

Abstract
Tissue plasminogen activator (t-PA) and plasminogen activator inhibitor 1 (PAI-1) directly influence thrombus formation and degradation and thereby risk for arterial thrombosis. Activation of the renin-angiotensin system has been linked to the production of PAI-1 expression via the angiotensin II type 1 receptor (AT1R). In addition, bradykinin can induce the release of t-PA through a B2 receptor mechanism. In the present study, we aimed to investigate the epistatic effects of polymorphisms in genes from the renin-angiotensin, bradykinin and fibrinolytic systems on plasma t-PA and PAI-1 levels in a large population-based sample (n=2,527). We demonstrated a strong significant interaction within genetic variations of the bradykinin B2 gene (p=0.002) and between ACE and bradykinin B2 (p=0.003) polymorphisms on t-PA levels in females. In males, polymorphisms in the bradykinin B2 and AT1R gene showed the most strong effect on t-PA levels (p=0.006). In both females as well as males, the bradykinin B2 gene interacted with AT1R gene on plasma PAI-1 levels (p=0.026 and p=0.039, respectively). In addition, the current study found a borderline significant interaction between PAI 4G5G and ACE I/D on plasma t-PA and PAI-1 levels. These results support the idea that the interplay between the renin-angiotensin, bradykinin, and fibrinolytic systems might play an important role in t-PA and PAI-1 biology.
Keywords: Plasminogen, thrombosis, gene-gene interaction, epistasis, epidemiology, fibrinolysis
The endogenous fibrinolytic system describes a complex process resulting in the hydrolytic cleavage of fibrin by plasmin to promote clot dissolution and generate fibrin degradation products. This system helps protect the circulation from intravascular fibrin formation and thrombosis that would otherwise lead to vessel occlusion and tissue ischemia[1;2]. The efficacy of plasminogen activation and fibrin degradation is largely determined by the balance between local concentrations of tissue-type plasminogen activator (t-PA) and it’s cognate inhibitor, plasminogen activator inhibitor type 1 (PAI-1). There is substantial evidence indicating that plasma t-PA and PAI-1 are regulated in part by the renin-angiotensin system (RAS)[3]. Angiotensin II promotes the synthesis of PAI-1 in vitro[4;5]. Furthermore, ACE inhibitors have been shown to reduce plasma PAI-1 levels in patients post-myocardial infarction[6], in hypertensive subjects[7], and in postmenopausal females[8]. Conversely, bradykinin is a potent stimulator for t-PA in humans[9], and ACE-inhibition augments bradykinin induced t-PA release from the arterial vasculature[10;11].
Interactions between the RAS, bradykinin and fibrinolytic systems on t-PA and PAI-1 levels may explain at least part of the relationship between those systems and cardiovascular disease and the beneficial effects of ACE-inhibition on thrombotic events. Indeed, prior studies demonstrated interactions between genes from the fibrinolytic system and RAS on levels of t-PA and PAI-1[12–14]. However, these studies were either performed in selected high-risk populations or had small sample sizes. In addition, no study has yet investigated the interplay between genes from the RAS, bradykinin, and fibrinolytic systems on plasma levels of t-PA and PAI-1 in a single study.
Therefore, the aim of the present study was to investigate the effects of polymorphisms in genes from the renin-angiotensin, bradykinin, and fibrinolytic systems on plasma levels of t-PA and PAI-1 in a large population-based sample, explicitly examining two-way epistatic interactions.
Results
Clinical characteristics of both females and males are listed in Table 1. Except for body mass index and total cholesterol levels, all characteristics are significantly different between females and males.
Table 1.
Baseline characteristics expressed as mean ± standard deviation divided by gender.
| Females (n=1338) | Males (n=1189) | P-value* | |
|---|---|---|---|
| Age (years) | 48 ± 12 | 50 ± 12 | 0.008 |
| Body mass index (kg/m2) | 25.8 ± 4.5 | 25.9 ± 3.4 | 0.447 |
| Waist-hip ratio | 0.82 ± 0.07 | 0.93 ± 0.07 | <0.001 |
| Current smoking | 32.2 % | 37.0 % | 0.011 |
| Diabetes | 2.7 % | 3.2 % | 0.453 |
| Systolic blood pressure (mmHg) | 122 ± 19 | 131 ± 17 | <0.001 |
| Diastolic blood pressure (mmHg) | 70 ± 9 | 76 ± 9 | <0.001 |
| Total cholesterol (mmol/l) | 5.60 ± 1.10 | 5.67 ± 1.17 | 0.071 |
| HDL-cholesterol (mmol/l) | 1.52 ± 40 | 1.18 ± 0.32 | <0.001 |
| Ln tissue type plasminogen activator (ng/ml) | 1.13 ± 0.71 | 1.32 ± 0.80 | <0.001 |
| Ln Plasminogen activator inhibitor 1 (ng/ml) | 4.13 ± 0.82 | 4.37 ± 0.79 | <0.001 |
P-value calculated with student’s t-test for continuous variables and Chi-square test for categorical variables
Figures 1 and 2 summarize the results of the statistical tests for epistasis in the form of network diagrams for females and males, respectively. Only significant results with suggestive or strong evidence for interaction are illustrated. Supplementary Tables 1 and 2 present the p-values for all statistical tests performed.
Fig. 1.
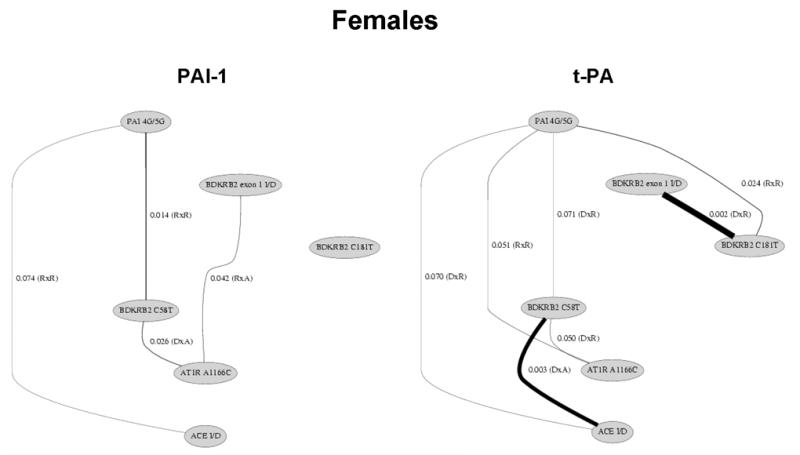
Polymorphisms of the renin-angiotensin, bradykinin, and fibrinolytic system show significant interactions on plasma t-PA and PAI-levels among females. Only the significant (p<0.05) and borderline significant (p<0.10) interactions are presented. The lines illustrate the interactions between genes and line thickness is inversely related to the significance level. The model of inheritance is given in parentheses. A: additive; D: dominant; R: recessive. For example, the BDKRB2 exon 1 I/D by BDKRB2 C181T interaction on t-PA levels involved a dominant by recessive effect with a p-value of 0.002.
Fig. 2.
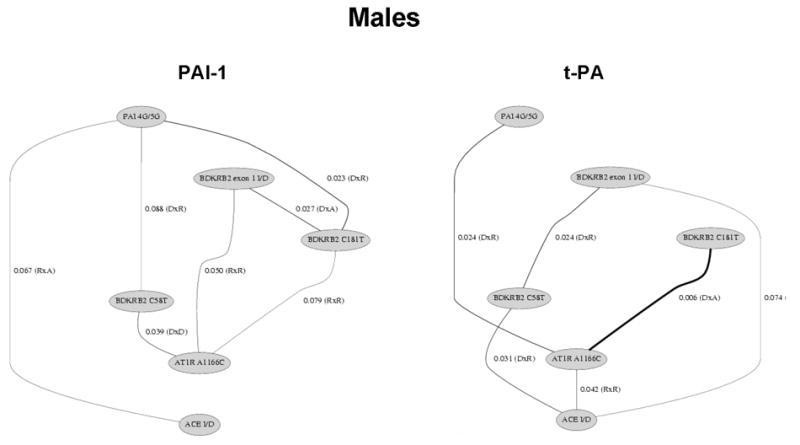
Polymorphisms of the renin-angiotensin, bradykinin, and fibrinolytic system show significant interactions on plasma t-PA and PAI-levels among males. Only the significant (p<0.05) and borderline significant (p<0.10) interactions are presented. The lines illustrate the interactions between genes and line thickness is inversely related to the significance level. The model of inheritance is given in parentheses. A: additive; D: dominant; R: recessive. For example, the AT1R A1166C by BDKRB2 C181T interaction on t-PA levels involved a dominant by additive effect with a p-value of 0.006.
In females, we found significant evidence for fibrinolytic-RAS, fibrinolytic-bradykinin, RAS-bradykinin, and bradykinin-bradykinin interaction effects on plasma t-PA levels. A total of seven gene-gene interactions were detected out of the 15 comparisons. We also found significant evidence for fibrinolytic-RAS, fibrinolytic-bradykinin, and RAS-bradykinin interaction effects on plasma PAI-1 levels. A total of six gene-gene interactions were detected out of the 15 comparisons.
In males, we found significant evidence for fibrinolytic-RAS, RAS-bradykinin, RAS-RAS, and bradykinin-bradykinin interaction effects on plasma t-PA levels. A total of six gene-gene interactions were detected out of the 15 comparisons. We also found significant evidence for fibrinolytic-RAS, fibrinolytic-bradykinin, RAS-bradykinin, and bradykinin-bradykinin interaction effects on plasma PAI-1 levels. A total of seven gene-gene interactions were detected out of the 15 comparisons.
Common to both traits and both genders were significant interactions between genes from the bradykinin system and the RAS, but the specific interaction detected differed between gender. Figure 3 illustrates the mean ln t-PA and mean ln PAI-1 levels by genotype combination in females and males. In each case, there is evidence for crossing of the lines that is characteristic of epistasis. That is, the effect of any one polymorphism on either t-PA or PAI-1 levels is dependent on the genotypes at the second polymorphism.
Fig. 3.
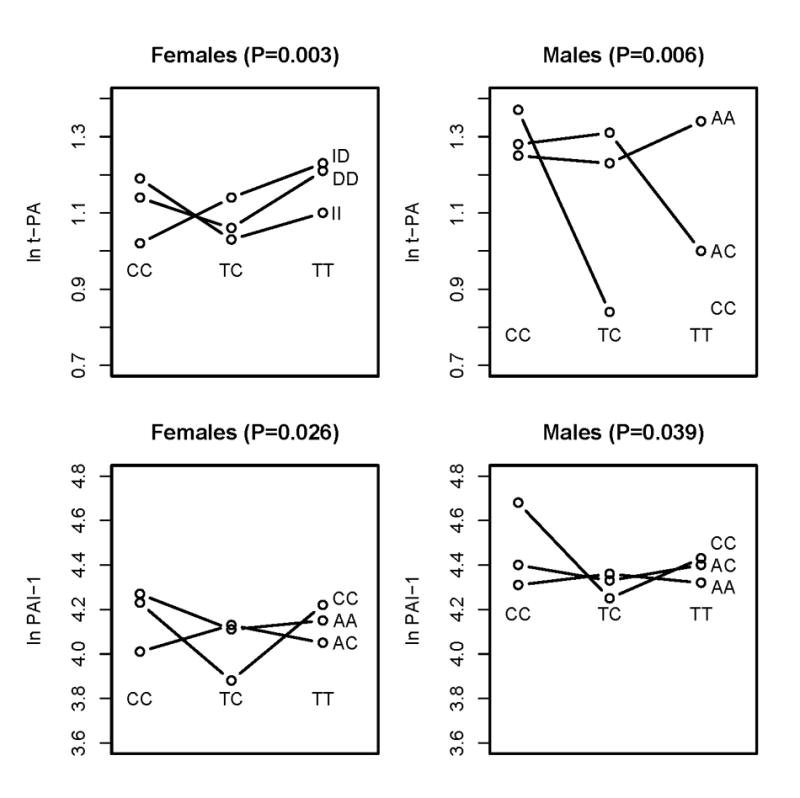
A consistent significant interaction is present between polymorphisms of the renin-angiotensin and bradykinin genes on plasma t-PA and PAI-1 levels among females and males. The four figures illustrate the interactions on the absolute values of the natural log transformed t-PA and PAI-1.
Thus, for both traits and both genders we found evidence for multiple epistatic effects using either suggestive or strong supporting statistical evidence. A total of 24 significant gene-gene interactions were observed out of 60 possible. Of the 24, 10 involved dominant-recessive interactions, seven involved recessive-recessive interactions, four involved dominant-additive interactions, two involved recessive-additive interactions and one involved a dominant-dominant interaction. It is important to note that the interactions reported here and summarized in Figures 1 and 2 are only the most significant of those examined; all results are shown in Supplemental Tables 1 and 2. For example, the BDKRB2 C58T by ACE I/D interaction on t-PA levels in females reported in Figures 1 and 3 involved a dominant by additive effect with a p-value of 0.003. However, the additive by dominant (p = 0.025), dominant by dominant (p = 0.007), and additive by additive (p = 0.018) were all statistically significant at an α=0.025 level. The magnitude of each interaction effect was on the order of 1% variation explained. This is consistent with a genetic architecture in which many polymorphisms account for the heritability of the trait. As such, it will be important explore higher-order models.
It is important to note that there are many differences between females and males (see Figures 1 and 2). For example, there was strong evidence for an interaction effect of the BDKRB2 exon 1 I/D and BDKRB2 C181T polymorphisms on t-PA levels in females (p = 0.002) but not in males (P > 0.10). As an additional example, we observed strong evidence for an interaction effect of the PAI-1 4G/5G and BDKRB2 C181T polymorphisms on PAI-1 levels in males (p = 0.023) but not in females (P > 0.10). These results are consistent with the gender-specific univariate effects of these polymorphisms [31]. One similarity in females and males is that in subjects homozygous for AT1R A1166C, PAI-1 levels are only lower in subjects heterozygous for BDKRB2 C58T in both females and males (Figure 3).
Discussion
The present study examined the relationships between genetic polymorphisms in the RAS, bradykinin, and fibrinolytic systems on plasma levels of t-PA and PAI-1 in a large population-based sample. We observed interactions between genetic polymorphisms of the three different systems both on plasma t-PA levels as well as on plasma PAI-1 levels. Interestingly, these interactions appear to be different among females and males.
Epistatic effects of RAS and bradykinin system on plasma t-PA and PAI-1 levels
The effect of RAS polymorphisms on t-PA and PAI-1 levels has been studied previously, but no data is available about the joint effects of RAS and bradykinin gene polymorphisms on markers of the fibrinolytic system. The I/D polymorphism of the ACE gene has been associated with PAI-1 levels in apparently healthy persons[13], in patients with hypertension[15], and in patients attending a metabolic ward[14]. In contrast, no relation between ACE I/D polymorphism on PAI-1 levels could be detected in the National Heart, Lung, and Blood Institute Family Heart Study[16]. In vitro data have shown that the angiotensin II induced PAI-1 expression may be mediated by the angiotensin II type 1 receptor[3;17].
In the present study, polymorphisms of the RAS and bradykinin genes showed epistatic effects on both PAI-1 as t-PA levels among females and males. In contrast to the aforementioned relation between RAS gene polymorphisms and the fibrinolytic system, less is known about the relation between bradykinin gene polymorphisms and t-PA and PAI-1 levels. Besides being an inflammatory mediator, bradykinin is a potent endothelial cell stimulant that can induce the acute release of t-PA from the endothelium through a B2 receptor mechanism[18]. ACE-inhibition augments the effects of bradykinin on the B2 receptor, which leads to t-PA release from the endothelium[10]. In addition, a recent study showed that ACE inhibition reduced PAI-1 expression in tubular epithelial cells by bradykinin action[19]. To our knowledge, no study investigated the effect of bradykinin gene polymorphisms or the interaction between RAS and bradykinin gene polymorphisms on t-PA and PAI-1 levels. In the present study, we demonstrate several significant interactions between genetic variations of the bradykinin B2 gene on t-PA levels. These findings confirm results from prior studies that bradykinin plays an important role in t-PA levels. In addition, a consistent interaction was present between the RAS and bradykinin system on plasma levels of t-PA and PAI-1 in both genders, although the nature of the interactions differed.
Epistatic effects of PAI 4G5G on plasma t-PA and PAI-1 levels
Despite the strong univariate relation between PAI 4G5G polymorphism and PAI levels[20], the PAI 4G/5G polymorphism did not interact very strongly with the RAS and bradykinin gene polymorphisms. However, a consistent interaction was present between the PAI 4G5G and the ACE I/D polymorphisms in both females and males. This epistatic effect between both systems has been described previously in patients attending a metabolic ward[14] and a normotensive unrelated population[21]. Angiotensin II may regulate PAI-1 levels in the general population and inhibition of renin-angiotensin system has been shown to reduce PAI-1 levels[8]. These data and the current results support the idea that the two systems interact. This therefore could potentially explain the beneficial effects of ACE inhibition on thrombotic events.
Gender differences in epistatic effects on t-PA/PAI-1 levels
It is well known that risk for cardiovascular disease differs among gender and several mechanisms have been proposed to account for this difference[22]. One of the main mechanisms might be the gender difference in fibrinolysis and thereby thrombotic state. The THROMBO investigators found gender differences in several thrombogenic factors predicting recurrent cardiac events in patients after acute myocardial infarction[23]. Several study groups have postulated that the cardioprotective effect of estrogen may be mediated, in part, by an increase in fibrinolytic potential. Subjects with lower estrogen have a lower fibrinolytic potential (higher PAI-1 levels) than subjects with a high estrogen status[24]. This relation between estrogen and fibrinolysis has been related to the capacity of estrogen to regulate the RAS system[25;26]. In addition, ACE-inhibition therapy has been shown to increase basal t-PA release in females but not in males[11]. Furthermore, side effects associated with ACE inhibition such as coughing occur more frequently in females than males. The mechanism of cough associated with ACE-inhibitor therapy is related to the bradykinin metabolism, which again is related to t-PA release in humans[27;28]. These studies and the current results suggest sex specific differences in epistatic effects of RAS, bradykinin and fibrinolytic system on plasma t-PA and PAI-1 levels. Future studies are needed to investigate whether gender-specific therapeutics need to be developed in order to protect against disease processes that show clear sex difference in their etiology and progression.
Future considerations
This study supports the idea that epistasis is a ubiquitous component of the genetic architecture of human traits [29]. Future studies need to address several issues to understand the complicated epistatic effects of polymorphisms in the RAS, bradykinin, and fibrinolytic genes. First, the nature and biological interpretation of the interactions need to be determined. Although this is very important, traversing the hierarchical divide between statistical epistasis, observed as deviations from additivity in a linear model that summarizes specific patterns of genotypes and phenotypes across multiple individuals in a sample, and biological epistasis, observed as biomolecular interactions at the cellular level in an individual, is very difficult and will likely only be possible when all etiological factors can be measured [30,31]. Second, in addition to the described systems, future studies need to consider other systems that might be implicated in arterial thrombosis. Third, this paper focused on two-way interactions, but high-order interactions are certainly possible, but may be hard to detect due to limits of the traditional statistical approaches[32]. Finally, we need to consider the context of the environment on the genetic effects (gene-environment interactions). This approach will lead to a better understanding of the genetic architecture of fibrinolytic factors and subsequently of the development of arterial thrombosis in human.
In conclusion, the results of this large population-based study support the idea that the interplay between the renin-angiotensin, bradykinin, and fibrinolytic systems might have an important role in t-PA and PAI-1 biology and thereby arterial thrombosis.
Materials and methods
Study population
The sample analyzed in this study was obtained from the ongoing prospective Prevention of Renal and Vascular ENd-stage Disease (PREVEND) study. The PREVEND study was designed to investigate prospectively the natural course of albuminuria and its relation to renal and cardiovascular disease in a large cohort drawn from the general population. Details of the study protocol have been described previously[33]. In summary, during 1997–1998, all 85,421 inhabitants of the city of Groningen, the Netherlands, from the ages of 28 to 75 years old were sent a one-page postal questionnaire (regarding demographics, use of medication, and pregnancy) and a vial to collect an early morning urine sample. A total of 40,856 subjects responded. After exclusion of subjects with type 1 diabetes mellitus, females who were possibly pregnant, and males and females not able or willing to participate, a total of 6,000 subjects with a urinary albumin concentration ≥ 10 mg l 1 and a random control sample of subjects with a urinary albumin concentration <10 mg l 1 (n=2,592) completed the screening protocol and formed the baseline PREVEND cohort (n=8,592). From this cohort, we selected a random sample of 2,527 subjects (1338 females and 1189 males) as being representative of the entire population from which the PREVEND cohort was selected. These individuals were used in the present study to explore t-PA and PAI-1 levels in the general population. In this group, t-PA and PAI-1 levels were missing in 39 (1.5%) and 42 (1.7%) subjects, respectively. The PREVEND study was approved by the local medical ethics committee and conducted in accordance with the guidelines of the declaration of Helsinki.
Laboratory measurements
Blood samples were obtained during the morning in all subjects to minimize variability due to circadian rhythms, but day-to-day variability of t-PA and PAI-1 levels was not addressed in the current study. Plasma levels of t-PA and PAI-1 were measured using an ELISA kit from Technoclone Gmbh (Vienna, Austria). The assays measured both free t-PA and PAI-1 as well as their complexes. In brief, diluted plasma samples were added to a 96-well microtiter plate coated with antibodies to either t-PA or PAI and incubated for two hours at 37 degrees Celsius. After washing, a peroxidase-labeled second antibody to either t-PA or PAI-1 was added to the wells and the plate was incubated for one hour at 37 degrees Celsius. After washing, substrate (ABTS) was added. The reaction was stopped after 30 minutes and the absorbance at 405 nm was measured. Each assay included calibrants (1.0–20 ng/ml for t-PA and 1.0–25 ng/ml for PAI-1) and two quality control samples. The lower limit of quantification was 1.0 ng/ml for t-PA and 1.5 ng/ml for PAI-1. Assays not meeting quality control standards were not included in the analyses.
Serum cholesterol was determined by Kodak Ektachem dry chemistry (Eastman Kodak, Rochester, NY, USA). HDL-cholesterol was measured with a homogeneous method (direct HDL, no. 7D67, AEROSET System; Abbott Laboratories).
Measurement of Genetic Polymorphisms
The AT1R A1166C (rs5186), the BDKRB2 C-58T (rs1799722) and C181T (rs1046248), and PAI 4G/5G (rs1799768) polymorphisms were analyzed using TaqMan-MGB probes and PCR primers, designed through the Assay-by-Design service (Applied Biosystems). TaqMan assays were carried out according to the manufacturers recommendations on an ABI 7900HT apparatus and results were analyzed with SDS 2.0 genotype calling software (Applied Biosystems).
The ACE I/D (rs4646994) and the BDKRB2 exon 1 insertion/deletion polymorphisms were also analyzed by PCR amplification, one of the primers having a fluorescent label, and subsequent determination of the lengths of the PCR products on a capillary sequencer. Primers were picked with the aid of the online Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). After PCR cycling, samples of the above assays were pooled and separated on a MegaBACE 1000 sequencer. Fragments were analyzed with Genetic Profiler 2.0 software (Amersham Biosciences).
Statistical Analysis
Continuous data are reported as mean ± standard deviation. Categorical data are presented as per group percentages. Differences in continuous characteristics between genders were evaluated by Student's t-test. Differences in categorical data between genders were compared with the χ2 test.
The primary goal of the statistical analysis was to test the null hypothesis that the effect of any one polymorphism on t-PA or PAI-1 levels is not dependent on one of the other polymorphisms. We used a two-way analysis of variance (ANOVA) to test this null hypothesis. A statistically significant interaction term was considered evidence in favor of the alternative hypothesis of epistasis or gene-gene interaction. All tests were carried out in females and males separately since the distributions of t-PA or PAI-1 have previously been shown to be gender-specific [34].
All genotypes were coded (AA=0, Aa=1, aa=2) to capture additive effects, (0=AA or Aa, 1=aa) to capture dominant effects, and (0=AA, 1=Aa or aa) to capture recessive effects. All possible pairs of genotypes were evaluated using ANOVA. The nine possible interaction effects tested included additive by additive, additive by dominant, additive by recessive, dominant by additive, dominant by dominant, dominant by recessive, recessive by additive, recessive by dominant, and recessive by recessive. For each pair of polymorphisms, all nine interactions were tested and the most significant result reported. We did not limit the analysis by eliminating rare genotypes since the sample size was sufficient in most cases to estimate the interaction effects.
Within each gender and for each trait we tested 15 possible pairs of polymorphisms for epistatic effects. Within each of the 15 comparisons we tested the nine possible interactions (e.g. additive by additive, etc.). Thus, there are multiple statistical tests at the polymorphism level (15 tests) and at the genotype coding level (nine tests). We present and use here two different levels of statistical significance for making inferences about the epistatic effects of the polymorphisms studied. First, we used an uncorrected significance level of α=0.10 as suggestive evidence for interaction. This liberal significance was selected for several reasons. First, the pathways, genes and polymorphisms selected for study are all strong candidates for influencing t-PA and PAI-1 biochemistry. Second, we used a more conservative significance level of α=0.025 as strong evidence for interaction. This more conservative significance level was derived from 1000 Monte Carlo simulations under the null hypothesis. For each of the 1000 simulations, we recorded the smallest p-values selected from the nine different interaction tests. From this null distribution we determined that a minimum p-value of approximately 0.025 or smaller was expected 10% of the time. All p-values for all tests performed are reported so the reader may make their own interpretations regarding biological significance. We did not perform corrections for multiple testing because the universal null hypothesis that is assumed for a Bonferroni-type correction does not apply to this study [35]. In addition, this is the first study to examine interactions among genes from the fibrinolytic, RAS, and bradykinin systems. As such, we are more concerned about type II errors (false-negatives) than type I errors (false-positives). This is especially true since ANOVA has less power to detect interactions than independent main effects.
Supplementary Material
Acknowledgments
This work was supported by NIH grant HL65234, HL67466, RR018787, and by grant E.013 of Netherlands Kidney Foundation. F.W. Asselbergs is a research fellow of the Netherlands Heart Foundation (2003T010) and the Dutch Inter University Cardiology Institute Netherlands (ICIN). We would like to thank Bill C. White for preparing Figures 1 and 2.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Meade TW, Ruddock V, Stirling Y, Chakrabarti R, Miller GJ. Fibrinolytic activity, clotting factors, and long-term incidence of ischaemic heart disease in the Northwick Park Heart Study. Lancet. 1993;342:1076–1079. doi: 10.1016/0140-6736(93)92062-x. [DOI] [PubMed] [Google Scholar]
- 2.Ridker PM, Vaughan DE, Stampfer MJ, Manson JE, Hennekens CH. Endogenous tissue-type plasminogen activator and risk of myocardial infarction. Lancet. 1993;341:1165–1168. doi: 10.1016/0140-6736(93)90998-v. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura S, Nakamura I, Ma L, Vaughan DE, Fogo AB. Plasminogen activator inhibitor-1 expression is regulated by the angiotensin type 1 receptor in vivo. Kidney Int. 2000;58:251–259. doi: 10.1046/j.1523-1755.2000.00160.x. [DOI] [PubMed] [Google Scholar]
- 4.Vaughan DE, Lazos SA, Tong K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J Clin Invest. 1995;95:995–1001. doi: 10.1172/JCI117809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oikawa T, Freeman M, Lo W, Vaughan DE, Fogo A. Modulation of plasminogen activator inhibitor-1 in vivo: a new mechanism for the anti-fibrotic effect of renin-angiotensin inhibition. Kidney Int. 1997;51:164–172. doi: 10.1038/ki.1997.20. [DOI] [PubMed] [Google Scholar]
- 6.Vaughan DE, Rouleau JL, Ridker PM, Arnold JM, Menapace FJ, Pfeffer MA. Effects of ramipril on plasma fibrinolytic balance in patients with acute anterior myocardial infarction. HEART Study Investigators. Circulation. 1997;96:442–447. doi: 10.1161/01.cir.96.2.442. [DOI] [PubMed] [Google Scholar]
- 7.Brown NJ, Agirbasli M, Vaughan DE. Comparative effect of angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor antagonism on plasma fibrinolytic balance in humans. Hypertension. 1999;34:285–290. doi: 10.1161/01.hyp.34.2.285. [DOI] [PubMed] [Google Scholar]
- 8.Brown NJ, Abbas A, Byrne D, Schoenhard JA, Vaughan DE. Comparative effects of estrogen and angiotensin-converting enzyme inhibition on plasminogen activator inhibitor-1 in healthy postmenopausal women. Circulation. 2002;105:304–309. doi: 10.1161/hc0302.102570. [DOI] [PubMed] [Google Scholar]
- 9.Brown NJ, Gainer JV, Stein CM, Vaughan DE. Bradykinin stimulates tissue plasminogen activator release in human vasculature. Hypertension. 1999;33:1431–1435. doi: 10.1161/01.hyp.33.6.1431. [DOI] [PubMed] [Google Scholar]
- 10.Labinjoh C, Newby DE, Pellegrini MP, Johnston NR, Boon NA, Webb DJ. Potentiation of bradykinin-induced tissue plasminogen activator release by angiotensin-converting enzyme inhibition. J Am Coll Cardiol. 2001;38:1402–1408. doi: 10.1016/s0735-1097(01)01562-5. [DOI] [PubMed] [Google Scholar]
- 11.Pretorius M, Luther JM, Murphey LJ, Vaughan DE, Brown NJ. Angiotensin-converting enzyme inhibition increases basal vascular tissue plasminogen activator release in women but not in men. Arterioscler Thromb Vasc Biol. 2005;25:2435–2440. doi: 10.1161/01.ATV.0000186185.13977.94. [DOI] [PubMed] [Google Scholar]
- 12.Moore JH, Smolkin ME, Lamb JM, Brown NJ, Vaughan DE. The relationship between plasma t-PA and PAI-1 levels is dependent on epistatic effects of the ACE I/D and PAI-1 4G/5G polymorphisms. Clin Genet. 2002;62:53–59. doi: 10.1034/j.1399-0004.2002.620107.x. [DOI] [PubMed] [Google Scholar]
- 13.Kim DK, Kim JW, Kim S, Gwon HC, Ryu JC, Huh JE, Choo JA, Choi Y, Rhee CH, Lee WR. Polymorphism of angiotensin converting enzyme gene is associated with circulating levels of plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. 1997;17:3242–3247. doi: 10.1161/01.atv.17.11.3242. [DOI] [PubMed] [Google Scholar]
- 14.Margaglione M, Grandone E, Vecchione G, Cappucci G, Giuliani N, Colaizzo D, Celentano E, Panico S, Di Minno G. Plasminogen activator inhibitor-1 (PAI-1) antigen plasma levels in subjects attending a metabolic ward: relation to polymorphisms of PAI-1 and angiontensin converting enzyme (ACE) genes. Arterioscler Thromb Vasc Biol. 1997;17:2082–2087. doi: 10.1161/01.atv.17.10.2082. [DOI] [PubMed] [Google Scholar]
- 15.Jeng JR, Harn HJ, Yueh KC, Jeng CY, Shieh SM. Plasminogen activator inhibitor-1 and angiotensin I converting enzyme gene polymorphism in patients with hypertension. Am J Hypertens. 1998;11:235–239. doi: 10.1016/s0895-7061(97)00476-7. [DOI] [PubMed] [Google Scholar]
- 16.Pankow JS, Arnett DK, Borecki IB, Hunt SC, Eckfeldt JH, Folsom AR, Djousse L. Lack of association between the angiotensin-converting enzyme insertion/deletion polymorphism and plasminogen activator inhibitor-1 antigen levels in the National Heart, Lung, and Blood Institute Family Heart Study. Blood Coagul Fibrinolysis. 2000;11:551–558. doi: 10.1097/00001721-200009000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Tsujino T, Naito Y, Kawasaki D, Okuda S, Sakoda T, Fujioka Y, Sugaya T, Ohyanagi M. Circadian expression of plasminogen activator inhibitor-1 in angiotensin II type 1a receptor knockout mice. Clin Exp Hypertens. 2005;27:159–168. [PubMed] [Google Scholar]
- 18.Brown NJ, Gainer JV, Murphey LJ, Vaughan DE. Bradykinin stimulates tissue plasminogen activator release from human forearm vasculature through B(2) receptor-dependent, NO synthase-independent, and cyclooxygenase-independent pathway. Circulation. 2000;102:2190–2196. doi: 10.1161/01.cir.102.18.2190. [DOI] [PubMed] [Google Scholar]
- 19.Okada H, Watanabe Y, Kikuta T, Kobayashi T, Kanno Y, Sugaya T, Suzuki H. Bradykinin decreases plasminogen activator inhibitor-1 expression and facilitates matrix degradation in the renal tubulointerstitium under angiotensin-converting enzyme blockade. J Am Soc Nephrol. 2004;15:2404–2413. doi: 10.1097/01.ASN.0000136132.20189.95. [DOI] [PubMed] [Google Scholar]
- 20.Margaglione M, Cappucci G, d'Addedda M, Colaizzo D, Giuliani N, Vecchione G, Mascolo G, Grandone E, Di Minno G. PAI-1 plasma levels in a general population without clinical evidence of atherosclerosis: relation to environmental and genetic determinants. Arterioscler Thromb Vasc Biol. 1998;18:562–567. doi: 10.1161/01.atv.18.4.562. [DOI] [PubMed] [Google Scholar]
- 21.Moore JH, Lamb JM, Brown NJ, Vaughan DE. A comparison of combinatorial partitioning and linear regression for the detection of epistatic effects of the ACE I/D and PAI-1 4G/5G polymorphisms on plasma PAI-1 levels. Clin Genet. 2002;62:74–79. doi: 10.1034/j.1399-0004.2002.620110.x. [DOI] [PubMed] [Google Scholar]
- 22.Jacobs AK. Women, ischemic heart disease, revascularization, and the gender gap: what are we missing? J Am Coll Cardiol. 2006;47:S63–S65. doi: 10.1016/j.jacc.2004.12.085. [DOI] [PubMed] [Google Scholar]
- 23.Kalaria VG, Zareba W, Moss AJ, Pancio G, Marder VJ, Morrissey JH, Weiss HJ, Sparks CE, Greenberg H, Dwyer E, Goldstein R, Watelet LF. Gender-related differences in thrombogenic factors predicting recurrent cardiac events in patients after acute myocardial infarction. The THROMBO Investigators. Am J Cardiol. 2000;85:1401–1408. doi: 10.1016/s0002-9149(00)00785-2. [DOI] [PubMed] [Google Scholar]
- 24.Gebara OC, Mittleman MA, Sutherland P, Lipinska I, Matheney T, Xu P, Welty FK, Wilson PW, Levy D, Muller JE. Association between increased estrogen status and increased fibrinolytic potential in the Framingham Offspring Study. Circulation. 1995;91:1952–1958. doi: 10.1161/01.cir.91.7.1952. [DOI] [PubMed] [Google Scholar]
- 25.Zhao YY, Zhou J, Narayanan CS, Cui Y, Kumar A. Role of C/A polymorphism at −20 on the expression of human angiotensinogen gene. Hypertension. 1999;33:108–115. doi: 10.1161/01.hyp.33.1.108. [DOI] [PubMed] [Google Scholar]
- 26.Velez DR, Guruju D, Vinukonda G, Prater A, Kumar A, Williams SM. Angiotensinogen promoter sequence variants in essential hypertension. Am J Hypertens. 2006 doi: 10.1016/j.amjhyper.2006.05.020. in press. [DOI] [PubMed] [Google Scholar]
- 27.Os I, Bratland B, Dahlof B, Gisholt K, Syvertsen JO, Tretli S. Female preponderance for lisinopril-induced cough in hypertension. Am J Hypertens. 1994;7:1012–1015. doi: 10.1093/ajh/7.11.1012. [DOI] [PubMed] [Google Scholar]
- 28.Trifilieff A, Da Silva A, Gies JP. Kinins and respiratory tract diseases. Eur Respir J. 1993;6:576–587. [PubMed] [Google Scholar]
- 29.Moore JH. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered. 2003;56:73–82. doi: 10.1159/000073735. [DOI] [PubMed] [Google Scholar]
- 30.Moore JH. A global view of epistasis. Nat Genet. 2005;37:13–14. doi: 10.1038/ng0105-13. [DOI] [PubMed] [Google Scholar]
- 31.Moore JH. Traversing the conceptual divide between biological and statistical epistasis: Systems biology and a more modern synthesis. Bioessays. 2005;27:637–46. doi: 10.1002/bies.20236. [DOI] [PubMed] [Google Scholar]
- 32.Thornton-Wells TA, Moore JH, Haines JL. Genetics, statistics and human disease: analytical retooling for complexity. Trends Genet. 2004;20:640–647. doi: 10.1016/j.tig.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 33.Hillege HL, Fidler V, Diercks GF, van Gilst WH, de Zeeuw D, van Veldhuisen DJ, Gans RO, Janssen WM, Grobbee DE, de Jong PE. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation. 2002;106:1777–1782. doi: 10.1161/01.cir.0000031732.78052.81. [DOI] [PubMed] [Google Scholar]
- 34.Asselbergs FW, Williams SM, Hebert PR, Coffey CS, Hillege HL, Navis G, Vaughan DE, van Gilst WH, Moore JH. The gender-specific role of polymorphisms from the fibrinolytic renin-angiotensin and bradykinin systems in determining plasma t-PA and PAI-1 levels. Thromb Haemost. 2006;96:471–7. [PubMed] [Google Scholar]
- 35.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology. 1990;1:43–46. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.