

Abstract
Lipocalin-type prostaglandin D2 synthase (L-PGDS) is a highly glycosylated protein found in several body fluids. Elevated L-PGDS levels have been observed in the serum of patients with renal impairment, diabetes mellitus, and hypertension. Recently, we demonstrated the ability of L-PGDS to induce apoptosis in a variety of cell types including epithelial cells, neuronal cells, and vascular smooth muscle cells (VSMCs). The aim of this study was to investigate the effect several site-directed mutations had on L-PGDS-induced apoptosis in order to identify potential sites of regulation. Point mutations created in a glycosylation site (Asn51), a protein kinase C phosphorylation site (Ser106), and the enzymatic active site (Cys65) all inhibited L-PGDS-induced apoptosis as determined by both terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) and caspase-3 activity. We also compared the L-PGDS isoforms present in GK rat serum to WKY control serum using two-dimensional gel electrophoresis and observed distinct differences which vanished after PNGase F glycolytic digestion. We conclude that post-translational modification of L-PGDS, by either glycosylation or phosphorylation, enhances its apoptotic activity and inhibits VSMC hyperproliferation and postulate that this process is altered in type 2 diabetes.
Introduction
Lipocalin-type prostaglandin D2 synthase (L-PGDS) is a highly glycosylated protein found in cerebrospinal fluid (CSF) (~25 mg/l), blood (~0.4 mg/l), urine (~1.5 mg/l), and seminal fluid (~150 mg/l) 1. Certain isoforms have been reported to be prominent in the CSF of patients with neurological disorders 2–4. In addition, a variety of cardiovascular implications have been associated with L-PGDS 1, 5–10 or it’s enzymatic product, prostaglandin (PG) D2 11–14. In fact, we recently observed accelerated cardiovascular disease, impaired glucose tolerance, and insulin resistance in L-PGDS knockout mice 15.
Goto-Kakizaki (GK) rats are a non-obese model for type 2 diabetes exhibiting defects in glucose-stimulated insulin secretion, peripheral insulin resistance, hyperglycemia, hypertension and hyperinsulinemia as early as four weeks after birth. Previously, we demonstrated that in vitro L-PGDS exposure inhibited the exaggerated growth phenotype of VSMCs isolated from diabetic GK rats via the stimulation of apoptosis 16. In addition, we determined that phorbol ester-induced apoptosis was mediated by L-PGDS phosphorylation and was accompanied by inhibition of the phosphatidylinositol 3-kinase (PI3-K) and protein kinase B (Akt) anti-apoptotic signaling pathways 17. We have also reported on the differential effects of L-PGDS on VSMC cell cycle progression, migration, and apoptosis in wildtype VSMCs versus those from a type 2 diabetic model and have demonstrated that L-PGDS retards cell cycle progression and migration of wildtype VSMCs 18.
The aim of this study was to investigate the several site-directed mutations had on L-PGDS-induced apoptosis in order to identify potential sites of regulation. Point mutations were created in potential glycosylation sites, a possible protein kinase C (PKC) phosphorylation site, and the active site of L-PGDS. Apoptosis was determined by both caspase-3 activity and terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL). L-PGDS enzymatic activity of the altered proteins was also investigated. Finally, we examined the L-PGDS isoforms present in the serum of GK rats and compared them to controls using two-dimensional gel electrophoresis. We report on the loss of L-PGDS-induced apoptotic activity with mutations in a glycosylation site (Asn51), a protein kinase C phosphorylation site (Ser106), and the enzymatic active site (Cys65). We propose that post-translational modification of L-PGDS augment its apoptotic activity and enhance its ability to inhibit VSMC hyperplasia which may help protect blood vessels against atherosclerosis.
Materials and Methods
Cell culture reagents, including fetal bovine serum, were purchased from Life Technologies (Grand Island, NY). Electrophoresis reagents were from Bio-Rad (Richmond, CA). Restriction endonucleases were from Roche Molecular Biochemicals (Indianapolis, IN). The QuikChange® Site-Directed Mutagenesis Kit was purchased from Stratagene (La Jolla, CA). The prokaryotic expression vector pRSET was purchased from Invitrogen (San Diego, CA). Bicinchoninic acid protein assay reagent was purchased from Pierce (Rockford, IL). The caspase3 activity apoptotic detection kit was purchased from R&D (Minneapolis, MN). L-PGDS enzymatic activity was determined using a PGD2 methyl-oxylamine hydrochloride (MOX) kit (Cayman Chemical Co., Ann Arbor, MI). All other reagents were of reagent grade or better and purchased from the Sigma Chemical Co (St. Louis, MO).
Cell Culture
VSMCs were isolated by collagenase digestion of the aortic media from male Wistar-Kyoto (WKY) rats and Goto-Kakizaki (GK) diabetic rats with body weights between 200–220 g, as described previously 16, 19. VSMCs prepared from these rats were not contaminated with fibroblasts or endothelial cells as evidenced by a greater than 99% positive immunostaining of smooth muscle α-actin with fluorescein isothiocyanate-conjugated α-actin antibody. Subcultures of VSMCs were maintained in α-MEM containing 10% FBS, and 50 U/ml penicillin and 50 μg/ml streptomycin (Life Technologies, Gaithersburg, MD) at 37°C in 5% CO2. Cells were synchronized in G0 by incubating in serum-free medium for 24 h. Cells were grown to confluency and studied at passages 5–6 for all experiments.
Cell Proliferation
Cell proliferation was assessed by counting cell number. After the specified incubation period, cells were washed twice with ice-cold PBS, harvested with trypsin, and centrifuged. Cell pellets were re-suspended in ice-cold PBS and counted using a hemocytometer.
Two-Dimensional Gel Electrophoresis
Protein samples (5.0 μg) were resolved by isoelectric focusing on 11-cm immobilized pH (3–10) gradient strips using a Protean IEF Cell as per the manufacturer’s instructions (Bio Rad, Hercules CA). Second dimension separation was performed by electrophoresis on a 10–20% gradient SDS-polyacrylamide gel in a Criterion™ Cell (Bio Rad). The gels were analyzed by immunoblotting with anti-L-PGDS antibody (1:1000) as previously described 18. Where indicated proteins were digested with PNGase F (New England Biolabs, Ipswich, MA) as per the manufacturer’s directions.
Site-directed Mutagenesis
Point mutations of two prospective glycosylation sites (Gly1 and Gly2), an apparent protein kinase C phosphorylation site (PKC), and the enzymatic active site (ACT) were created using the QuikChange® Site-Directed Mutagenesis Kit as per the manufacturer’s instructions. Gly1 (Asn51), Gly2 (Asn78), and the potential PKC phosphorylation site (Ser106) were all exchanged for alanine, and the ACT (Cys65) mutation was created by exchanging Cys65 with a glycine residue (Figure 3). All mutations were confirmed by sequence analysis and the mutated L-PGDS genes subcloned into the prokaryotic expression vector pRSETA for bacterial overexpression. Mutant L-PGDS proteins were purified as previously described 20.
Figure 3. Effect of Mutation on L-PGDS-induced apoptosis.
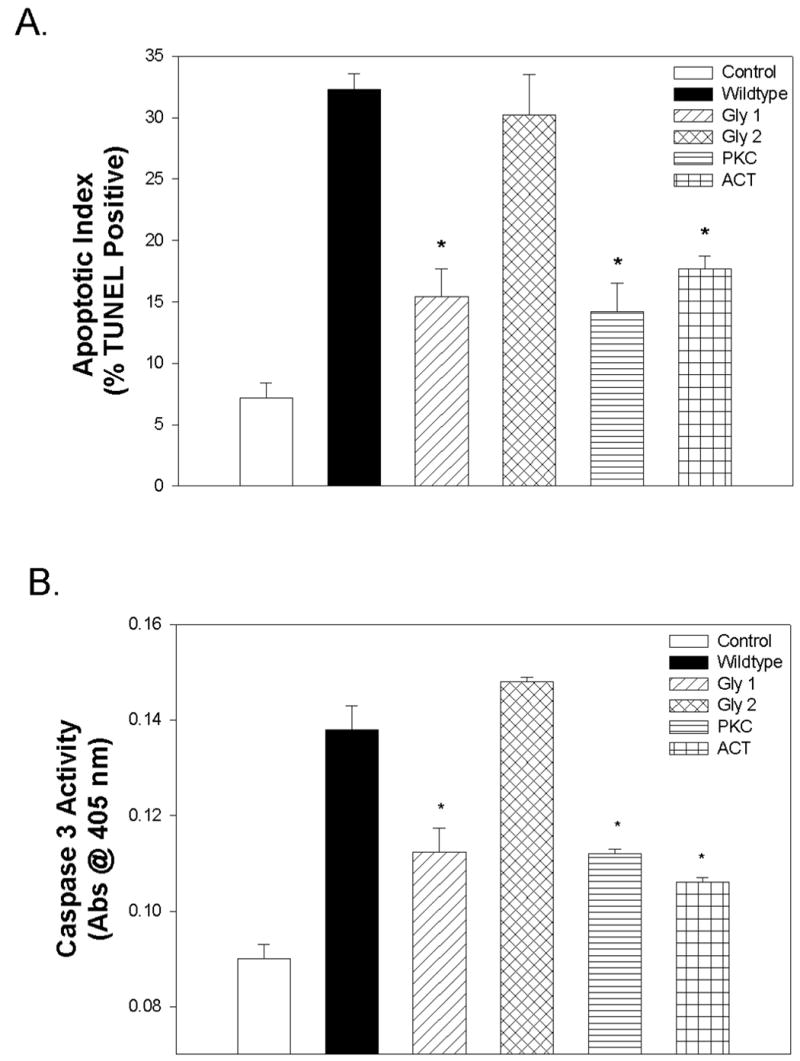
VSMCs were cultured as described in Materials and Methods. At approximately 100% confluency the cells, where indicated, were treated with L-PGDS (50 μg/ml) for 18h, and the apoptotic index calculated by TUNEL assay (Panel A) or Caspase3 assay (Panel B). Results are the mean ± SEM of three different experiments each performed in duplicate. Asterisks (*) represents a P<0.05 versus GK controls.
L-PGDS Protein Expression and Purification
Both wild type and mutant L-PGDS were isolated and purified from E. coli over-expressing the respective proteins. The cDNA for human L-PGDS was obtained from the ATCC (#2519499), verified by sequence analysis (data not shown), and inserted in-frame into the BamHI site of vector pRSETA by standard techniques, yielding plasmid pRSETA/PGDS. This vector is part of the XpressJ protein expression system (Invitrogen, Carlsbad, CA) which is designed for high-level prokaryotic expression controlled by a T7 promoter. Expression of the inserted gene is via the induction of T7 polymerase with 2.0 mM isopropyl-β-thio-D-galactopyranoside (IPTG) in BL21(DE3) E. coli after cells reach mid-log phase. Purification of the protein was facilitated by the polyhistidine tag at the amino terminus of the L-PGDS fusion protein and the ProBondJ resin included in the kit. Finally, the polyhistidine tag was removed via enterokinase digestion, and the resulting 21 kDa protein confirmed by western analysis (data not shown) using antibodies against L-PGDS as previously described 18.
Apoptotic Activity
Apoptosis was quantitated by either TUNEL staining or by the measurement of caspase3 activity as per the manufacturer’s instructions, as previously described 20, 21.
L-PGDS Enzymatic Activity
L-PGDS enzymatic activity was determined using a PG D2-MOX enzyme immunoassay kit. Cells were treated as described and lysed by freeze-thawing in 0.1M Tris-HCl (pH 7.5) and 1mM phenylmethylsulfonyl fluoride. Equal amounts of whole-cell lysates were used as the enzyme source. The reaction mixture contained 0.1 M Tris-HCl (pH 7.5), 1.0 mM β–mercaptoethanol, 40 :M PG H2 as the substrate, and cell extracts in a final volume of 50 μl. The reaction was initiated by the addition of the substrate to the reaction mixture, incubated for 1 min. at 25°C, and terminated by heating to 100°C for 10 min. The resulting PG D2 product in the reaction mixture was quantitated as per the manufacturer’s instructions.
Protein Assay
Protein content of cell extracts were determined with bicinchoninic acid 22.
Statistics
Results are expressed as means +/− S.E.M. of at least three independent experiments each performed in duplicate at different times. Paired student’s ‘t’ tests were used to compare experimental preparations with controls. A value of P≤0.05 was deemed significant.
RESULTS
Two-dimensional gel electrophoresis of L-PGDS isoforms
Since L-PGDS is known to have many isoforms with varying degrees of glycosylation we decided to examine the various species found in the serum of control (WKY) and diabetic (GK) rats. The isoelectric pattern of affinity purified L-PGDS was distinctly different in WKY when compared to GK (Fig. 1A). Figure 1B was generated by superimposing the two gel images in Photoshop® CS and highlighting the differences in orange. It was clear that three isoforms at an isoelectric point of approximately seven and above were present in WKY and absent in GK. We then decided to digest both L-PGDS pools with PNGase F to remove the glycosylated residues. The results can be seen in Fig. 1C where the major acidic isoforms of WKY-derived L-PGDS had been lost and appear more closely aligned with the isoforms isolated from GK serum.
Figure 1. Two-dimensional gel electrophoresis of L-PGDS Isoforms.
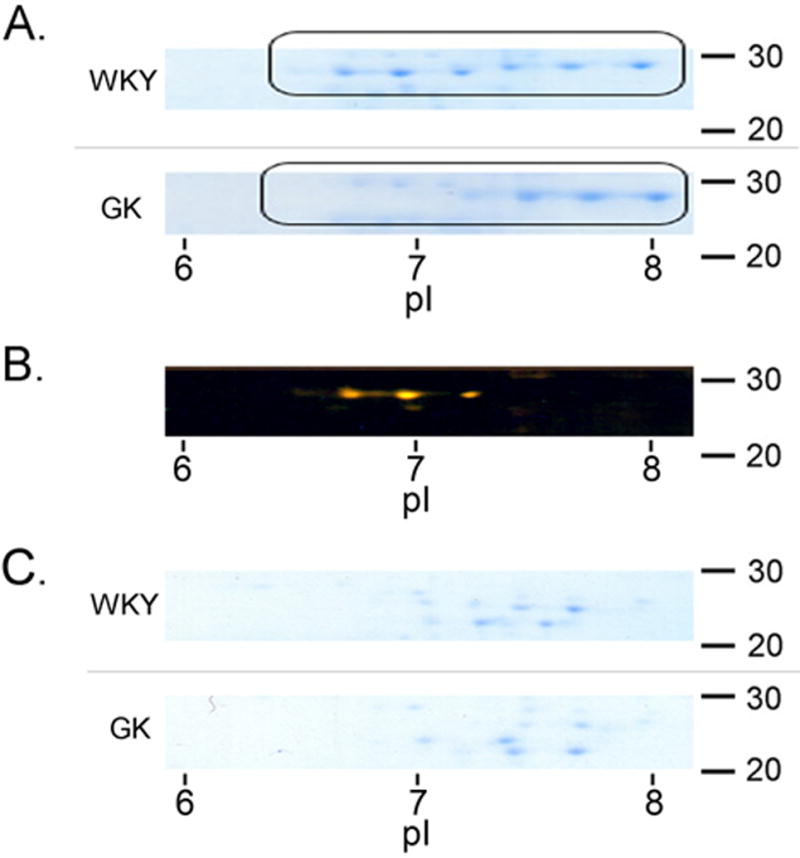
Protein samples (5.0 μg) were resolved by isoelectric focusing as described in Materials and Methods. After electrophoresis, the gels were probed for L-PGDS and analyzed by using enhanced chemiluminescence (Panel A). Isoforms were compared using Photoshop® CS overlaying which highlights differences in orange (Panel B). Proteins were digested with PNGase F to remove glycosylated residues (Panel C). Molecular weights (right) and isoelectric points (below) are provided for reference.
Site-directed mutagenesis of L-PGDS
In view of the fact that there were obvious glycosylation differences between L-PGDS isolated from WKY and GK we chose to create mutations in two potential glycosylation sites that were identified by Protein Blast analysis (Fig. 2). Asparagine residues 51 (Gly1) and 78 (Gly2) were exchanged to alanine. We also decided to alter the enzymatic active site and a potential site of protein kinase C phosphorylation by exchanging a glycine for cysteine 65 (Act), and an alanine for serine 106 (PKC), respectively (Fig. 2). All point mutations were confirmed by sequence analysis (data not shown) and the mutant proteins overexpressed via bacterial expression system and purified by affinity chormatography as described in Materials and Methods.
Figure 2. Site-directed Mutagenesis of L-PGDS.
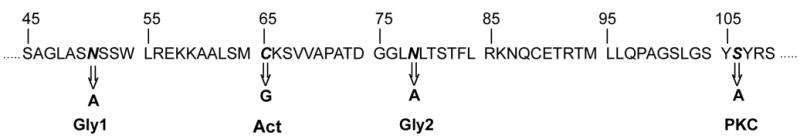
Point mutations in L-PGDS were created at two prospective glycosylation sites (Gly1 and Gly2), an apparent protein kinase C phosphorylation site (PKC), and the enzymatic active site (ACT) as described in Materials and Methods. Gly1 (Asn51), Gly2 (Asn78), and the potential PKC phosphorylation site (Ser106) were all exchanged for alanine, and the ACT (Cys65) mutation was created by exchanging Cys65 with a glycine residue.
Effect of mutation on L-PGDS-induced apoptosis
The mutated L-PGDS proteins were assayed for their ability to induce apoptosis. As seen in Figure 3A, mutation at the Gly1, PKC, and Act sites significantly inhibited the ability of L-PGDS to induce apoptosis as measured by TUNEL assay. Similar results were obtained using caspase3 activity as the apoptotic indicator (Fig. 3B).
Effect of mutation on L-PGDS enzymatic activity
In order to determine the effect of mutation on enzymatic activity we decided to measure the in vitro prostaglandin (PG) D2 formation. Mutation at the Gly1, PKC, and Act sites significantly inhibited the ability of L-PGDS to catalyze the conversion of PG H2 into PG D2 (Fig. 4).
Figure 4. Effect of Mutation on L-PGDS Enzymatic Activity.
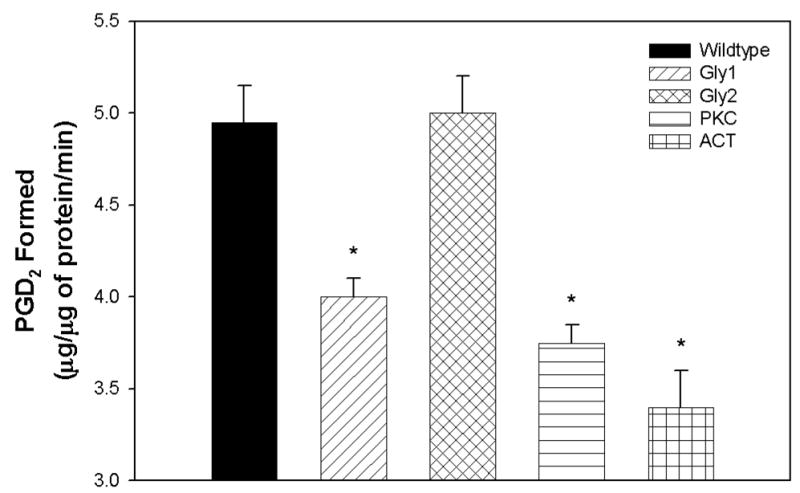
VSMCs were cultured as described in Materials and Methods. At approximately 100% confluency the PG D2 concentration of both the culture medium, and the cell lysate was determined by the conversion of PG D2 into a stable ‘MOX’ product. Results are the mean ± SEM of three experiments performed in duplicate and expressed as μg of PG D2 formed/ μg of protein /ml. Asterisks (*) represent a P<0.05 when compared to wildtype L-PGDS.
Effect of hyperglycemia on L-PGDS-mediated cell proliferation
Given our previous observations that L-PGDS induced apoptosis and inhibited the hyperproliferation of GK VSMCs, we decided to examine the effect of L-PGDS on proliferation under the hyperglycemic condition normally observed in type 2 diabetes. Figure 5 demonstrates that the inhibition of hyperproliferation was limited to growth at 5 mM glucose. When GK VSMCs were cultured in medium supplemented with 20 mM glucose no significant inhibition of cell growth was observed (Fig. 5).
Figure 5. Effect of Hyperglycemia on L-PGDS-mediated Cell Proliferation.
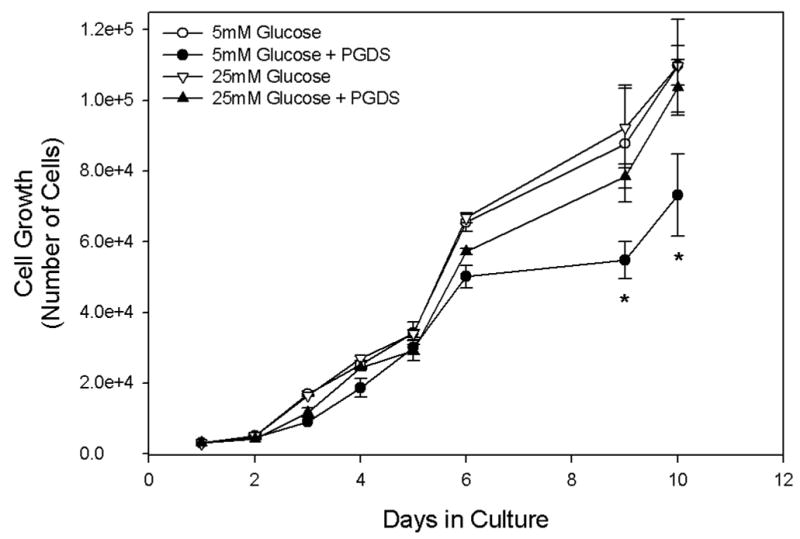
Diabetic GK VSMCs were cultured as described in Materials and Methods in the presence or absence of L-PGDS (50 μg/ml) in media containing either unmodified (5 mM) or high (20 mM) glucose. Cells were plated at a density of 1x104 cells/ml in 35 mm dishes and counted using a hemocytometer after the indicated time. Results are the mean ± S.E.M. of at least three experiments, each performed in duplicate. Asterisks (*) represent a P<0.01 when compared to cells cultured in high glucose.
DISCUSSION
It is widely accepted that prolonged hyperglycemia significantly contributes to the cardiovascular disease present in type 2 diabetics via the alteration VSMC growth, proliferation, and migration. In the current study we have demonstrated the loss of L-PGDS’s ability to inhibit the hyperproliferation of GK VSMCs under hyperglycemic conditions (Fig. 5). This may explain why the excess L-PGDS observed in the serum of diabetics is unable to protect against VSMC hypertrophy and the subsequent arterial thickening leading to atherosclerosis. It will be interesting to determine if hyperglycemia has a similar effect on the inhibition of VSMC migration and cell cycle progression which we observed in the presence of L-PGDS 18.
Post-translational modifications of L-PGDS have been reported in the CSF of patients with neurological disorders 2, 3. Although conflicting data does exist, Puchades et al. have reported a decrease in the ratio of acidic/basic isoforms in Alzheimer’s disease (AD) as well as other forms of dementia 23. Indeed, we have previously reported on the induction of apoptosis by L-PGDS in neuronal PC12 cells 21, offering one possible etiology for AD. In accordance with the Puchades data, we observed a similar loss of the acidic (glycosylated) isoforms in L-PGDS isolated from the serum of diabetic GK rats when compared to WKY controls (Fig. 1). It appears that Asn51 is an important site of L-PGDS glycosylation. Mutation of this residue to an alanine, which prevents glycosylation, consequently impedes the ability of L-PGDS to stimulate apoptosis (Figs. 3A & 3B). Substitution of Asn78, another potential glycosylation site, had no effect on apoptotic activity. Surprisingly, L-PGDS isolated from diabetic GK rat serum was actually less glycosylated than control L-PGDS, even under the hyperglycemic conditions present in GK. Actually, defects in glycosylation are becoming increasingly associated with a range of human diseases, including insulin resistance 24. For example, alterations in the activity of several glycosylation enzymes result in altered levels of sialic acid and fucose in insulin-resistant patients and animals 25–27. It is quite possible that the L-PGDS glycosylation enzyme becomes defective under these conditions or that the protein is de-glycosylated leading to a loss of its apoptotic ability. In fact, we have shown that L-PGDS knockout mice become insulin resistant 15, so it seems feasible that defects in L-PGDS glycosylation could contribute to the insulin resistance observed in type 2 diabetes.
Fujimori et al. have demonstrated that phorbol esters such as 12-O-tetradecanoylphorbol-13-acetate induce L-PGDS expression 28. Our group concluded that phorbol ester-induced apoptosis was mediated by L-PGDS phosphorylation and activation by PKC and is accompanied by inhibition of the PI3-K/PKB anti-apoptotic signaling pathways. The significance of L-PGDS phosphorylation is demonstrated by the mutation of Ser106 to an alanine, thus blocking L-PGDS phosphorylation, which inhibits L-PGDS apoptotic activity (Figs. 3A & 3B). We believe that the importance of both post-translational modifications is related to the alteration of enzymatic activity since the levels of PG D2 directly parallel the apoptotic activity (Fig. 4) as well as mutation of the enzymatic active site (Fig. 3).
We conclude that alteration of L-PGDS by post-translational modification, either by glycosylation at Asn51 or phosphorylation at Ser106, augment its apoptotic activity and inhibit VSMC hyperproliferation. Furthermore, we believe that this process is altered in type 2 diabetes, presumably resulting from hyperglycemia, and may be partially responsible for the increased cardiovascular disease observed in this population.
Acknowledgments
We wish to thank Linda A. Razzano for her technical assistance generating and purifying mutant proteins. This laboratory is supported by an American Diabetes Association Career Development Award, an NIH R01HL06 7953-01A2 grant, and the Winthrop-University Hospital Department of Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Urade Y, Hayaishi O. Prostaglandin D synthase: structure and function. Vitam Horm. 2000;58:89–120. doi: 10.1016/s0083-6729(00)58022-4. [DOI] [PubMed] [Google Scholar]
- 2.Hiraoka A, Seiki K, Oda H, et al. Electrophoresis. 2001;22(16):3433–7. doi: 10.1002/1522-2683(200109)22:16<3433::AID-ELPS3433>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 3.Lescuyer P, Gandini A, Burkhard PR, Hochstrasser DF, Sanchez JC. Prostaglandin D2 synthase and its post-translational modifications in neurological disorders. Electrophoresis. 2005;26(23):4563–70. doi: 10.1002/elps.200500292. [DOI] [PubMed] [Google Scholar]
- 4.Melegos DN, Freedman MS, Diamandis EP. Prostaglandin D synthase concentration in cerebrospinal fluid and serum of patients with neurological disorders. Prostaglandins. 1997;54(1):463–74. doi: 10.1016/s0090-6980(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 5.Inoue T, Takayanagi K, Morooka S, et al. Thromb Haemost. 2001;85(1):165–70. [PubMed] [Google Scholar]
- 6.Hirawa N, Uehara Y, Ikeda T, et al. Nephron. 2001;87(4):321–7. doi: 10.1159/000045937. [DOI] [PubMed] [Google Scholar]
- 7.Eguchi Y, Eguchi N, Oda H, et al. Its accumulation in the coronary circulation of angina patients. Proc Natl Acad Sci U S A. 1997;94(26):14689–94. doi: 10.1073/pnas.94.26.14689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirawa N, Uehara Y, Yamakado M, et al. Hypertension. 2002;39(2 Pt 2):449–54. doi: 10.1161/hy0202.102835. [DOI] [PubMed] [Google Scholar]
- 9.Rogers MS, Rohan RM, Birsner AE, D'Amato RJ. Genetic loci that control vascular endothelial growth factor-induced angiogenesis. Faseb J. 2003 doi: 10.1096/fj.03-0246fje. [DOI] [PubMed] [Google Scholar]
- 10.Taba Y, Sasaguri T, Miyagi M, et al. Circ Res. 2000;86(9):967–73. doi: 10.1161/01.res.86.9.967. [DOI] [PubMed] [Google Scholar]
- 11.Matsuoka T, Hirata M, Tanaka H, et al. Science. 2000;287(5460):2013–7. doi: 10.1126/science.287.5460.2013. [DOI] [PubMed] [Google Scholar]
- 12.Braun M, Schror K. Prostaglandin D2 relaxes bovine coronary arteries by endothelium-dependent nitric oxide-mediated cGMP formation. Circ Res. 1992;71(6):1305–13. doi: 10.1161/01.res.71.6.1305. [DOI] [PubMed] [Google Scholar]
- 13.Eguchi N, Minami T, Shirafuji N, et al. Proc Natl Acad Sci U S A. 1999;96(2):726–30. doi: 10.1073/pnas.96.2.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whittle BJ, Moncada S, Vane JR. Comparison of the effects of prostacyclin (PGI2), prostaglandin E1 and D2 on platelet aggregation in different species. Prostaglandins. 1978;16(3):373–88. doi: 10.1016/0090-6980(78)90216-2. [DOI] [PubMed] [Google Scholar]
- 15.Ragolia L, Palaia T, Hall CE, Maesaka JK, Eguchi N, Urade Y. Accelerated glucose intolerance, nephropathy, and atherosclerosis in prostaglandin D2 synthase knock-out mice. J Biol Chem. 2005;280(33):29946–55. doi: 10.1074/jbc.M502927200. [DOI] [PubMed] [Google Scholar]
- 16.Ragolia L, Palaia T, Paric E, Maesaka JK. Prostaglandin D2 synthase inhibits the exaggerated growth phenotype of spontaneously hypertensive rat vascular smooth muscle cells. J Biol Chem. 2003;278(24):22175–81. doi: 10.1074/jbc.M302769200. [DOI] [PubMed] [Google Scholar]
- 17.Ragolia L, Palaia T, Paric E, Maesaka JK. Elevated L-PGDS activity contributes to PMA-induced apoptosis concomitant with downregulation of PI3-K. Am J Physiol Cell Physiol. 2003;284(1):C119–26. doi: 10.1152/ajpcell.00247.2002. [DOI] [PubMed] [Google Scholar]
- 18.Ragolia L, Palaia T, Koutrouby TB, Maesaka JK. Inhibition of cell cycle progression and migration of vascular smooth muscle cells by prostaglandin D2 synthase: resistance in diabetic Goto-Kakizaki rats. Am J Physiol Cell Physiol. 2004;287(5):C1273–81. doi: 10.1152/ajpcell.00230.2004. [DOI] [PubMed] [Google Scholar]
- 19.Ragolia L, Begum N. The effect of modulating the glycogen-associated regulatory subunit of protein phosphatase-1 on insulin action in rat skeletal muscle cells. Endocrinology. 1997;138(6):2398–404. doi: 10.1210/endo.138.6.5194. [DOI] [PubMed] [Google Scholar]
- 20.Maesaka JK, Palaia T, Frese L, Fishbane S, Ragolia L. Prostaglandin D(2) synthase induces apoptosis in pig kidney LLC-PK1 cells. Kidney Int. 2001;60(5):1692–8. doi: 10.1046/j.1523-1755.2001.00989.x. [DOI] [PubMed] [Google Scholar]
- 21.Ragolia L, Palaia T, Frese L, Fishbane S, Maesaka JK. Prostaglandin D2 synthase induces apoptosis in PC12 neuronal cells. Neuroreport. 2001;12(12):2623–8. doi: 10.1097/00001756-200108280-00008. [DOI] [PubMed] [Google Scholar]
- 22.Smith PK, Krohn RI, Hermanson GT, et al. Anal Biochem. 1985;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 23.Puchades M, Hansson SF, Nilsson CL, Andreasen N, Blennow K, Davidsson P. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer's disease. Brain Res Mol Brain Res. 2003;118(1–2):140–6. doi: 10.1016/j.molbrainres.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Parry S, Hadaschik D, Blancher C, et al. Glycomics investigation into insulin action. Biochim Biophys Acta. 2006 doi: 10.1016/j.bbagen.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 25.Cohen-Forterre L, Andre J, Mozere G, Peyroux J, Sternberg M. Kidney sialidase and sialyltransferase activities in spontaneously and experimentally diabetic rats. Influence of insulin and sorbinil treatments. Biochem Pharmacol. 1990;40(3):507–13. doi: 10.1016/0006-2952(90)90549-z. [DOI] [PubMed] [Google Scholar]
- 26.Rellier N, Ruggiero-Lopez D, Lecomte M, Lagarde M, Wiernsperger N. In vitro and in vivo alterations of enzymatic glycosylation in diabetes. Life Sci. 1999;64(17):1571–83. doi: 10.1016/s0024-3205(99)00094-6. [DOI] [PubMed] [Google Scholar]
- 27.Wiese TJ, Dunlap JA, Yorek MA. Effect of L-fucose and D-glucose concentration on L-fucoprotein metabolism in human Hep G2 cells and changes in fucosyltransferase and alpha-L-fucosidase activity in liver of diabetic rats. Biochim Biophys Acta. 1997;1335(1–2):61–72. doi: 10.1016/s0304-4165(96)00123-7. [DOI] [PubMed] [Google Scholar]
- 28.Fujimori K, Kadoyama K, Urade Y. Protein kinase C activates human lipocalin-type prostaglandin D synthase gene expression through de-repression of notch-HES signaling and enhancement of AP-2 beta function in brain-derived TE671 cells. J Biol Chem. 2005;280(18):18452–61. doi: 10.1074/jbc.M411755200. [DOI] [PubMed] [Google Scholar]