us? Invest. Ophthalmol. Vis. Sci. 2006;47:4181–4187. doi: 10.1167/iovs.06-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambati B.K., et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cotsarelis G., Cheng S.Z., Dong G., Sun T.T., Lavker R.M. Existence of slow-cycling limbal epithelial basal cells that can be preferentially stimulated to proliferate: implications on epithelial stem cells. Cell. 1989;57:201–209. doi: 10.1016/0092-8674(89)90958-6. [DOI] [PubMed] [Google Scholar]
- 8.Tsubota K., et al. Treatment of severe ocular-surface disorders with corneal epithelial stem-cell transplantation. N. Engl. J. Med. 1999;340:1697–1703. doi: 10.1056/NEJM199906033402201. [DOI] [PubMed] [Google Scholar]
- 9.Di Girolamo N., Chui J., Coroneo M.T., Wakefield D. Pathogenesis of pterygia: role of cytokines, growth factors, and matrix metalloproteinases. Prog. Retin. Eye Res. 2004;23:195–228. doi: 10.1016/j.preteyeres.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Fini M.E. Keratocyte and fibroblast phenotypes in the repairing cornea. Prog. Retin. Eye Res. 1999;18:529–551. doi: 10.1016/s1350-9462(98)00033-0. [DOI] [PubMed] [Google Scholar]
- 11.Lavker R.M., Tseng S.C., Sun T.T. Corneal epithelial stem cells at the limbus: looking at some old problems from a new angle. Exp. Eye Res. 2004;78:433–446. doi: 10.1016/j.exer.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Bishop P.N. Structural macromolecules and supramolecular organisation of the vitreous gel. Prog. Retin. Eye Res. 2000;19:323–344. doi: 10.1016/s1350-9462(99)00016-6. [DOI] [PubMed] [Google Scholar]
- 13.Fruttiger M. Development of the mouse retinal vasculature: angiogenesis versus vasculogenesis. Invest. Ophthalmol. Vis. Sci. 2002;43:522–527. [PubMed] [Google Scholar]
- 14.Fruttiger M., et al. PDGF mediates a neuron-astrocyte interaction in the developing retina. Neuron. 1996;17:1117–1131. doi: 10.1016/s0896-6273(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 15.Bringmann A., et al. Muller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Bok D. The retinal pigment epithelium: a versatile partner in vision. J. Cell Sci. Suppl. 1993;17:189–195. doi: 10.1242/jcs.1993.supplement_17.27. [DOI] [PubMed] [Google Scholar]
- 17.Crabb J.W., et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rakoczy P.E., Yu M.J., Nusinowitz S., Chang B., Heckenlively J.R. Mouse models of age-related macular degeneration. Exp. Eye Res. 2006;82:741–752. doi: 10.1016/j.exer.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Michels S., Schmidt-Erfurth U., Rosenfeld P.J. Promising new treatments for neovascular age-related macular degeneration. Expert Opin. Investig. Drugs. 2006;15:779–793. doi: 10.1517/13543784.15.7.779. [DOI] [PubMed] [Google Scholar]
- 20.Dorrell M.I., Uusitalo-Jarvinen H., Aguilar E., Friedlander M. Ocular neovascularization; basic mechanisms and therapeutic advances. Surv. Ophthalmol. 2007;52(Suppl. 1):S3–S19. doi: 10.1016/j.survophthal.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 21.Wong T.Y., et al. Diabetic retinopathy in a multi-ethnic cohort in the United States. Am. J. Ophthalmol. 2006;141:446–455. doi: 10.1016/j.ajo.2005.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harding S. Diabetic retinopathy. Clin. Evid. . 2006;15:900–907. [PubMed] [Google Scholar]
- 23.Cogan D.G. Congenital anomalies of the retina. Birth Defects Orig. Artic. Ser. 1971;7:41–51. [PubMed] [Google Scholar]
- 24.Penn J.S., Tolman B.L., Lowery L.A. Variable oxygen exposure causes preretinal neovascularization in the newborn rat. Invest. Ophthalmol. Vis. Sci. 1993;34:576–585. [PubMed] [Google Scholar]
- 25.Iandiev I., et al. Glial cell reactivity in a porcine model of retinal detachment. Invest. Ophthalmol. Vis. Sci. 2006;47:2161–2171. doi: 10.1167/iovs.05-0595. [DOI] [PubMed] [Google Scholar]
- 26.Smith L.E., et al. Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- 27.Shing Y., et al. Heparin affinity: purification of a tumor-derived capillary endothelial cell growth factor. Science. 1984;223:1296–1299. doi: 10.1126/science.6199844. [DOI] [PubMed] [Google Scholar]
- 28.Folkman J. The role of angiogenesis in tumor growth. Semin. Cancer Biol. 1992;3:65–71. [PubMed] [Google Scholar]
- 29.Ferrara N., Houck K., Jakeman L., Leung D.W. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr. Rev. 1992;13:18–32. doi: 10.1210/edrv-13-1-18. [DOI] [PubMed] [Google Scholar]
- 30.Fett J.W., et al. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry. 1985;24:5480–5486. doi: 10.1021/bi00341a030. [DOI] [PubMed] [Google Scholar]
- 31.Antonelli-Orlidge A., Saunders K.B., Smith S.R., D’Amore P.A. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc. Natl. Acad. Sci. U. S. A. 1989;86:4544–4548. doi: 10.1073/pnas.86.12.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brem R.B., et al. Immunolocalization of integrins in the human retina. Invest. Ophthalmol. Vis. Sci. 1994;35:3466–3474. [PubMed] [Google Scholar]
- 33.Beutler B., Cerami A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature. 1986;320:584–588. doi: 10.1038/320584a0. [DOI] [PubMed] [Google Scholar]
- 34.Ishikawa F., et al. Identification of angiogenic activity and the cloning and expression of platelet-derived endothelial cell growth factor. Nature. 1989;338:557–562. doi: 10.1038/338557a0. [DOI] [PubMed] [Google Scholar]
- 35.Ezekowitz R.A., Mulliken J.B., Folkman J. Interferon alfa-2a therapy for life-threatening hemangiomas of infancy. N. Engl. J. Med. 1992;326:1456–1463. doi: 10.1056/NEJM199205283262203. [DOI] [PubMed] [Google Scholar]
- 36.D’Amato R.J., Loughnan M.S., Flynn E., Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 1994;91:4082–4085. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tolsma S.S., et al. Peptides derived from two separate domains of the matrix protein thrombospondin-1 have anti-angiogenic activity. J. Cell Biol. 1993;122:497–511. doi: 10.1083/jcb.122.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Derynck R. Transforming growth factor-alpha. Mol. Reprod. Dev. 1990;27:3–9. doi: 10.1002/mrd.1080270104. [DOI] [PubMed] [Google Scholar]
- 39.O’Reilly M.S., et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 40.Brooks P.C., Silletti S., von Schalscha T.L., Friedlander M., Cheresh D.A. Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell. 1998;92:391–400. doi: 10.1016/s0092-8674(00)80931-9. [DOI] [PubMed] [Google Scholar]
- 41.Wakasugi K., et al. A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 2002;99:173–177. doi: 10.1073/pnas.012602099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otani A., et al. A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 2002;99:178–183. doi: 10.1073/pnas.012601899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dawson D.W., et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285:245–248. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- 44.D’Amore P.A. Mechanisms of retinal and choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 1994;35:3974–3979. [PubMed] [Google Scholar]
- 45.Leavesley D.I., Schwartz M.A., Rosenfeld M., Cheresh D.A. Integrin beta 1- and beta 3-mediated endothelial cell migration is triggered through distinct signaling mechanisms. J. Cell Biol. 1993;121:163–170. doi: 10.1083/jcb.121.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks P.C., Clark R.A., Cheresh D.A. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 47.Friedlander M., et al. Definition of two angiogenic pathways by distinct alpha v integrins. Science. 1995;270:1500–1502. doi: 10.1126/science.270.5241.1500. [DOI] [PubMed] [Google Scholar]
- 48.Kim S., Harris M., Varner J.A. Regulation of integrin alpha vbeta 3-mediated endothelial cell migration and angiogenesis by integrin alpha5beta1 and protein kinase A. J. Biol. Chem. 2000;275:33920–33928. doi: 10.1074/jbc.M003668200. [DOI] [PubMed] [Google Scholar]
- 49.Friedlander M., et al. Involvement of integrins alpha v beta 3 and alpha v beta 5 in ocular neovascular diseases. Proc. Natl. Acad. Sci. U. S. A. 1996;93:9764–9769. doi: 10.1073/pnas.93.18.9764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elner S.G., Elner V.M. The integrin superfamily and the eye. Invest. Ophthalmol. Vis. Sci. 1996;37:696–701. [PubMed] [Google Scholar]
- 51.Fini M.E., Girard M.T. Expression of collagenolytic/gelatinolytic metalloproteinases by normal cornea. Invest. Ophthalmol. Vis. Sci. 1990;31:1779–1788. [PubMed] [Google Scholar]
- 52.Brown D., Hamdi H., Bahri S., Kenney M.C. Characterization of an endogenous metalloproteinase in human vitreous. Curr. Eye Res. 1994;13:639–647. doi: 10.3109/02713689408999899. [DOI] [PubMed] [Google Scholar]
- 53.Jones B.E., et al. Characterization and novel activation of 72-kDa metalloproteinase in retinal interphotoreceptor matrix and Y-79 cell culture medium. Exp. Eye Res. 1994;59:257–269. doi: 10.1006/exer.1994.1107. [DOI] [PubMed] [Google Scholar]
- 54.Alexander J.P., Bradley J.M., Gabourel J.D., Acott T.S. Expression of matrix metalloproteinases and inhibitor by human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 1990;31:2520–2528. [PubMed] [Google Scholar]
- 55.Arias-Salgado E.G., et al. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13298–13302. doi: 10.1073/pnas.2336149100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weick M., Wiedemann P., Reichenbach A., Bringmann A. Resensitization of P2Y receptors by growth factor-mediated activation of the phosphatidylinositol-3 kinase in retinal glial cells. Invest. Ophthalmol. Vis. Sci. 2005;46:1525–1532. doi: 10.1167/iovs.04-0417. [DOI] [PubMed] [Google Scholar]
- 57.Alavi A., Hood J.D., Frausto R., Stupack D.G., Cheresh D.A. Role of Raf in vascular protection from distinct apoptotic stimuli. Science. 2003;301:94–96. doi: 10.1126/science.1082015. [DOI] [PubMed] [Google Scholar]
- 58.Miller J.W., et al. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am. J. Pathol. 1994;145:574–584. [PMC free article] [PubMed] [Google Scholar]
- 59.Aiello L.P. Vascular endothelial growth factor and the eye: biochemical mechanisms of action and implications for novel therapies. Ophthalmic Res. 1997;29:354–362. doi: 10.1159/000268033. [DOI] [PubMed] [Google Scholar]
- 60.Aiello L.P., et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 61.Alon T., et al. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat. Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- 62.Amano S., Rohan R., Kuroki M., Tolentino M., Adamis A.P. Requirement for vascular endothelial growth factor in wound- and inflammation-related corneal neovascularization. Invest. Ophthalmol. Vis. Sci. 1998;39:18–22. [PubMed] [Google Scholar]
- 63.Hanneken A., et al. Altered distribution of basic fibroblast growth factor in diabetic retinopathy. Arch. Ophthalmol. 1991;109:1005–1011. doi: 10.1001/archopht.1991.01080070117048. [DOI] [PubMed] [Google Scholar]
- 64.Kitaoka T., Morse L.S., Schneeberger S., Ishigooka H., Hjelmeland L.M. Expression of FGF5 in choroidal neovascular membranes associated with ARMD. Curr. Eye Res. 1997;16:396–399. doi: 10.1076/ceyr.16.4.396.10685. [DOI] [PubMed] [Google Scholar]
- 65.Rousseau B., Larrieu-Lahargue F., Bikfalvi A., Javerzat S. Involvement of fibroblast growth factors in choroidal angiogenesis and retinal vascularization. Exp. Eye Res. 2003;77:147–156. doi: 10.1016/s0014-4835(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 66.Mordenti J., et al. Efficacy and concentration-response of murine anti-VEGF monoclonal antibody in tumor-bearing mice and extrapolation to humans. Toxicol. Pathol. 1999;27:14–21. doi: 10.1177/019262339902700104. [DOI] [PubMed] [Google Scholar]
- 67.Drolet D.W., et al. Pharmacokinetics and safety of an anti-vascular endothelial growth factor aptamer (NX1838) following injection into the vitreous humor of rhesus monkeys. Pharm. Res. 2000;17:1503–1510. doi: 10.1023/a:1007657109012. [DOI] [PubMed] [Google Scholar]
- 68.Seo M.S., et al. Dramatic inhibition of retinal and choroidal neovascularization by oral administration of a kinase inhibitor. Am. J. Pathol. 1999;154:1743–1753. doi: 10.1016/S0002-9440(10)65430-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hammes H.P., Brownlee M., Jonczyk A., Sutter A., Preissner K.T. Subcutaneous injection of a cyclic peptide antagonist of vitronectin receptor-type integrins inhibits retinal neovascularization. Nat. Med. 1996;2:529–533. doi: 10.1038/nm0596-529. [DOI] [PubMed] [Google Scholar]
- 70.Luna J., Tobe T., Mousa S.A., Reilly T.M., Campochiaro P.A. Antagonists of integrin alpha v beta 3 inhibit retinal neovascularization in a murine model. Lab. Invest. 1996;75:563–573. [PubMed] [Google Scholar]
- 71.Boger D.L., Goldberg J., Silletti S., Kessler T., Cheresh D.A. Identification of a novel class of small-molecule antiangiogenic agents through the screening of combinatorial libraries which function by inhibiting the binding and localization of proteinase MMP2 to integrin alpha(V)beta(3). . J. Am. Chem. Soc. 2001;123:1280–1288. doi: 10.1021/ja003579+. [DOI] [PubMed] [Google Scholar]
- 72.Silletti S., Kessler T., Goldberg J., Boger D.L., Cheresh D.A. Disruption of matrix metalloproteinase 2 binding to integrin alpha vbeta 3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc. Natl. Acad. Sci. U. S. A. 2001;98:119–124. doi: 10.1073/pnas.011343298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Das A., McGuire P.G. Retinal and choroidal angiogenesis: pathophysiology and strategies for inhibition. Prog. Retin. Eye Res. 2003;22:721–748. doi: 10.1016/j.preteyeres.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 74.Gupta A.R., et al. Strain-dependent anterior segment neovascularization following intravitreal gene transfer of basic fibroblast growth factor (bFGF). J. Gene Med. 2001;3:252–259. doi: 10.1002/1521-2254(200105/06)3:3<252::AID-JGM185>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 75.Crum R., Szabo S., Folkman J. A new class of steroids inhibits angiogenesis in the presence of heparin or a heparin fragment. Science. 1985;230:1375–1378. doi: 10.1126/science.2416056. [DOI] [PubMed] [Google Scholar]
- 76.Clark A.F., et al. Inhibition of intraocular tumor growth by topical application of the angiostatic steroid anecortave acetate. Invest. Ophthalmol. Vis. Sci. 1999;40:2158–2162. [PubMed] [Google Scholar]
- 77.Penn J.S., Rajaratnam V.S., Collier R.J., Clark A.F. The effect of an angiostatic steroid on neovascularization in a rat model of retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 2001;42:283–290. [PubMed] [Google Scholar]
- 78.Qi J.H., et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat. Med. 2003;9:407–415. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 79.Grant M.B., Caballero S., Millard W.J. Inhibition of IGF-I and b-FGF stimulated growth of human retinal endothelial cells by the somatostatin analogue, octreotide: a potential treatment for ocular neovascularization. Regul. Pept. 1993;48:267–278. doi: 10.1016/0167-0115(93)90356-d. [DOI] [PubMed] [Google Scholar]
- 80.Danesi R., et al. Inhibition of experimental angiogenesis by the somatostatin analogue octreotide acetate (SMS 201-995). Clin. Cancer Res. 1997;3:265–272. [PubMed] [Google Scholar]
- 81.Smith L.E., et al. Essential role of growth hormone in ischemia-induced retinal neovascularization. Science. 1997;276:1706–1709. doi: 10.1126/science.276.5319.1706. [DOI] [PubMed] [Google Scholar]
- 82.Bressler N.M., et al. Verteporfin therapy for subfoveal choroidal neovascularization in age-related macular degeneration: four-year results of an open-label extension of 2 randomized clinical trials: TAP Report No. 7. Arch. Ophthalmol. 2005;123:1283–1285. doi: 10.1001/archopht.123.9.1283. [DOI] [PubMed] [Google Scholar]
- 83.Rosenfeld P.J., et al. Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2006;355:1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- 84.Dorrell M.I., Aguilar E., Scheppke L., Barnett F.H., Friedlander M. Combination angiostatic therapy completely inhibits ocular and tumor angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:967–972. doi: 10.1073/pnas.0607542104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fischer A.J., et al. Different aspects of gliosis in retinal Muller glia can be induced by CNTF, insulin, and FGF2 in the absence of damage. Mol. Vis. 2004;10:973–986. [PubMed] [Google Scholar]
- 86.Dyer M.A., Cepko C.L. Control of Muller glial cell proliferation and activation following retinal injury. Nat. Neurosci. 2000;3:873–880. doi: 10.1038/78774. [DOI] [PubMed] [Google Scholar]
- 87.Honjo M., et al. Expression of ciliary neurotrophic factor activated by retinal Muller cells in eyes with NMDA- and kainic acid-induced neuronal death. Invest. Ophthalmol. Vis. Sci. 2000;41:552–560. [PubMed] [Google Scholar]
- 88.Strauss O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 89.Campochiaro P.A. Cytokine production by retinal pigmented epithelial cells. Int. Rev. Cytol. 1993;146:75–82. doi: 10.1016/s0074-7696(08)60380-0. [DOI] [PubMed] [Google Scholar]
- 90.Folkman J. Angiogenesis. Annu. Rev. Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 91.Korbling M., Estrov Z. Adult stem cells for tissue repair - a new therapeutic concept? . N. Engl. J. Med. 2003;349:570–582. doi: 10.1056/NEJMra022361. [DOI] [PubMed] [Google Scholar]
- 92.Cahill M.T., Freedman S.F., Toth C.A. Macular translocation with 360 degrees peripheral retinectomy for geographic atrophy. Arch. Ophthalmol. 2003;121:132–133. doi: 10.1001/archopht.121.1.132. [DOI] [PubMed] [Google Scholar]
- 93.Lai J.C., et al. Visual outcomes following macular translocation with 360-degree peripheral retinectomy. Arch. Ophthalmol. 2002;120:1317–1324. doi: 10.1001/archopht.120.10.1317. [DOI] [PubMed] [Google Scholar]
- 94.Huang J.C., Ishida M., Hersh P., Sugino I.K., Zarbin M.A. Preparation and transplantation of photoreceptor sheets. Curr. Eye Res. 1998;17:573–585. [PubMed] [Google Scholar]
- 95.Ishida M., Lui G.M., Yamani A., Sugino I.K., Zarbin M.A. Culture of human retinal pigment epithelial cells from peripheral scleral flap biopsies. Curr. Eye Res. 1998;17:392–402. doi: 10.1080/02713689808951220. [DOI] [PubMed] [Google Scholar]
- 96.Dandekar S.S., et al. Autofluorescence imaging of choroidal neovascularization due to age-related macular degeneration. Arch. Ophthalmol. 2005;123:1507–1513. doi: 10.1001/archopht.123.11.1507. [DOI] [PubMed] [Google Scholar]
- 97.Tao W., et al. Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2002;43:3292–3298. [PubMed] [Google Scholar]
- 98.Bush R.A., et al. Encapsulated cell-based intraocular delivery of ciliary neurotrophic factor in normal rabbit: dose-dependent effects on ERG and retinal histology. Invest. Ophthalmol. Vis. Sci. 2004;45:2420–2430. doi: 10.1167/iovs.03-1342. [DOI] [PubMed] [Google Scholar]
- 99.Tao W. Application of encapsulated cell technology for retinal degenerative diseases. Expert Opin. Biol. Ther. 2006;6:717–726. doi: 10.1517/14712598.6.7.717. [DOI] [PubMed] [Google Scholar]
- 100.Klimanskaya I., et al. Derivation and comparative assessment of retinal pigment epithelium from human embryonic stem cells using transcriptomics. Cloning Stem Cells. 2004;6:217–245. doi: 10.1089/clo.2004.6.217. [DOI] [PubMed] [Google Scholar]
- 101.Lund R.D., et al. Human embryonic stem cell-derived cells rescue visual function in dystrophic RCS rats. Cloning Stem Cells. 2006;8:189–199. doi: 10.1089/clo.2006.8.189. [DOI] [PubMed] [Google Scholar]
- 102.Jin H., et al. A homing mechanism for bone marrow-derived progenitor cell recruitment to the neovasculature. J. Clin. Invest. 2006;116:652–662. doi: 10.1172/JCI24751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Otani A., et al. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nat. Med. 2002;8:1004–1010. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- 104.Otani A., et al. Rescue of retinal degeneration by intravitreally injected adult bone marrow-derived lineage negative hematopoietic stem cells. J. Clin. Invest. 2004;114:765–774. doi: 10.1172/JCI200421686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Krause D.S. Plasticity of marrow-derived stem cells. Gene Ther. 2002;9:754–758. doi: 10.1038/sj.gt.3301760. [DOI] [PubMed] [Google Scholar]
- 106.Blau H., Brazelton T., Keshet G., Rossi F. Something in the eye of the beholder. Science. 2002;298:361–362. doi: 10.1126/science.298.5592.361c. [DOI] [PubMed] [Google Scholar]
- 107.Barber C.L., Iruela-Arispe M.L. The ever-elusive endothelial progenitor cell: identities, functions and clinical implications. Pediatr. Res. 2006;59:26R–32R. doi: 10.1203/01.pdr.0000203553.46471.18. [DOI] [PubMed] [Google Scholar]
- 108.Grant M.B., et al. The contribution of adult hematopoietic stem cells to retinal neovascularization. Adv. Exp. Med. Biol. 2003;522:37–45. doi: 10.1007/978-1-4615-0169-5_5. [DOI] [PubMed] [Google Scholar]
- 109.Espinosa-Heidmann D.G., Caicedo A., Hernandez E.P., Csaky K.G., Cousins S.W. Bone marrow-derived progenitor cells contribute to experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2003;44:4914–4919. doi: 10.1167/iovs.03-0371. [DOI] [PubMed] [Google Scholar]
- 110.Sengupta N., et al. The role of adult bone marrow-derived stem cells in choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 2003;44:4908–4913. doi: 10.1167/iovs.03-0342. [DOI] [PubMed] [Google Scholar]
- 111.Csaky K.G., et al. Recruitment of marrow-derived endothelial cells to experimental choroidal neovascularization by local expression of vascular endothelial growth factor. Exp. Eye Res. 2004;78:1107–1116. doi: 10.1016/j.exer.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 112.Dorrell M.I., Otani A., Aguilar E., Moreno S.K., Friedlander M. Adult bone marrow-derived stem cells utilize R-cadherin to target sites of neovascularization in the developing retina. Blood. 2004;103:3420–3427. doi: 10.1182/blood-2003-09-3012. [DOI] [PubMed] [Google Scholar]
- 113.Ritter M.R., et al. Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy. J. Clin. Invest. 2006;116:3266–3276. doi: 10.1172/JCI29683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Quaini F., et al. Chimerism of the transplanted heart. N. Engl. J. Med. 2002;346:5–15. doi: 10.1056/NEJMoa012081. [DOI] [PubMed] [Google Scholar]
- 115.Asahara T., et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 116.Hill J.M., et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 117.Werner N., et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N. Engl. J. Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 118.Kawamoto A., et al. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation. 2001;103:634–637. doi: 10.1161/01.cir.103.5.634. [DOI] [PubMed] [Google Scholar]
- 119.Grunewald M., et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–189. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 120.Bailey A.S., et al. Myeloid lineage progenitors give rise to vascular endothelium. Proc. Natl. Acad. Sci. U. S. A. 2006;103:13156–13161. doi: 10.1073/pnas.0604203103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nakazawa T., et al. Monocyte chemoattractant protein–1 mediates retinal detachment–induced photoreceptor apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:2425–2430. doi: 10.1073/pnas.0608167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. . Sarraf, D. 2001. A slide-script program: diabetes and eye disease [slide]. American Academy of Ophthalmology. San Francisco, California, USA. [Google Scholar]