

Abstract
In eukaryotic mismatch repair (MMR) MSH2-MSH6 initiates the repair of base-base and small insertion/deletion mismatches while MSH2-MSH3 repairs larger insertion/deletion mismatches. In this study we showed that the msh2Δ1 mutation, containing a complete deletion of the conserved mismatch recognition Domain I of MSH2, conferred a separation of function phenotype with respect to MSH2-MSH3 and MSH2-MSH6 functions. Strains bearing the msh2Δ1 mutation were nearly wild-type in MSH2-MSH6-mediated MMR and in suppressing recombination between DNA sequences predicted to form mismatches recognized by MSH2-MSH6. However, these strains were completely defective in MSH2-MSH3-mediated MMR and recombination functions. This information encouraged us to analyze the contributions of Domain I to the mismatch binding specificity of MSH2-MSH3 in genetic and biochemical assays. We found that Domain I in MSH2 contributed a non-specific DNA binding activity while Domain I of MSH3 appeared important for mismatch binding specificity and for suppressing non-specific DNA-binding. These observations reveal distinct requirements for the MSH2 DNA binding Domain I in the repair of DNA mismatches and suggest that the binding of MSH2-MSH3 to mismatch DNA involves protein-DNA contacts that appear very different from those required for MSH2-MSH6 mismatch binding.
Keywords: MSH2-MSH6, MSH2-MSH3, mismatch recognition, mismatch repair, genetic recombination
Introduction
DNA mismatch repair (MMR) systems act to maintain genome stability by excising base-base and insertion-deletion loop mismatches generated during DNA replication and genetic recombination.1, 2 MMR proteins also prevent chromosomal rearrangements by suppressing recombination between divergent DNA sequences. In E. coli, base-base and insertion/deletion mismatches of up to four nucleotides (nt) arising during DNA replication are recognized by MutS. ATP-dependent recruitment of MutL to mismatch-MutS complexes is thought to be important to transmit a mismatch recognition signal to key downstream repair factors including MutH, an endonuclease that nicks newly replicated DNA strand at unmethylated GATC sites. These actions result in the unwinding of DNA by UvrD helicase from the nick towards the mismatch site. This is followed by excision of the unwound newly synthesized strand by one of four single-strand specific exonucleases, ExoI, ExoVII, ExoX, or RecJ. The resulting gap is repaired by DNA polymerase III and DNA ligase in conjunction with single-strand DNA binding protein (SSB).
A critical question in the MMR field is how mismatch recognition is accomplished. Crystallographic analysis of MutS bound to unpaired T or G·T mismatches revealed that the MutS homodimer binds to mismatch DNA in an asymmetric fashion.3, 4 While each MutS subunit contains two DNA binding domains, I and IV, only Domain I from subunit A and Domain IV from subunit B make extensive interactions with the DNA backbone (Figure 1(b)). For Domain I of subunit A, two residues (Phe-39 and Glu-41 in Taq MutS, Phe-36 and Glu-38 in E. coli) directly interact with the mismatch DNA. For Domain IV of subunit B, several residues have been identified that provide sequence non-specific interactions to stabilize the DNA backbone. In contrast, the other DNA binding domains only make sequence non-specific contacts with the sugar-phosphate backbone.
Figure 1.
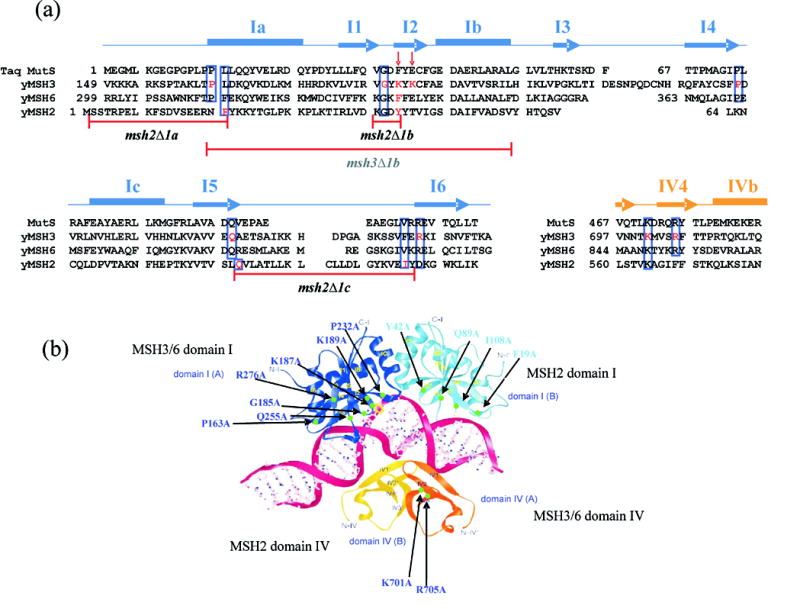
Mutations in the MSH proteins analyzed in this study. (a) Structure-based sequence alignment of yeast MSH2, MSH3, and MSH6. The secondary structures (arrow = β sheet, rectangle = α helix) of Taq MutS are depicted above the aligned sequences.3 Residues involved in DNA recognition in Taq MutS crystal structure homologous to residues in the MSH proteins are boxed in blue. Residues in MSH2, MSH3, and MSH6 that were mutated in this study are highlighted in red. Red arrows indicate the critical phenylalanine and glutamate residues in Taq MutS described in the text. (b) Ribbon diagrams of DNA-binding domains of Taq MutS complexed with +1 mismatched DNA.3 Domains I and IV of subunit A are colored in dark blue and orange. Domains I and IV of subunit B are colored in light blue and yellow. The DNA phosphate backbones are shown as red lines and sugars and bases are shown as ball-and-stick figures. Regions mutated in this study are highlighted in light green and the amino acid changes are noted. Changes in MSH2 are colored in light blue; changes in MSH3 are colored in dark blue. Figure 1b is adapted with permission from Wei Yang, National Institutes of Health, Bethesda, MD, USA.
In both of the crystal structures, a phenylalanine residue (Phe-39 in Taq MutS, Phe-36 in E. coli MutS) base stacks with the mispaired thymidine. The DNA containing the mismatch is kinked by 60° towards the major groove. The major groove is narrowed, whereas the minor groove in which the phenylalanine interacts with the DNA mispair is expanded. Domain IV of subunit B stabilizes the narrowed major groove.3 Bending of the DNA upon MutS binding has been confirmed by Wang et al.5 in solution and atomic force microscopy. Wang et al.3, however, suggest that the bent state does not represent a specific MutS-mismatch DNA interaction, but that upon binding to mismatch DNA, the MutS-mismatch complex undergoes additional conformational changes to form a recognition complex in which mismatch DNA is unbent.
As described above, a phenylalanine residue located in Domain I of the A subunit of MutS (Phe-39 in Taq and Phe-36 in E. coli) is thought to play a critical role in mismatch recognition by base stacking with an unpaired/mismatched thymidine nucleotide. Consistent with this, mutations that alter this residue confer a strong mutator phenotype.6 The glutamate residue (Glu-41 in Taq, Glu-38 in E. coli) located in Domain I of subunit A is also thought to be important for mismatch binding; in the MutS structures, the carboxyl oxygen of the glutamate residue makes a hydrogen bond with the N3 position of the unpaired/mismatched thymidine. Biochemical analyses revealed that this residue plays a critical role in discriminating between mismatch and homoduplex DNA,7 and that the hydrogen bond between the glutamate and the mismatch DNA is thought to be important in forming a stable MutS-ATP-DNA complex that activates downstream repair factors.8
In eukaryotes, MMR is initiated through the actions of two heterodimeric MutS homologs, MSH2-MSH3 and MSH2-MSH6.1, 2 Like MutS, MSH2-MSH6 and MSH2-MSH3 bend DNA substrates containing mismatch DNA (Refs. 5, 9; J. A. S., unpublished observations). These two complexes recognize different types of mismatches; MSH2-MSH6 primarily acts in a pathway that repairs base-base and single insertion/deletion mismatches, while MSH2-MSH3 acts mainly to repair insertion/deletion loop mismatches 2 to 13 nt in size. MSH2-MSH3 also binds to DNA flap structures predicted to form during genetic recombination.10 Genetic and biochemical studies have suggested that MSH6 and MSH2 are the functional homologs of MutS subunits A and B, respectively. In support of this designation, mutation of the Phe-337 residue to alanine in yeast MSH6, corresponding to Phe-39 in Taq MutS, caused a mutator phenotype similar to that seen in msh6Δ strains.11, 12 In addition, yeast (MSH2-msh6F337A) and human (MSH2-msh6F432A) complexes containing a mutation in this phenylalanine residue displayed defects in both mismatch-specific and non-specific DNA binding.11–14 In contrast, yeast and human mutants containing a mutation in the corresponding position in MSH2 (Tyr-42) displayed a wild-type-like phenotype.11, 12, 14 S. cerevisiae strains bearing the E339A mutation in MSH6 (equivalent to Glu-41 in Taq MutS) displayed a weak mutator phenotype. Consistent with this, the MSH2-msh6E339A complex displayed a reduction in both mismatch-specific and non-specific DNA binding. The analogous mutation could not be analyzed in MSH2 because the glutamate residue is not conserved.12 Together, these observations suggest that MSH2-MSH6-mismatch complexes are likely to share key features seen in the MutS-mismatch crystal structures.
Three lines of evidence suggest that binding of MSH2-MSH3 to mismatch DNA is likely to occur through different protein-DNA contacts than those observed for MutS and MSH2-MSH6. First, as described above, MSH2-MSH3 and MSH2-MSH6 confer very different mismatch substrate specificities; MSH2-MSH6 binds to base-base and +1 loop mismatches efficiently, but not to larger loop mismatches in vitro, whereas MSH2-MSH3 binds to 2–12 nt loop mismatches quite efficiently, but not to base-base mismatches.2, 10 Second, the phenylalanine and glutamate residues that play critical roles in MutS and MSH2-MSH6 mismatch recognition are not conserved in MSH3; instead two lysine residues are present in yeast (Lys-187 and Lys-189) and a lysine and an arginine are present in human MSH3 (Lys-246 and Arg-248). Third, unlike MSH2-MSH6, MSH2-MSH3 binds to and participates in the removal of 3’ non-homologous tails present in DNA flap structures.10, 15 Biochemical analysis showed that MSH2-MSH3 specifically binds at the double-strand/single-strand junction of a 3’ flap substrate, causing a conformational change in the DNA structure.10 These observations suggest that mismatch recognition by MSH2-MSH3 is likely to involve protein-DNA contacts that are different from those seen between MutS and mismatch DNA.
Previously, we made a deletion of DNA binding Domain I of MSH216 (msh2Δ1, deletion of amino acids 2–133), based on the Taq MutS crystal structure,3 and were struck by the observation that this mutation disrupted MSH2-MSH3 functions in non-homologous tail removal, but appeared functional for MSH2-MSH6 functions in MMR. This observation provided us with an opportunity to better understand the genetic requirements for MSH2-MSH3 binding to DNA mismatches and compare them with those required for MSH2-MSH6. As shown below, we found that the msh2Δ1 mutation conferred a clean separation of function phenotype with respect to MSH2-MSH3 and MSH2-MSH6 in MMR and genetic recombination assays. In addition, genetic and biochemical analysis of the DNA binding domains of these complexes revealed distinct contributions of these domains in the repair of specific mismatches and suggest that the crystallographic analysis of MutS bound to mismatch DNA cannot be used to accurately model MSH2-MSH3-mismatch DNA interactions.
Results
The msh2Δ1 is functional in MSH2-MSH6 mediated MMR, but defective in MSH2-MSH3 mediated MMR
We investigated the effect of mutations in Domain I of MSH2 and MSH3 (Figure 1, msh2Δ1, msh3Δ1) on the repair of loop mismatches formed during DNA slippage.17 The msh2Δ1 (deletion of amino acids 2–133) and msh3Δ1 (Δ149–306) alleles contain complete deletions of Domain I (Figure 1). We employed a sensitive microsatellite instability assay developed by the Petes laboratory in which DNA sequences of various repeat units were inserted in frame into the coding region of URA3. Cells bearing these reporters are Ura+. DNA slippage events that primarily occur during DNA replication can disrupt the URA3 reading frame, leading to a Ura−, 5-FOAr phenotype.17
As outlined in the Introduction, MSH2-MSH3 acts to repair larger loop mispairs while MSH2-MSH6 repairs base-base mismatches and small loop mispairs.2, 17 This specificity is best demonstrated in the microsatellite instability assay, where msh3Δ strains displayed rates of microsatellite instability for 4, 5, and 8 nt repeat tracts (9 – 60 fold above wild-type) comparable to that seen in msh2Δ strains (9 – 60 fold; in all cases msh3Δ vs. msh2Δ, p > 0.65); the msh6 mutation, however, conferred little or no increased rate of instability (1.0 to 1.4 fold above wild-type; in all cases msh6Δ vs. wild-type, p > 0.24) for these tract lengths (Table 3). When the repeat tracts were decreased to 1–2 nt, msh3Δ showed a greater increase in instability (53 to 180 fold higher than wild-type) compared to msh6Δ (2.1 to 20 fold; msh3Δ vs. msh6Δ, p < 0.001), but neither strain displayed defects as large as those observed in msh2Δ (360 to 4800; msh2Δ vs. msh3Δ and msh2Δ vs. msh6Δ, p < 0.001) or msh3Δ msh6Δ strains (400 to 3100 fold). These results also illustrated the overlapping specificities for MSH2-MSH3 and MSH2-MSH6 for small (1–2 nt) loop mispairs.17, 18
TABLE 3.
Rates of microsatellite instability in mismatch repair defective strains
| nt in repeat unit | genotype | average rate of tract alteration (X 10−6)a | relative rate |
|---|---|---|---|
| 1 | wild-type | 4.2 (3.2–6.2)* | 1.0 |
| msh2Δ | 20,000 (14,000–23,000) | 4,800 | |
| msh3Δ | 740 (560–1,200) | 180 | |
| msh2Δ1 | 760 (500–1,500) | 190 | |
| msh6Δ | 83 (46–120) | 20 | |
| msh3Δ msh6Δ | 13,000 (9,700–20,000) | 3,100 | |
| msh2Δ1 msh3Δ | 1,800 (1,200–2,400) | 440 | |
| msh2Δ1 msh6Δ | 16,000 (11,000–21,000) | 3,700 | |
| 2 | wild-type | 1.5 (0.7–2.0) | 1.0 |
| msh2Δ | 540 (420–890) | 360 | |
| msh3Δ | 79 (37–120) | 53 | |
| msh2Δ1 | 85 (52–140) | 57 | |
| msh6Δ | 3.1 (1.7–9.0) | 2.1 | |
| msh3Δ msh6Δ | 600 (290–870) | 400 | |
| msh2Δ1 msh3Δ | 120 (90–150) | 79 | |
| msh2Δ1 msh6Δ | 550 (380–980) | 370 | |
| 4 | wild-type | 2.1 (1.6–3.0) | 1.0 |
| msh2Δ | 120 (74–170) | 57 | |
| msh3Δ | 130 (72–160) | 62 | |
| msh2Δ1 | 110 (75–130) | 53 | |
| msh6Δ | 2.2 (1.1–3.4) | 1.0 | |
| 5 | wild-type | 5.9 (4.2–9.7) | 1.0 |
| msh2Δ | 77 (66–110) | 13 | |
| msh3Δ | 75 (61–110) | 13 | |
| msh2Δ1 | 77 (58–110) | 13 | |
| msh6Δ | 8.0 (4.4–12) | 1.4 | |
| 8 | wild-type | 3.5 (2.8–4.6) | 1.0 |
| msh2Δ | 30 (19–35) | 8.6 | |
| msh3Δ | 30 (25–49) | 8.6 | |
| msh2Δ1 | 29 (24–37) | 8.3 | |
| msh6Δ | 3.9 (1.7–8.0) | 1.1 |
Rates are in tract alterations per cell division.17
Numbers in parentheses indicate 95% confidence intervals. Microsatellite instability rates and 95% confidence intervals were calculated as described in the Materials and Methods from 14–21 individual cultures. The indicated strains were transformed with the reporter plasmids containing the indicated repeat unit (Table 1).
We measured rates of microsatellite instability in strains bearing the msh2Δ1 allele and other mismatch repair mutations. As shown in Table 3 and Figure 2, the effect of the msh2Δ1 mutation on microsatellite instability was similar to that seen for msh3Δ, providing evidence that only the MSH2-MSH3 repair pathway was compromised in msh2Δ1strains. To test if the MSH2-MSH6 pathway was functional in msh2Δ1, we made msh2Δ1 msh3Δ double mutants and measured the rates of microsatellite instability for the repeat units of 1 and 2 nt (Table 3 and Figure 2). The rate of microsatellite instability for the msh2Δ1 msh3Δ double mutant was somewhat elevated compared with either single mutant strain (msh3Δ vs. msh2Δ1 msh3Δ, p = 0.002 for 1 nt., p = 0.056 for 2 nt.), but was significantly lower than msh2Δ(msh2Δ1 msh3Δ vs. msh2Δ, p < 0.001), indicating that the msh2Δ1-MSH6 complex was nearly completely functional. In contrast, for the msh2Δ1 msh6Δ double mutant, the rate of microsatellite instability was elevated 3,700 fold for the repeat unit of 1 nt and 370 fold for the repeat unit of 2 nt. These increases were comparable to those seen in msh3Δ msh6Δ or msh2Δ strains. With the 4 nt repeat unit, msh2Δ, msh3Δ and msh2Δ1 all showed similar levels of microsatellite instability, while msh6Δ had no effect (Table 3 and Figure 2), consistent with the idea that repair of these slippage events is solely MSH2-MSH3-dependent.
Figure 2.
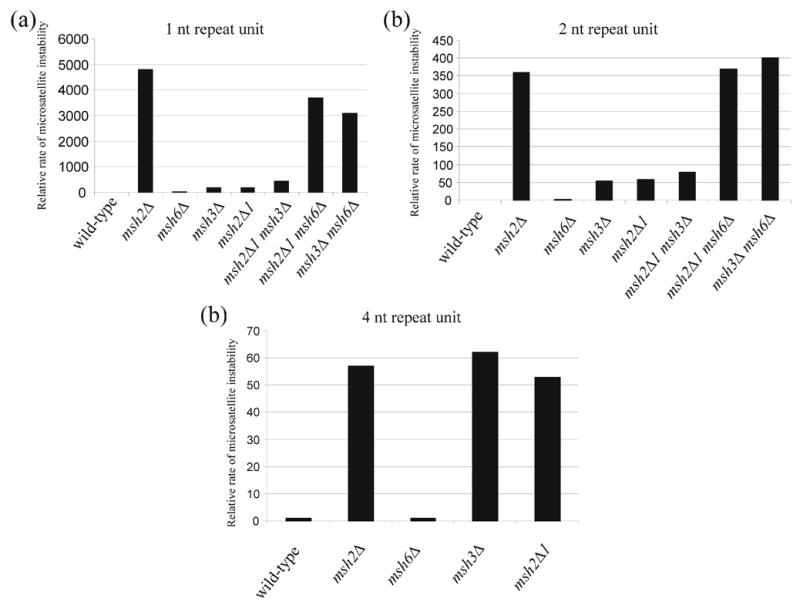
Relative rates of microsatellite instability in wild-type and msh strains. Data in Table 3 are plotted for the indicated repeat units. In all panels, data are presented relative to the appropriate wild-type strain (Panel A, 1X = 4.2 x 10−6, Panel B, 1X = 1.5 x 10−6, Panel C, 1X = 2.1 x 10−6).
The msh2Δ1 mutation suppresses recombination for homeologous substrates specific to MSH2-MSH6, but does not suppress recombination specific to MSH2-MSH3
MMR proteins have been shown to inhibit recombination between partially divergent (homeologous) sequences. This activity is thought to be initiated by the binding of MSH proteins to DNA mismatches present in heteroduplex DNA, followed by recruitment of factors such as SGS1 that unwind the recombination intermediate.19 Consistent with this is the finding that the MSH2-MSH3 and MSH2-MSH6 mismatch binding specificities are maintained in genetic recombination.20 Using an intron-based intramolecular recombination assay involving a HIS3 reporter, Nicholson et al.20 showed that recombination between substrates predicted to form base-base (cβ2/cβ7, cβ2/cβ2-ns) and 1 nt loop mismatches (cβ2/cβ2-1L) in heteroduplex DNA was suppressed by both MSH2-MSH3 and MSH2-MSH6; a null mutation in MSH3 or MSH6 increased the recombination rates of these types of homeologous substrates to some extent but not to that seen in msh2Δ strains (Table 4). However, recombination between substrates predicted to form larger loop mismatches (cβ2/cβ2-4L and cβ2/cβ2-12L) was suppressed only by MSH2-MSH3; the effect of the msh3Δ mutation on recombination of these substrates was comparable to that seen in msh2Δ strain, while the effect of a msh6Δ mutation was negligible.
TABLE 4.
Rates of homeologous recombination in mismatch repair defective strains
| substrate | genotype | rate of His+ recombinants (X 10−6) | normalized homeologous rate** | mutant/wild-type*** |
|---|---|---|---|---|
| Cβ2/Cβ2 (homologous) | wild-type | 2.7 (2.5–3.8)* | ||
| msh2Δ | 5.5 (4.7–9.2) | |||
| msh3Δ | 6.6 (6.2–9.6) | |||
| msh2Δ1 | 7.0 (5.5–8.8) | |||
| msh6Δ | 2.0 (1.3–2.4) | |||
| msh2Δ1 msh3Δ | 9.6 (9.0–11) | |||
| msh2Δ1 msh6Δ | 10 (8.9–12) | |||
| Cβ2/Cβ7 (9% diverged) | wild-type | 0.087 (0.057–0.11) | 0.032 | 1.0 |
| msh2Δ | 4.4 (3.8–7.2) | 0.80 | 25 | |
| msh3Δ | 0.61 (0.5–0.79) | 0.093 | 2.9 | |
| msh2Δ1 | 0.51 (0.48–0.71) | 0.073 | 2.3 | |
| msh6Δ | 0.40 (0.36–0.44) | 0.20 | 6.4 | |
| msh2Δ1 msh3Δ | 1.1 (0.89–1.6) | 0.11 | 3.4 | |
| msh2Δ1 msh6Δ | 7.1 (5.5–8.4) | 0.70 | 22 | |
| Cβ2/Cβ2-ns (1.3% diverged) | wild-type | 0.16 (0.15–0.22) | 0.060 | 1.0 |
| msh2Δ | 8.7 (5.2–10) | 1.6 | 26 | |
| msh3Δ | 1.8 (1.2–2.4) | 0.28 | 4.6 | |
| msh2Δ1 | 1.8 (1.5–2.8) | 0.26 | 4.3 | |
| msh6Δ | 2.2 (1.9–2.4) | 1.1 | 19 | |
| msh2Δ1 msh3Δ | 4.9 (4.3–6.1) | 0.51 | 8.5 | |
| msh2Δ1 msh6Δ | 16 (12–17) | 1.5 | 26 | |
| Cβ2/Cβ2-1L | wild-type | 0.29 (0.25–0.49) | 0.11 | 1.0 |
| msh2Δ | 11 (9.6–15) | 2.1 | 19 | |
| msh3Δ | 3.6 (2.5–4.8) | 0.54 | 5 | |
| msh2Δ1 | 4.3 (2.8–5.6) | 0.61 | 5.6 | |
| msh6Δ | 0.63 (0.48–0.83) | 0.32 | 3 | |
| msh2Δ1 msh3Δ | 6.2 (6.0–7.7) | 0.64 | 6 | |
| msh2Δ1 msh6Δ | 21 (16–24) | 2.1 | 19 | |
| Cβ2/Cβ2-4L | wild-type | 0.25 (0.15–0.30) | 0.091 | 1.0 |
| msh2Δ | 6.7 (6.5–8.4) | 1.2 | 13 | |
| msh3Δ | 7.6 (6.6–9.1) | 1.2 | 13 | |
| msh2Δ1 | 7.4 (4.7–7.7) | 1.1 | 12 | |
| msh6Δ | 0.27 (0.21–0.34) | 0.14 | 1.5 | |
| Cβ2/Cβ2-12L | wild-type | 0.49 (0.40–0.60) | 0.18 | 1.0 |
| msh2Δ | 5.7 (3.8–6.6) | 1.0 | 5.7 | |
| msh3Δ | 5.7 (5.1–10) | 0.86 | 4.8 | |
| msh2Δ1 | 6.2 (4.9–7.4) | 0.88 | 4.9 | |
| msh6Δ | 0.51 (0.42–0.74) | 0.26 | 1.4 |
Numbers in parentheses indicate 95% confidence intervals. Homeologous recombination rates and 95% confidence intervals were calculated as described in the Materials and Methods from 14–21 individual cultures. The genotype of the strains is shown in Table 2.
homeologous rate normalized to homologous rate in strain containing the same msh genotype.
mutant homeologous rate relative to wild-type homeologous rate.20
We used the HIS3::intron assay to determine if the MSH2-MSH3 defect seen in msh2Δ1 strains extended to other MSH2-MSH3-dependent pathways such as antirecombination. We were also interested in determining if msh2Δ1 can fully suppress recombination of homeologous substrates specific to MSH2-MSH6. The recombination rates for various homeologous substrates (base-base and 1–12 nt loop mismatches, Table 2) in msh2Δ1 strains containing null mutations in MSH2, MSH3, and MSH6 are shown in Table 4. Since rates for the homologous substrates (Cβ2/Cβ2) vary depending on genetic background, the rates for homeologous substrates were normalized to those observed in isogenic strains containing homologous substrate.20 For all types of homeologous substrates, the recombination rates in msh2Δ1 strains were similar to those seen in msh3Δ strains (Table 4), suggesting that the recombination of the homeologous substrates specific to MSH2-MSH3 was affected in msh2Δ1 strains. Consistent with this, the homeologous recombination rate of the msh2Δ1 msh3Δ double mutant in the Cβ2/Cβ7 substrate (Table 4) was similar to that seen in either single mutant (msh3Δ, 2.9 fold higher relative to wild-type; msh2Δ1, 2.3 fold; msh2Δ1 msh3Δ, 3.4 fold; msh2Δ1 msh3Δ vs. msh3Δ, p = 0.05) but significantly lower than for msh2Δ (msh2Δ1 msh3Δ vs. msh2Δ, p = 0.005). For the msh2Δ1 msh6Δ double mutant, the recombination rate was elevated significantly compared with either single mutant strain (p < 0.001) and was similar to that seen in msh2Δ (msh2Δ1 msh6Δ vs. msh2Δ, p = 0.07). These results suggest that the msh2Δ1 mutation fully suppressed recombination between homeologous substrates that form mismatches recognized by MSH2-MSH6, but did not suppress recombination between substrates that form mismatches recognized by MSH2-MSH3.
TABLE 2.
Strains used to measure homeologous recombination using the HIS3::intron recombination assay
| Strain | Genotype | Recombination substrates |
|---|---|---|
| SJR769 | wild-type | cβ2/cβ2: homologous substrates |
| SJR770 | wild-type | cβ2/cβ7: predicted to form 9% base-base mismatches |
| GCY615 | wild-type | cβ2/cβ2-ns: predicted to form four (1.3%) base-base mismatches |
| SJR767 | wild-type | cβ2/cβ2-1L: predicted to form four 1-nt loops |
| GCY559 | wild-type | cβ2/cβ2-4L: predicted to form four 4-nt loops |
| SJR768 | wild-type | cβ2/cβ2-12L: predicted to form two 12-nt loops |
| EAY1605 | msh2Δ | cβ2/cβ2 |
| EAY1607 | msh2Δ | cβ2/cβ7 |
| EAY1609 | msh2Δ | cβ2/cβ2-ns |
| EAY1611 | msh2Δ | cβ2/cβ2-1L |
| EAY1613 | msh2Δ | cβ2/cβ2-4L |
| EAY1615 | msh2Δ | cβ2/cβ2-12L |
| EAY1617 | msh3Δ | cβ2/cβ2 |
| EAY1619 | msh3Δ | cβ2/cβ7 |
| EAY1621 | msh3Δ | cβ2/cβ2-ns |
| EAY1623 | msh3Δ | cβ2/cβ2-1L |
| EAY1625 | msh3Δ | cβ2/cβ2-4L |
| EAY1627 | msh3Δ | cβ2/cβ2-12L |
| EAY1629 | msh6Δ | cβ2/cβ2 |
| EAY1631 | msh6Δ | cβ2/cβ7 |
| EAY1633 | msh6Δ | cβ2/cβ2-ns |
| EAY1635 | msh6Δ | cβ2/cβ2-1L |
| EAY1637 | msh6Δ | cβ2/cβ2-4L |
| EAY1639 | msh6Δ | cβ2/cβ2-12L |
| EAY1641 | msh2Δ1 | cβ2/cβ2 |
| EAY1643 | msh2Δ1 | cβ2/cβ7 |
| EAY1645 | msh2Δ1 | cβ2/cβ2-ns |
| EAY1647 | msh2Δ1 | cβ2/cβ2-1L |
| EAY1649 | msh2Δ1 | cβ2/cβ2-4L |
| EAY1651 | msh2Δ1 | cβ2/cβ2-12L |
| EAY1653 | msh2Δ1 msh3Δ | cβ2/cβ2 |
| EAY1655 | msh2Δ1 msh3Δ | cβ2/cβ7 |
| EAY1657 | msh2Δ1 msh3Δ | cβ2/cβ2-ns |
| EAY1659 | msh2Δ1 msh3Δ | cβ2/cβ2-1L |
| EAY1661 | msh2Δ1 msh6Δ | cβ2/cβ2 |
| EAY1663 | msh2Δ1 msh6Δ | cβ2/cβ7 |
| EAY1665 | msh2Δ1 msh6Δ | cβ2/cβ2-ns |
| EAY1667 | msh2Δ1 msh6Δ | cβ2/cβ2-1L |
The SJR769, SJR770, SJR767, GCY559, GCY615 strains bearing the above recombination substrates are described.20 The mismatches predicted to form in heteroduplex DNA during genetic recombination are indicated for each substrate. Integration vectors used to make the indicated msh strains were: pEAI98 (msh2Δ::hisG-URA3-hisG), pEAI88 (msh3Δ::hisG-URA3-hisG), pEAI108 (msh6Δ::hisG-URA3-hisG), and pEAI244 (msh2Δ1::KanMX).16, 31 Note that the intact hisG-URA3-hisG cassette is present in these msh2Δ, msh3Δ and msh6Δ mutant derivatives.
Structure-function analysis of MSH2 Domain I
The above observations encouraged us to identify key regions and residues in MSH2 Domain I critical for MSH2-MSH3-mediated MMR. Results from this work should also provide important information on the residues required for MSH2-MSH3 mismatch specificity, for which little is known. We made a series of deletion and point mutations in Domain I of MSH2 using the Taq MutS crystal structure as a guide and investigated the effect of these mutations on the repair of loop mispairs (Figure 1 and Table 5). Domain I of MSH2 is equivalent to Domain I of subunit B of MutS which makes only limited interactions with the sugar-phosphate backbone and does not directly contact the mismatched DNA (Figure 1). The critical phenylalanine (Phe-39 in Taq) is not conserved in MSH2, but is substituted with tyrosine (Tyr-42). Although the phenylalanine residue in subunit B of Taq MutS does not contact the mismatch, we were curious whether this tyrosine in MSH2 played a role in MSH2-MSH3-mediated MMR. Previously, msh2Y42A conferred a wild-type-like phenotype in the canavanine resistance assay, a mutator assay specific to MSH2-MSH6, and a weak mutator phenotype in lys2::insE-A14 reversion assay, a mutator assay that detects one base pair deletions in the run of 14 adenine residues inserted into the LYS2 gene.11, 12, 21 Similarly, we found that the msh2Y42A mutation conferred a wild-type-like phenotype in the microsatellite instability assay, suggesting that this residue alone does not appear to play a critical role in MSH2-MSH3 mediated MMR (Table 5). Other amino acids in Domain I of MSH2 (Phe-19, Gln-89, and Ile-108) that are highly conserved among MutS/MSH proteins were shown to directly contact the sugar-phosphate backbone in subunit A of MutS.3 These mutants also displayed a microsatellite instability phenotype similar to wild-type (Figure 1 and Table 5).
TABLE 5.
Rates of microsatellite instability in strains bearing small deletion and point mutations in Domains I and IV of MSH2, MSH3, and MSH6
| nt in repeat unit | genotype | avg. rate of tract alteration (X10−6) | relative rate |
|---|---|---|---|
| 1 | wild-type | 4.2 (3.2–6.2) | 1.0 |
| msh2Δ | 20,000 (14,000–23,000) | 4,800 | |
| msh3Δ | 740 (560–1,200) | 180 | |
| msh2Δ1 | 760 (500–1,500) | 190 | |
| msh2Δ1a | 760 (520–1,000) | 190 | |
| msh2Δ1b | 15,000 (11,000–35,000) | 3,600 | |
| msh2Δ1c | 20,000 (19,000–33,000) | 4,800 | |
| msh2F19A | 15 (6.8–21) | 3.5 | |
| msh2Y42A | 4.3 (2.8–8.3) | 1.0 | |
| msh2Q89A | 8.9 (5.7–20) | 2.1 | |
| msh2I108A | 15 (10–30) | 3.5 | |
| msh3Δ | 740 (560–1,200) | 180 | |
| msh3Δ1 | 780 (470–1,400) | 190 | |
| msh3Δ1a | 770 (710–1,700) | 180 | |
| msh3Δ1b | 420 (340–820) | 100 | |
| msh3P163A | 3.5 (2.2–6.1) | 0.83 | |
| msh3G185A | 7.5 (4.2–9.3) | 1.8 | |
| msh3K187A | 17 (12–25) | 4.1 | |
| msh3K189A | 38 (27–100) | 9.0 | |
| msh3K187A,K189A | 320 (260–420) | 76 | |
| msh3P232A | 13 (5.3–22) | 3.1 | |
| msh3Q255A | 8.3 (2.0–14) | 2.0 | |
| msh3R276A | 400 (250–730) | 95 | |
| msh3K701A | 3.4 (2.8–5.2) | 0.81 | |
| msh3R705A | 3.8 (2.5–7.9) | 0.90 | |
| msh6Δ | 83 (46–120) | 20 | |
| msh6F337A | 69 (25–80) | 16 | |
| 2 | wild-type | 1.5 (0.7–2.0) | 1.0 |
| msh2Δ | 540 (420–890) | 360 | |
| msh2Δ1 | 85 (52–140) | 57 | |
| msh3Δ | 79 (37–120) | 53 | |
| msh3K187A | 10 (5.5–20) | 7.0 | |
| msh3K189A | 22 (10–34) | 15 | |
| msh3K187A,K189A | 80 (72–100) | 53 | |
| msh3R276A | 44 (39–62) | 29 | |
| 4 | wild-type | 2.1 (1.6–3.0) | 1.0 |
| msh2Δ | 120 (74–170) | 57 | |
| msh2Δ1 | 110 (75–130) | 53 | |
| msh3Δ | 130 (72–160) | 62 | |
| msh3K187A | 7.5 (3.2–14) | 3.6 | |
| msh3K189A | 31 (16–93) | 15 | |
| msh3K187A,K189A | 140 (110–190) | 67 | |
| msh3R276A | 74 (47–110) | 35 |
Numbers in parentheses indicate 95% confidence intervals. Microsatellite instability rates and 95% confidence intervals were calculated as described in the Materials and Methods. The indicated strains were transformed with the reporter plasmids containing the indicated repeat unit (Table 1).
In an attempt to narrow down the critical region(s) within Domain I, three additional deletion mutants of Domain I of MSH2 were made; msh2Δ1a (amino acid 2–19 deleted), msh2Δ1b (39–42), and msh2Δ1c (89–109). These regions were selected to minimize disruption of the secondary structure of the protein (W. Yang, personal communication). The rate of microsatellite instability for msh2Δ1a was similar to that of msh3Δ, indicating the importance of this N-terminal sequence in MSH2-MSH3 mediated MMR (Table 5). The deleted region in msh2Δ1b (Lys-39, Gly-40, Asp-41, and Tyr-42) spans the amino acids homologous to those in MutS subunit A that are in direct contact with the mismatched DNA (Phe-39 in Taq) and sugar-phosphate backbone (Gly-37 and Asp-38 in Taq). The msh2Δ1b and msh2Δ1c strains showed high rates of microsatellite instability comparable to that of msh2Δ (Table 5); this was likely due to disruption of the overall stability of the MSH2 subunit.
msh2Δ1 confers a defect in DNA binding in MSH2-MSH3
Consistent with its role in repairing large loop mispairs in vivo, MSH2-MSH3 specifically binds to large DNA loop substrates as measured in gel mobility shift assays.10, 22–24 We investigated in gel mobility shift assays whether msh2Δ1-MSH3 was defective in binding to large DNA loop substrates (Figure 3). Consistent with previous work10, MSH2-MSH3 bound to a +8 loop substrate with highest affinity but more weakly to a +1 loop substrate. Weak binding to homoduplex DNA was also observed; this likely represents a non-specific DNA binding activity.10 For msh2Δ1-MSH3, this non-specific DNA binding activity was greatly reduced; it bound poorly to the homoduplex and +1 loop substrates at all protein concentrations. msh2Δ1-MSH3 bound to the +8 loop substrate at high protein concentrations, suggesting that it retained binding specificity for the large loop substrates. Titration experiments to calculate approximate Kd values also indicated a higher affinity for the +8 loop substrate (Table 6). These observations are consistent with Domain I in MSH2 playing a role in non-specific DNA binding that is critical for MSH2-MSH3 function.
Figure 3.
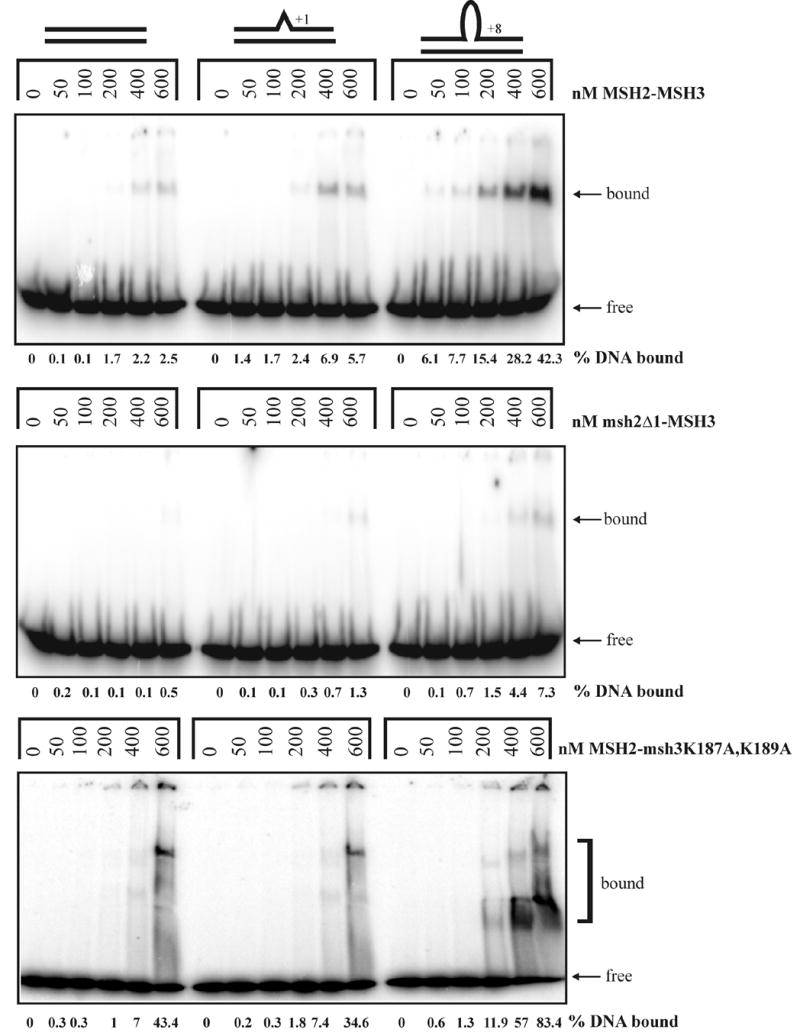
Gel mobility shift assays of MSH2-MSH3 (top panel), msh2Δ1-MSH3 (second panel), and MSH2-msh3K187A,K189A (bottom panel) incubated with homoduplex, +1 loop, and +8 loop substrates as described in the Materials and Methods. The percentage of labeled DNA substrate that was bound is indicated below each gel.
TABLE 6.
Apparent Kd values* for MSH2-MSH3, msh2Δ1-MSH3, and MSH2-msh3K187A, K189A binding to DNA substrates
| homoduplex substrate
|
+1 substrate
|
+8 substrate
|
|
|---|---|---|---|
| MSH2-MSH3** | 220 nM +/− 26 | 110 nM +/− 5 | 50 nM +/− 6 |
| msh2Δ1-MSH3 | > 300 nM | > 300 nM | 168 nM +/− 33 |
| MSH2-msh3K187A,K189A | 131 nM +/− 10 | 126 nM +/− 3 | 86 nM +/− 15 |
The values represent the protein concentration at 50% maximal binding for each substrate and are an average of at least 3 independent experiments (± SEM).
Data from Surtees and Alani.10
We have previously shown that MSH2-MSH3 binds at the 5’ double-strand/single-strand DNA junction of the +8 loop substrate (Ref. 10; Figure 4). We performed an in situ 1,10-phenanthroline-copper (OP-Cu) footprinting experiment with msh2Δ1-MSH3 to determine if it binds the same location on this substrate (Figure 4). OP-Cu is a small chemical nuclease that interacts with the minor groove of DNA, making single-strand nicks. For this experiment, we electrophoresed the protein-DNA complexes through a native gel and then treated the entire gel with OP-Cu. This allowed us to separate the protein-DNA complex from the free DNA, which was important given the weak DNA-binding activity of msh2Δ1-MSH3. We found that msh2Δ1-MSH3 exhibited a footprint on the +8 loop substrate that was nearly identical to that of MSH2-MSH3 (Figure 4 and data not shown). This strongly supports the idea that msh2Δ1-MSH3 retains its specificity for looped substrates. The binding defect for this protein complex appeared to be in its general, non-specific DNA binding activity.
Figure 4.
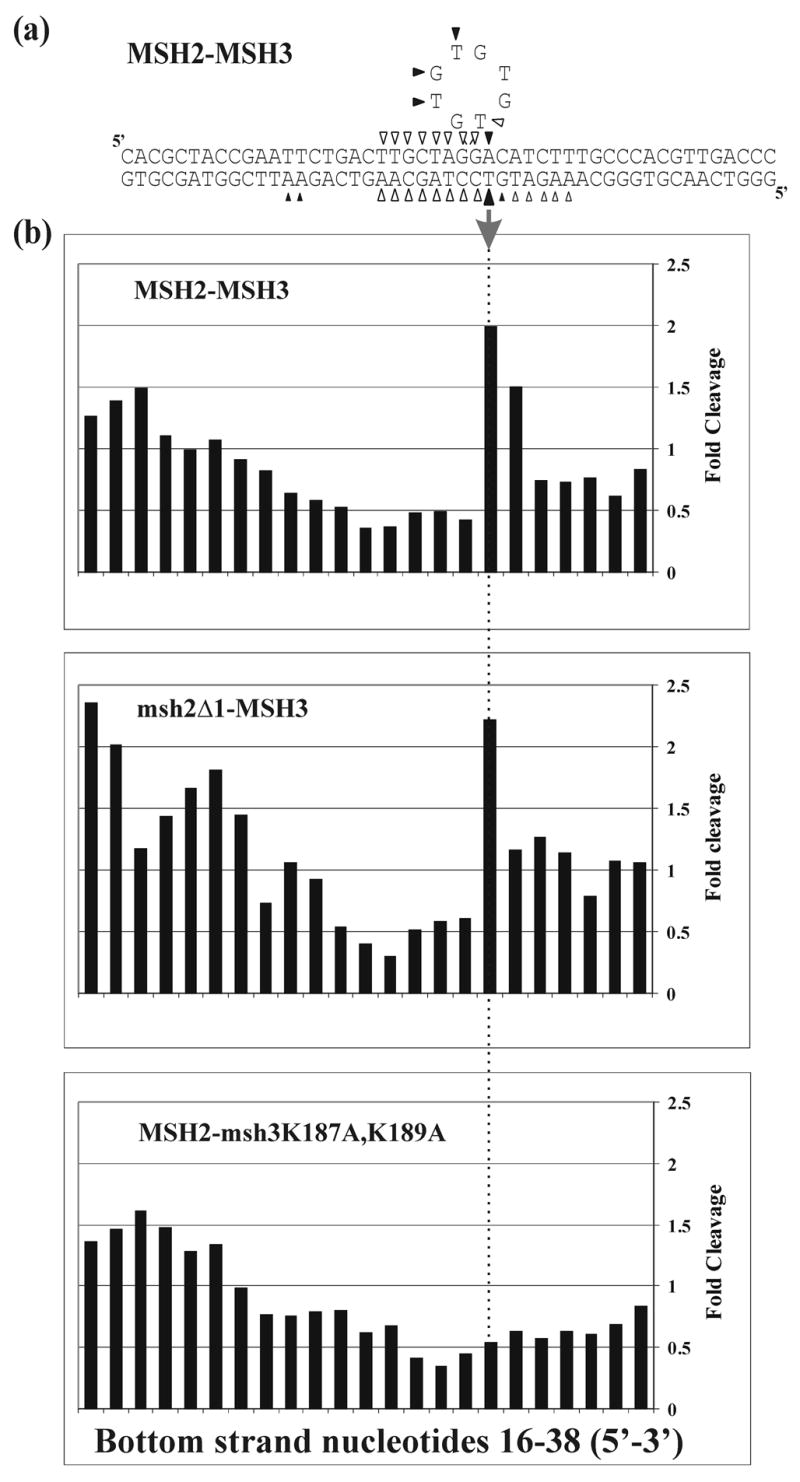
In situ footprinting of DNA-protein complexes with 1, 10-phenanthroline-copper (OP-Cu). MSH2-MSH3-DNA complexes, msh2Δ1-MSH3-DNA or MSH2-msh3K187A,K189A-DNA complexes were separated by gel electrophoresis and treated with OP-Cu as described in Materials and Methods. (a) Summary of OP-Cu cleavage of top and bottom strands of the +8 loop substrate in the presence of MSH2-MSH3 based on at least four independent experiments. Bands that were protected from cleavage are indicated by the empty symbols. Enhanced cleavage is indicated by the filled symbols. Smaller symbols indicate weaker protection or enhancement. MSH2-MSH3 binding creates a “signature” enhancement on the bottom strand of the substrate, as indicated by the filled arrow. (b) Histograms of representative protection patterns of the +8 loop bottom strand substrate. Only the central portion of each substrate is shown. The signal of each band of bound DNA was normalized to the equivalent band produced in the absence of protein. Values greater than 1 represent enhanced cleavage; values lower than 1 represent protection from cleavage. The arrow shows the signature enhancement observed upon MSH2-MSH3 binding to a +8 loop substrate.
MSH3 Lys-187 and Lys-189 residues play critical roles in MSH2-MSH3 mediated MMR
The finding that msh2Δ1-MSH3 bound to the +8 loop substrate at high protein concentrations and showed a footprinting pattern similar to that of wild-type MSH2-MSH3 encouraged us to test whether Domain I of MSH3 was critical for mismatch binding specificity. We made a series of deletion and point mutations in Domain I of MSH3 using the Taq MutS crystal structure as a guide and investigated the effect of these mutations on the repair of loop mispairs (Figures 1 and 5(a) and Table 5). The first amino acid of Domain I of Taq MutS (and MSH2) corresponds to amino acid 149 in MSH3. Therefore we made two versions of a Domain I deletion mutant in MSH3 (Figure 1); a Domain I (amino acids 149–306) deletion mutant based on the amino acid sequence alignment between S. cerevisiae MSH3 and the Taq MutS (designated msh3Δ1, this is analogous to msh2Δ1), and a larger deletion of the 5’ end of MSH3 corresponding to amino acids 2–306 (designated msh3Δ1a). We also made a smaller deletion mutant (amino acids 162–202, msh3Δ1b) containing a deletion in a conserved MutS/MSH region (Figure 1). The msh3Δ1a and msh3Δ1 displayed high rates of microsatellite instability that were similar to that seen in msh3Δ (Figure 5(a) and Table 5). Microsatellite instability in msh3Δ1b strains was intermediate between wild-type and msh3Δ (msh3Δ1b vs. wild-type, p < 0.001; msh3Δ1b vs. msh3Δ, p = 0.042). Together, these results suggest that the Domain I of MSH3 plays a critical role in the repair of loop mispairs (see below).
Figure 5.
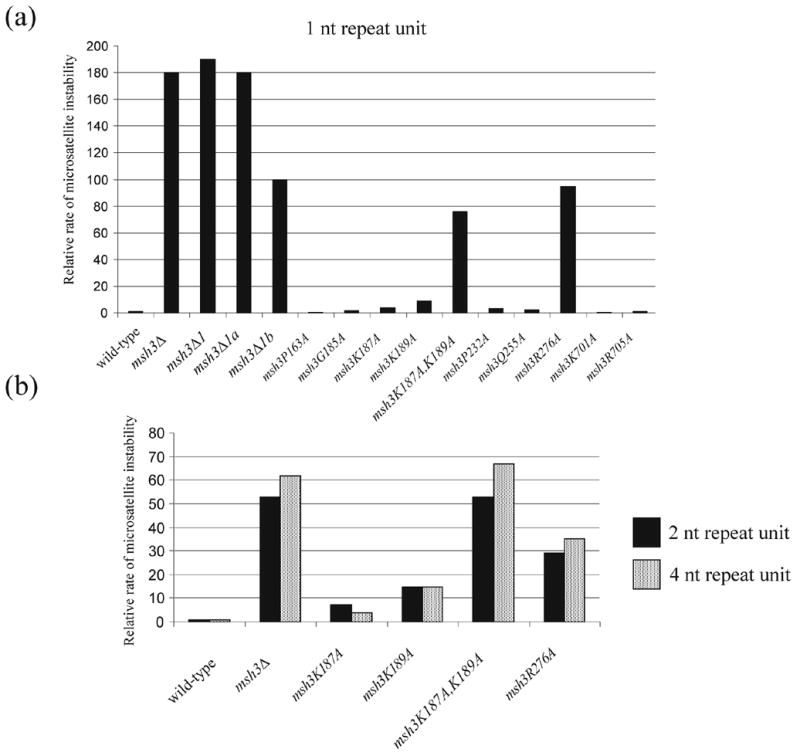
Relative rates of microsatellite instability (data from Table 5) in msh3 strains bearing point mutations in Domains I and IV. In all panels, data are presented relative to the appropriate wild-type strain. (a) 1 nt repeat, wild-type = 4.2 x 10−6; (b) 2 nt repeat, wild-type = 1.5 x 10−6; (c) 4 nt repeat, wild-type = 2.1 x 10−6.
In MutS, a phenylalanine residue (Phe-39 in Taq or Phe-36 in E. coli) in subunit A was shown to stabilize the protein-DNA complex by base stacking with an unpaired/mismatched thymidine residue. In addition, a glutamate residue (Glu-41 in Taq or Glu-38 in E. coli) in the same subunit of MutS also has been shown to play critical roles in mismatch discrimination and activating downstream repair events by making hydrogen bonds with the unpaired/mismatched thymidine.3, 4, 7, 8 These phenylalanine and glutamate residues are conserved in MSH6 (Phe-337 and Glu-339) and are important for MSH2-MSH6 mismatch specific and non-specific DNA binding (Ref. 11–14; Table 5). In yeast MSH3 these positions contain lysine residues (Lys-187 and Lys-189; Figure 1). We mutated each of these lysine residues to alanines and examined the effect of single and double mutants in the microsatellite instability assay.
As shown in Figure 5 and Table 5, rates of microsatellite instability in msh3K187A and msh3K189A mutants were elevated four and nine fold relative to wild-type, respectively. The msh3K187A,K189A double mutant, however, displayed a dramatically elevated rate of microsatellite instability. This mutant showed differential effects on the rates of microsatellite instability depending on the repeat unit sizes; the msh3K187A,K189A showed an intermediate mutator phenotype for the repeat unit size of 1 nt (msh3K187A,K189A vs. wild-type, p < 0.001; msh3K187A,K189A vs. msh3Δ, p < 0.001) but showed a msh3Δ-like phenotype for repeat unit sizes of 2 and 4 nt (Figure 5 and Table 5; msh3K187A,K189A vs. msh3Δ, p > 0.55). These observations suggest that the two lysine residues in MSH3 are essential for MSH2-MSH3-mediated MMR.
The Arg-110 residue in Taq MutS (Arg-108 in E. coli MutS) was shown to make a direct contact with the sugar-phosphate backbone surrounding the mismatched DNA.3,4 When the corresponding residue in MSH6 (Arg-412) was mutated, a strong mutator phenotype was observed. Furthermore, overexpression of the msh6-R412G allele conferred a dominant negative phenotype and the MSH2-msh6R412G complex displayed defects in mismatch binding.12, 13 We made the corresponding change in MSH3 (R276A) and found that msh3R276A mutant displayed elevated rates of microsatellite instability for the repeat unit sizes of 1, 2, and 4 nt (Figure 5 and Table 5). This suggests that like MSH6 Arg-412 in MSH2-MSH6 MMR, Arg-276 in MSH3 plays a critical role in MSH2-MSH3 MMR. Other point mutations in Domain I of MSH3, such as P232A and Q255A, conferred small effects on the rate of microsatellite instability (Table 5). In contrast, when the corresponding residues in MSH6 (P370 and Q393) were mutated to alanines, high and intermediate mutator phenotypes were observed, respectively.12 Also, the MSH2-msh6Q393R complex was defective in mismatch binding and overexpression of msh6-Q393R conferred a dominant negative mutator phenotype.13
In Taq MutS Domain IV of subunit B is thought to stabilize the bent DNA backbone of the mismatch substrate.3 Specifically, a lysine residue (Lys-471 in Taq) and an arginine residue (Arg-475 in Taq) in Domain IV of Taq and E. coli MutS are highly conserved among MutS/MSH proteins and were shown to make direct contacts with the sugar-phosphate backbone. Two amino acid residues (Lys-701 and Arg-705) in Domain IV of MSH3 were mutated individually to alanine and the effect of these mutations on the rate of microsatellite instability was measured. When the corresponding lysine residues in MSH2 (Lys-564) and MSH6 (Lys-848) were mutated, high mutator and wild-type phenotypes were seen, respectively.12 These mutations in MSH3 (msh3K701A and msh3R705A) conferred wild-type-like phenotypes (Table 5).
The defect of msh3K187A,K189A in repairing the loop mispairs in vivo is due to its altered DNA-binding activity
We purified MSH2-msh3K187A,K189A to test whether it displays a defect in binding to the large DNA loop substrates. As shown in gel mobility shift assays, MSH2-msh3K187A,K189A exhibited stronger binding to the homoduplex DNA and the +1 loop substrate than MSH2-MSH3 under these conditions (Figure 3). The increased binding of MSH2-msh3K187A,K189A to homoduplex and the +1 loop substrates suggested that this complex displays an enhanced non-specific DNA-binding activity. This suggestion was supported by the smeary appearance of the shifts in the presence of MSH2-msh3 K187A,K189A, in contrast to the discrete bands observed with MSH2-MSH3. This indicates alternative conformations of the protein-DNA complex or, more likely, unstable MSH2-msh3K187A,K189A binding to the +8 loop substrate. Nonetheless, the overall binding of MSH2-msh3K187A,K189A to all the DNA substrates tested was increased relative to MSH2-MSH3, presumably as a result of increased non-specific DNA binding activity.
To further characterize the DNA-binding specificity of this mutant protein complex, we performed titration experiments with homoduplex, +1 loop, and +8 loop substrates in the absence of competitor DNA to calculate approximate Kd values (Table 6). MSH2-msh3K187A,K189A displayed an approximately 2-fold lower Kd for the homoduplex substrate than MSH2-MSH3, and an increased Kd for the +8 loop substrate. Although these values indicated less than a twofold preference for the +8 loop substrate, MSH2-msh3K197A,K189A reproducibly bound more strongly to the looped substrate. These results suggest that Lys-187 and Lys-189 in MSH3 play critical roles in mismatch repair by suppressing non-specific DNA-binding activity and increasing the substrate specificity for large loop mispairs. This is in contrast to the equivalent MSH2-msh6F337A protein complex, which is highly compromised for all DNA-binding activity.11, 12
We performed in situ OP-Cu footprinting of MSH2-msh3K187A,K189A bound to the +8 loop substrate (Figure 4). When MSH2-MSH3 or msh2Δ1-MSH3 bound to this substrate, we observed a signature footprint pattern on the bottom DNA strand (Figure 4). MSH2-msh3K187A,K189A binding did not produce this signature. Instead, it produced an extended pattern of protection so that the protection center overlapped the center of the substrate, which is similar to that seen by MSH2-MSH3 and MSH2-MSH6 bound to the +1 insertion substrate (J.A.S., unpublished observations). This was in stark contrast to the asymmetric protection by MSH2-MSH3 (or msh2Δ1-MSH3), which is centered over the 5’ side of the loop. Importantly, MSH2-msh3K187A,K189A binding did not produce the signature enhancement observed with MSH2-MSH3 (Figure 4(b)). All three shifted complexes observed with MSH2-msh3K187A,K189A bound to the +8 loop substrate and exhibited the same pattern of protection (Figure 3).
Discussion
In this study we showed that the MSH2 DNA binding Domain I was critical for MSH2-MSH3 but not MSH2-MSH6-dependent MMR. This conclusion was made because msh2Δ1, msh3Δ and msh2Δ1 msh3Δ mutants displayed similar rates of microsatellite instability for 1 and 2 nucleotide short repeat tracts and msh2Δ1 strains were defective in repressing homeologous recombination for MSH2-MSH3 but not MSH2-MSH6-specific recombination substrates. These results are consistent with previous studies showing that mismatches generated during recombination are recognized by the same MSH factors that act in MMR.16, 25, 26 Biochemical and genetic analyses showed that MSH2 Domain I is important for the non-specific DNA binding activity of MSH2-MSH3 and that MSH3 Domain I is likely to be important for MSH2-MSH3 mismatch binding specificity. Our observations suggest that mismatch recognition by MSH2-MSH3 is unlikely to follow the paradigm established by crystallographic analysis of MutS and structure-function studies of MSH2-MSH6.3, 4, 11, 12
How is mismatch recognition accomplished by MSH2-MSH3? This question has been difficult to address because a crystal structure of MSH2-MSH3 bound to DNA containing loop mismatches is unavailable and the phenylalanine and glutamate residues that contact mismatch DNA in MutS are absent in this complex (Figure 1). Mutational analysis of Domain I residues that are either homologous to Taq MutS or are conserved within MSH2 and MSH3 subfamilies has not led to the identification of key residues required for MSH2-MSH3-specific mismatch recognition (Table 5). In contrast, much more is known about mismatch recognition by MSH2-MSH6 mainly because, like MutS, MSH6 contains phenylalanine (337) and glutamate (339) residues at sites predicted to interact with the mismatch, and mutational analysis has shown that these residues are important in MSH2-MSH6 for mismatch and general DNA binding (Figure 1; Table 5; Refs. 11, 12, 14).
We analyzed MSH2-MSH3 mismatch binding specificity by creating point mutations and small deletion mutations in the putative mismatch recognitions domains of MSH2 and MSH3. Of particular interest was msh3K187A,K189A, which contains mutations in MSH3 that mapped to the phenylalanine and glutamate positions in MutS (Figure 1). This allele conferred strong defects in MSH2-MSH3-mediated MMR that were not observed in msh3K187A and msh3K189A single mutants (Figure 5, Table 5). Strikingly, the msh3K187A,K189A mutant showed defects in microsatellite instability that were dependent on repeat unit size; this mutant showed an intermediate mutator phenotype for the repeat unit size of one nucleotide but a null-like phenotype for larger repeats. The strong mutator phenotype conferred by the msh3K187A,K189A mutation, combined with biochemical studies indicating that mismatch binding specificity was reduced but not eliminated in MSH2-msh3K187A,K189A, suggest that the MSH3 K187 and K189 residues participate in, but are not the sole determinants of, MSH2-MSH3 mismatch recognition. This work also suggests that multiple residues in Domain I of MSH2 and MSH3 participate in MSH2-MSH3 mismatch recognition.
We found that the DNA binding domains that contribute to general and mismatch-specific DNA binding appear to be organized differently in MSH2-MSH3 and MSH2-MSH6. As described above, the critical phenylalanine and glutamate counterparts in MSH3 Domain I, K187 and K189, appeared to be important for MSH2-MSH3 binding to large loop mispairs and for repressing non-specific binding while MSH2 Domain I appeared to be required for general DNA binding (Figure 3). For MSH2-MSH6, MSH6 Domain I Phe-337 appeared important for both non-specific and mismatch-specific DNA binding while MSH2 Domain I appeared dispensable. Previous biochemical analysis suggested that MSH2-MSH3 specifically recognized loop mismatches and branched DNA structures by binding to double-strand/single-strand junctions.10 One possibility is that the K187 and K189 residues in MSH3 contribute to mismatch recognition by interacting through ionic interactions with the sugar-phosphate backbone near the double-strand/single strand junction rather than stacking with mismatched bases as was observed for MutS.3, 4 We find this attractive because 1, 10-phenanthroline-copper (OP-Cu) and potassium permanganate footprinting analyses of MSH2-MSH3 bound to loop mismatches showed that MSH2-MSH3 conferred protection at the double-strand/single-strand junction and enhanced cleavage of single-stranded DNA present in loop mismatches.10 This signature footprint appears to be lost when K187 and K189 are replaced with alanines (Figure 4).
Our mutational analysis provides an explanation for why scientists have been unsuccessful in altering the mismatch binding specificities of MSH2-MSH3 and MSH2-MSH6 by simply swapping small sets of conserved residues in Domains I of MSH3 and MSH6 (Refs. 11, 12; T. Goldfarb, J. Haber, and E. A., unpublished data). In addition to showing that MSH2 Domain I functions differently in MSH2-MSH3 and MSH2-MSH6 pathways, we found several mutations in conserved residues of MSH proteins that confer different phenotypes in MSH2-MSH3 and MSH2-MSH6-specific pathways. For example, msh3P232A and msh3Q255A conferred wild-type-like phenotypes in MSH2-MSH3-mediated MMR (Table 5), whereas msh6P370A and msh6Q393A conferred high and intermediate mutator phenotypes, respectively, in MSH2-MSH6-mediated MMR.12 Interestingly, there was only one case where mutations in MSH3 (msh3R276A) and MSH6 (msh6R412A) that correspond to equivalent positions in MutS conferred similar defects in MMR (Table 6; Refs. 12, 13). For these alleles the homologous arginine in MutS was shown to contact the sugar-phosphate backbone in the vicinity of the mismatch. These observations suggest that structural approaches will be required to better understand the mismatch recognition properties of MSH2-MSH3.
Materials and Methods
Yeast strains and plasmids
msh2, msh3, and msh6 mutant derivatives of EAY74516, EAY740, SJR769, SJR770, SJR767, GCY559, and GCY615 20 parental strains were constructed by single step gene replacement using the lithium acetate method (Tables 1, 2; Ref. 27). Single step integration plasmids used to make the msh2, msh3, and msh6 alleles are listed in Table 1. When specified, the hisG-URA3-hisG cassette inserted into the target locus was “popped out” to delete URA3.28 For the Domain I and point mutation alleles, single step integration plasmids were constructed to include the LEU2 or KANMX markers at least 300 bp downstream of the stop codon of the mutated target gene. Insertion of the selectable marker did not affect the function of the wild-type target gene in microsatellite instability and homeologous recombination assays.
TABLE 1.
Strains and plasmids used to measure DNA slippage
| Strain | Genotype | Integration plasmid(s) |
|---|---|---|
| EAY745 | wild-type | |
| EAY1565 | msh2Δ::LEU2 | pEAI220 |
| EAY854 | msh3Δ::hisG | pEAI88 |
| EAY855 | msh6Δ::hisG | pEAI108 |
| EAY1398 | msh2Δ1::LEU2 | pEAI169 |
| EAY1566 | msh2Δ1::LEU2 msh3ΔΔ::hisG | pEAI169, pEAI88 |
| EAY1567 | msh2Δ1::LEU2 msh6Δ::hisG | pEAI169, pEAI108 |
| EAY856 | msh3Δ::hisG msh6Δ::hisG | pEAI88, pEAI108 |
| EAY1568 | msh2Δ1a::LEU2 | pEAI221 |
| EAY1569 | msh2Δ1b::LEU2 | pEAI222 |
| EAY1570 | msh2Δ1c::LEU2 | pEAI223 |
| EAY1571 | msh3Δ 1::LEU2 | pEAI224 |
| EAY1572 | msh3Δ1a::LEU2 | pEAI225 |
| EAY1573 | msh3Δ1b::LEU2 | pEAI226 |
| EAY1574 | msh2F19A::LEU2 | pEAI227 |
| EAY1575 | msh2Y42A::LEU2 | pEAI228 |
| EAY1576 | msh2Q89A::LEU2 | pEAI229 |
| EAY1577 | msh2I108A::LEU2 | pEAI230 |
| EAY1578 | msh3P163A::LEU2 | pEAI231 |
| EAY1579 | msh3G185A::LEU2 | pEAI232 |
| EAY1580 | msh3K187A::LEU2 | pEAI233 |
| EAY1581 | msh3K189A::LEU2 | pEAI234 |
| EAY1582 | msh3K187A, K189A::LEU2 | pEAI235 |
| EAY1583 | msh3P232A::LEU2 | pEAI236 |
| EAY1584 | msh3Q255A::LEU2 | pEAI237 |
| EAY1585 | msh3R276A::LEU2 | pEAI238 |
| EAY1586 | msh3K701A::LEU2 | pEAI239 |
| EAY1587 | msh3R705A::LEU2 | pEAI240 |
| EAY1588 | msh6F337A::LEU2 | pEAI243 |
| DNA slippage vectors | repeat unit | nucleotide sequence in repeat unit |
| pMD28 | 1 | (G)18 |
| pSH44 | 2 | (GT)16.5 |
| pBK1 | 4 | (CAGT)16 |
| pBK3 | 5 | (CAACG)15 |
| pBK10 | 8 | (CAATCGGT)10 |
Strains used to measure DNA slippage events were transformed with the indicated reporter plasmids. Plasmids are described in greater detail in Sia et al.17 Strains are derived from EAY745 16 (MATa Δho HMLalpha Δhmr::ADE1 ade1–100 leu2–3,112 lys5 trp1::hisG ura3–52 ade3::GAL::HO MSH2-HA4::LEU2) except for the msh3Δ::hisG mutant derivatives described in Table 5 which were derived from EAY740 (MATa Δho HMLalpha Δhmr::ADE1 ade1–100 leu2–3,112 lys5 trp1::hisG ura3–52 ade3::GAL::HO).
Microsatellite instability and homeologous recombination assays
Strains listed in Table 1 were transformed with reporter plasmids pMD28, pSH44, pBK1, pBK3, and pBK10 17 using the lithium acetate method.27 These plasmids contain microsatellite sequences inserted in frame into the coding region of URA3; ura3 chromosomal mutants bearing these plasmids are Ura+. DNA slippage events in these strains that disrupt the reading frame of the URA3 reporter cause a Ura−, 5- FOAR phenotype. DNA slippage events were measured in strains containing the indicated reporter plasmid. These strains were struck to single colonies onto minimal medium29 lacking tryptophan. 7–21 single colonies (but in almost all cases 14–21) from each strain were inoculated into 3 ml of medium lacking tryptophan, leucine, and threonine and grown to stationary phase. Multiple transformants were tested to confirm phenotypes. Appropriate dilutions of cells were plated onto selective (lacking tryptophan, leucine, and threonine but containing 5-FOA) and permissive (lacking tryptophan, leucine, and threonine) minimal medium.29 Plates were incubated for 5 days at 30 °C. The rate of microsatellite instability was calculated for each culture according to the formula μ = f/ln(N·μ), where f is the microsatellite instability frequency of 5-FOAr cells in each culture and N is the population size. The rate was determined from the median μ value obtained from all cultures analyzed for each genotype. The 95% confidence interval was also determined from this analysis.21
All yeast strains and recombination substrates used to measure homeologous recombination are listed in Table 2. This assay was performed by streaking strains onto YPD (1% yeast extract, 2% bacto-peptone, and 2% dextrose) plates. 14–21 single colonies per strain were then inoculated into 5 ml of YPGG (1% yeast extract, 2% bacto-peptone, 4% galactose, and 2% glycerol) medium and grown for 2 days at 30 °C. Appropriate dilutions of cells were plated onto synthetic galactose medium lacking histidine (selective) and onto synthetic complete medium (permissive). Plates were incubated for 4 days at 30 °C and then scored for frequency of His+ colonies. The rate of homeologous recombination was calculated as described.20
Pair-wise Kruskall-Wallis tests were performed between all strains with each reporter assay using MINITAB (http://www.minitab.com). Differences were considered significant when p < 0.05.
Expression and purification of MSH2-MSH3
MSH2-MSH3 was overexpressed and purified from a protease-deficient strain of S. cerevisiae (EAY33) containing pMMR8 (MSH2) and pMMR20 (MSH3) as described.10 msh2Δ1-MSH3 and MSH2-msh3K187A,K189A complexes were purified using the corresponding mutant derivatives of pMMR8 and pMMR20. msh2Δ1-MSH3 was purified in protease-deficient yeast derivatives of EAY33 deleted for MSH2.10 Both complexes were purified with yields similar to wild type, suggesting that the deletion mutation did not disrupt protein stability. Protein concentrations were determined in the Bradford assay30 using BSA (Sigma-Aldrich) as a standard.
Gel mobility shift assays
The 49-mer DNA substrates used in gel mobility shift assays were homoduplex (LS1/LS2), +1 insertion (LS2/LS6) and +8 loop (LS2/LS8). Substrates were prepared as described.10 The standard DNA binding assay (10 μl) contained 50 nM 5’ end-labeled [32P] substrate in 20 mM HEPES (pH 7.5), 100 mM NaCl, 1 mM DTT, 40 μg/ml BSA, 2 mM MgCl2 and 50 ng sonicated salmon sperm (Roche) competitor DNA. Reactions were assembled on ice with MSH2-MSH3, msh2Δ1-MSH3 or MSH2-msh3K187A,K189A added last, and then incubated at room temperature for 5 minutes. Samples were electrophoresed through 4% non-denaturing polyacrylamide (29:1) gels in 45 mM Tris borate, 0.5 mM EDTA (pH 8.0) at 130V for 45 minutes in a water-cooled gel electrophoresis apparatus. Gels were dried and exposed to a PhosphorImager screen (Molecular Dynamics) and quantified with ImageQuant (Amersham). For titration experiments, the 5’ end-labeled [32P] substrate was reduced to 1 nM and the competitor DNA was excluded. Protein concentrations ranged from 0 – 400 nM.
In situ OP-Cu footprinting
In situ 1,10-phenanthroline-copper (OP-Cu) footprinting was performed as described.10 Briefly, binding reactions were performed in 10 μl volumes containing 1–2 pmol 32P-labeled DNA substrate as described above for the gel mobility shift assays. Protein-DNA complexes were separated by electrophoresis through a 6% non-denaturing polyacrylamide gel. The gel was immersed in 10 mM Tris-HCl (pH 8.0), then treated with 20 ml solution A (2 mM OP-Cu, 0.45 mM CuSO4; Sigma-Aldrich) and 20 ml solution B (1:200 dilution of 3-mercaptopropionic acid in ddH20; Sigma-Aldrich). After 15 minutes at room temperature, digestion was stopped by the addition of 20 ml solution C (28 mM neocuproine; Sigma-Aldrich). Protein-DNA complexes and free DNA bands were located by autoradiography of the wet gel and excised, crushed and eluted overnight at 37°C in 0.5 M ammonium acetate, 1 mM EDTA (pH 8.0). The extracted DNA was phenol:chloroform extracted, precipitated with ethanol, dried and resuspended in 6 μl loading dye (98% formamide, 10 mM EDTA (pH 8.0), 0.025% xylene cyanol, 0.025% bromphenol blue). Samples were separated by electrophoresis on 10% denaturing urea-polyacrylamide gels. Sequencing standards were created by dimethyl sulfate (Sigma-Aldrich) modification of DNA substrates and cleavage with NaOH to generate G>A DNA ladders. The gels were dried on DE81 paper and exposed to a PhosphorImager screen for quantification (ImageQuant, Amersham). The footprint patterns were quantified by normalizing each band relative to the total counts per lane.
Acknowledgments
We thank Elaine Sia and Tom Petes for providing the plasmids and yeast strains for the DNA slippage studies and Sue Jinks-Robertson for providing the yeast strains required to measure homeologous recombination in the HIS3::intron assay. We are also grateful to T. Goldfarb for initiating this work, the Alani laboratory for helpful comments on the manuscript, and Wei Yang for suggesting deletion endpoints in Domains I of MSH2 and MSH3. S.D.L. was partially supported by a Field of Genetics and Development NIH training grant. J.A.S. is a Research Fellow of the National Cancer Institute of Canada supported with funds provided by the Terry Fox Run and E. A. was supported by NIH research grant GM-53085.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- 2.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 3.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 4.Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, de Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G x T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Yang Y, Schofield MJ, Du C, Fridman Y, Lee SD, Larson ED, Drummond JT, Alani E, Hsieh P, Erie DA. DNA bending and unbending by MutS govern mismatch recognition and specificity. Proc Natl Acad Sci USA. 2003;100:14822–14827. doi: 10.1073/pnas.2433654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto A, Schofield MJ, Biswas I, Hsieh P. Requirement for Phe36 for DNA binding and mismatch repair by Escherichia coli MutS protein. Nucleic Acids Res. 2000;28:3564–3569. doi: 10.1093/nar/28.18.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schofield MJ, Nayak S, Scott TH, Du C, Hsieh P. Interaction of Escherichia coli MutS and MutL at a DNA mismatch. J Biol Chem. 2001;276:28291–28299. doi: 10.1074/jbc.M103148200. [DOI] [PubMed] [Google Scholar]
- 8.Lebbink JH, Georgijevic D, Natrajan G, Fish A, Winterwerp HH, Sixma TK, de Wind N. Dual role of MutS glutamate 38 in DNA mismatch discrimination and in the authorization of repair. EMBO J. 2006;25:409–419. doi: 10.1038/sj.emboj.7600936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kijas AW, Studamire B, Alani E. msh2 separation-of-function mutations confer defects in the initiation steps of mismatch repair. J Mol Biol. 2003;331:123–138. doi: 10.1016/s0022-2836(03)00694-6. [DOI] [PubMed] [Google Scholar]
- 10.Surtees JA, Alani E. Mismatch repair factor MSH2-MSH3 binds and alters the conformation of branched DNA structures predicted to form during genetic recombination. J Mol Biol. 2006;360:523–536. doi: 10.1016/j.jmb.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 11.Bowers J, Sokolsky T, Quach T, Alani E. A mutation in the MSH6 subunit of the Saccharomyces cerevisiae MSH2- MSH6 complex disrupts mismatch recognition. J Biol Chem. 1999;274:16115–16125. doi: 10.1074/jbc.274.23.16115. [DOI] [PubMed] [Google Scholar]
- 12.Drotschmann K, Yang W, Brownwell FE, Kool ET, Kunkel TA. Asymmetric recognition of DNA local distortion. Structure-based functional studies of eukaryotic Msh2–Msh6. J Biol Chem. 2001;276:46225–46229. doi: 10.1074/jbc.C100450200. [DOI] [PubMed] [Google Scholar]
- 13.Das Gupta R, Kolodner RD. Novel dominant mutations in Saccharomyces cerevisiae MSH6. Nat Genet. 2000;24:53–56. doi: 10.1038/71684. [DOI] [PubMed] [Google Scholar]
- 14.Dufner P, Marra G, Raschle M, Jiricny J. Mismatch recognition and DNA- dependent stimulation of the ATPase activity of hMutSalpha is abolished by a single mutation in the hMSH6 subunit. J Biol Chem. 2000;275:36550–36555. doi: 10.1074/jbc.M005987200. [DOI] [PubMed] [Google Scholar]
- 15.Sugawara N, Paques F, Colaiacovo M, Haber JE. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc Natl Acad Sci USA. 1997;94:9214–9219. doi: 10.1073/pnas.94.17.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldfarb T, Alani E. Distinct roles for the Saccharomyces cerevisiae mismatch repair proteins in heteroduplex rejection, mismatch repair and nonhomologous tail removal. Genetics. 2005;169:563–574. doi: 10.1534/genetics.104.035204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sia E, Kokoska R, Dominska M, Greenwell P, Petes T. Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol Cell Biol. 1997;17:2851–2858. doi: 10.1128/mcb.17.5.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsischky GT, Filosi N, Kane MF, Kolodner RD. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 19.Surtees JA, Argueso JL, Alani E. Mismatch repair proteins: key regulators of genetic recombination. Cytogen Genome Res. 2004;107:146–159. doi: 10.1159/000080593. [DOI] [PubMed] [Google Scholar]
- 20.Nicholson A, Hendrix M, Jinks-Robertson S, Crouse GF. Regulation of mitotic homeologous recombination in yeast. Functions of mismatch repair and nucleotide excision repair genes. Genetics. 2000;154:133–146. doi: 10.1093/genetics/154.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–2865. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson T, Guerrette S, Fishel R. Dissociation of mismatch recognition and ATPase activity by hMSH2–hMSH3. J Biol Chem. 1999;274:21659–21664. doi: 10.1074/jbc.274.31.21659. [DOI] [PubMed] [Google Scholar]
- 23.Habraken Y, Sung P, Prakash L, Prakash S. Binding of insertion/deletion DNA mismatches by the heterodimer of yeast mismatch repair proteins MSH2 and MSH3. Curr Biol. 1996;6:1185–1187. doi: 10.1016/s0960-9822(02)70686-6. [DOI] [PubMed] [Google Scholar]
- 24.Palombo F, Iaccarino I, Nakajima E, Ikejima M, Shimada T, Jiricny J. hMutSbeta, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Curr Biol. 1996;6:1181–1184. doi: 10.1016/s0960-9822(02)70685-4. [DOI] [PubMed] [Google Scholar]
- 25.Junop MS, Yang W, Funchain P, Clendenin W, Miller JH. In vitro and in vivo studies of MutS, MutL and MutH mutants: correlations of mismatch repair and DNA recombination. DNA repair. 2003;2:387–405. doi: 10.1016/s1568-7864(02)00245-8. [DOI] [PubMed] [Google Scholar]
- 26.Calmann MA, Nowosielska A, Marinus MG. Separation of mutation avoidance and antirecombination functions in an Escherichia coli mutS mutant. Nucleic Acids Res. 2005;33:1193–1200. doi: 10.1093/nar/gki263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gietz RD, Schiestl RH. Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast. 1991;7:253–263. doi: 10.1002/yea.320070307. [DOI] [PubMed] [Google Scholar]
- 28.Alani E, Kleckner N. A new type of fusion analysis applicable to many organisms: protein fusions to the URA3 gene of yeast. Genetics. 1987;117:5–12. doi: 10.1093/genetics/117.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose MD, Winston F, Hieter P. Methods in yeast genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1990. [Google Scholar]
- 30.Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 31.Studamire B, Price G, Sugawara N, Haber JE, Alani E. Separation-of- function mutations in Saccharomyces cerevisiae MSH2 that confer mismatch repair defects but do not affect nonhomologous-tail removal during recombination. Mol Cell Biol. 1999;19:7558–7567. doi: 10.1128/mcb.19.11.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]