

Abstract
Two peptides, based on the sequences of the complementarity-determining regions (CDR) 1 and 3 of a pathogenic murine monoclonal anti-DNA autoatibody that bears the 16/6 idiotype (Id), were shown to either prevent or treat an already established systemic lupus erythematosus (SLE) in two murine models of lupus. Two additional peptides based on the human monoclonal anti-DNA, 16/6 Id were synthesized. This study was undertaken in order to investigate the ability of the CDR-based peptides to immunomodulate SLE-associated responses of peripheral blood lymphocytes (PBL) of SLE patients. PBL of 24 of the 62 SLE patients tested proliferated in vitro following stimulation with the human 16/6 Id. Peptides based on the CDRs of both the human and murine anti-DNA autoantibodies inhibited efficiently and specifically the 16/6 Id-induced proliferation and IL-2 production. The latter inhibitions correlated with an up-regulated production (by 2·5–3·5-fold) of the immunosuppressive cytokine, TGF-β. Overall, the results of our study demonstrate that the CDR-based peptides are capable of down-regulating in vitro autoreactive T cell responses of PBL of SLE patients. Thus, these peptides are potential candidates for a novel specific treatment of SLE patients.
Keywords: CDR-based peptides, IL-2, immunomodulation, systemic lupus erythematosus, TGF-β
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of a variety of autoantibodies, impairment of B and T cell functions, cytokines production and immune complex deposition accompanied by systemic clinical manifestations (e.g. neurological, dermal, haematological, musculoskeletal and renal) [1]. The exact pathogenesis of the disease, as well as the autoantigen(s) in SLE, are not yet defined. The common idiotype designated 16/6 Id was detected on anti-DNA antibodies of about 50% of SLE patients [2,3], and its presence was shown to correlate with disease activity [4]. Moreover, peripheral blood lymphocytes (PBL) obtained from SLE patients responded specifically to 16/6 Id stimulation [3–5]. Previous studies from our laboratory demonstrated the induction of experimental SLE in naive (not SLE-prone) mice by immunization with the human 16/6 Id monoclonal antibody (MoAb) or with the murine anti-DNA 16/6 Id, 5G12 MoAb [6,7]. The immunized mice developed high levels of autoantibodies, including antibodies bearing the 16/6 Id, as well as SLE-related clinical manifestations (e.g. leukopenia, thrombocytopenia and renal impairment) [6–8]. Autoantibodies isolated from the diseased mice including the 5G12 MoAb were shown to be highly homologous to anti-DNA MoAb isolated from the SLE-prone (NZB × NZW)F1 mice [9], supporting further the importance of the 16/6 Id in SLE.
Two peptides, based on the sequences of the complementarity-determining regions (CDR) of the pathogenic murine anti-DNA 16/6 Id (5G12), namely mCDR1 and mCDR3, were designed and synthesized [10]. Those peptides were shown to be immunodominant T-cell epitopes in non-autoimmune (e.g. BALB/c) and lupus-prone (NZB × NZW) F1 mice [10–12]. Furthermore, treatment with these peptides ameliorated the SLE-like clinical manifestations and decreased autoantibody production of both, spontaneous (NZB × NZW) F1 and 16/6 Id-induced SLE [10,12–12]. Experiments performed with single amino acid substituted analogues for mCDR1(39 analogues) and for mCDR3 (17 analogues) as well as with truncated peptides (either at the N and/or at the C-terminus), indicated that the original mCDR1 and mCDR3 were the best immunomodulators of SLE associated responses [15,16].
To investigate further the role of the CDR-based peptides in the treatment of human SLE we have synthesized two additional peptides, based on the sequences of CDRs of the human anti-DNA 16/6 Id, namely hCDR1 and hCDR3. In the present study, we have examined the ability of the murine and the newly synthesized human CDR-based peptides to inhibit the 16/6 Id specific stimulation (proliferation) of PBL obtained from patients with SLE. We demonstrate here that the peptides inhibited specifically the 16/6 Id-induced proliferation. This inhibition correlated with a reduction in IL-2 secretion and with an up-regulated production of the immunosuppressive cytokine, TGF-β.
Patients, Materials And Methods
Patients
Sixty-two patients, nine males (14·5%) and 53 females (85·5%), with SLE participated in our study. The mean age at diagnosis was 32·95 ± 12·92 (range 12–61) years and the mean follow-up period was 10·98 ± 10·76 (range 1–32) years. All patients fulfilled at least four of the American College of Rheumatology (ACR) revised diagnostic criteria for SLE [17]. Patients were recruited from three Israeli Medical Centers (Kaplan, Rehovot; Ichilov, Tel Aviv; Asaf-Harofeh, Rishon Lezion). Disease activity was determined according to the SLEDAI lupus activity index [18]. A control group of 36 sex- and age-matched healthy control volunteers was studied concomitantly with the SLE patients. All participants signed an informed consent form prior to the initiation of the study. The study was approved by the Ethical Committee of the Medical Center.
Monoclonal antibody
The human anti-DNA MoAb that bears the 16/6 Id (IgG1/k) has been characterized previously [19]. The MoAb was secreted by hybridoma cells that were grown in culture and were purified by using a protein G-sepharose column (Pharmacia, Fine Chemicals, Uppsala, Sweden) [14].
Synthetic peptides
Synthetic peptides based on the CDR1 and CDR3 of the murine monoclonal anti-DNA 16/6 Id and of the human anti-DNA 16/6 Id MoAbs [7,19] were prepared as described previously [20]. The amino acid sequences of the human and murine CDR-based peptides are presented in Table 1. As shown in the Table 1, the peptides based on the CDR of the human and murine anti-DNA antibodies share a significant number of amino acids at identical positions. The reversed peptides that were synthesized in the reversed order of mCDR1 and mCDR3 (revmCDR1; revmCDR3) were used as control.
Table 1.
Amino acid sequences of peptides based on the CDRs of murine and human monoclonal anti-DNA, 16/6Id autoantibodies
| mCDR1 | T | G | Y | Y | M | Q | W | V | K | Q | S | P | E | K | S | L | E | W | I | G | ||
| hCDR1 | G | Y | Y | W | S | W | I | R | Q | P | P | G | K | G | E | E | W | I | G | |||
| mCDR3 | Y | Y | C | A | R | F | L | W | E | P | Y | A | M | D | Y | W | G | Q | G | S | ||
| hCDR3 | Y | Y | C | A | R | G | L | L | R | G | G | W | N | D | V | D | Y | Y | G | M | D | V |
| Reversed mCDR1 | G | I | W | E | L | S | K | E | P | S | Q | K | V | W | Q | M | Y | Y | G | T | ||
| Reversed mCDR3 | S | G | Q | G | W | Y | D | M | A | Y | P | E | W | L | F | R | A | C | Y | Y |
Peptides based on the CDR1 and CDR3 of the murine autoantibody were designated mCDR1 and mCDR3, respectively, and peptides based on the CDRs of the human autoantibody were designated hCDR1 and hCDR3.
Proliferative responses
PBL were isolated from heparinized venous blood by Ficoll-Hypaque (Pharmacia) density-gradient centrifugation [21]. All assays were performed in triplicate in flat-bottomed microtitre plates (Falcon, Becton Dickinson, Oxmard, CA, USA) in which 2 × 105 PBL were cultured in enriched RPMI-1640 as described [3]. The PBL were exposed to various concentrations (0·1–40 µg/well) of the human anti-DNA 16/6 Id MoAb with and without the addition of the various CDR-based peptides. Phytohaemagglutinin (PHA; 2 µg/well) was used as a control for culture conditions at each experiment. The cultures were incubated in 7·5% CO2 at 37°C for 6 days. Eighteen hours before the cells were harvested, [3H]thymidine (0·5 µ Ci of 5 Ci/mmol) (Nuclear Research Center, Negev, Israel) was added to all cultures. Results are expressed as the mean thymidine incorporation in counts per minute (cpm) of triplicate culture ± s.d., or as stimulation index (SI; the ratio of mean cpm at the optimal concentration of the human 16/6 Id to the mean cpm in the presence of medium alone). SI ≥ 2 was considered a positive response [3]. Inhibition (the ratio of mean cpm in the presence of the 16/6 Id and various CDR-based peptides to the mean cpm with the 16/6 Id without the CDR-based peptide) above 50% was considered positive.
Assessment of cytokine production
Supernatants were collected 48 h following the initiation of the cultures and stored at − 70°C. IL-2 was determined by using the IL-2-dependent CTLL line as described previously [22]. TGF-β was determined by an enzyme-linked immunosorbant assay (ELISA). Briefly, plates were coated with the recombinant human TGF-β1 sRII/Fc chimera (R&D Systems Inc., Minneapolis, MN, USA). For detection we used biotin-labelled anti-human TGF-β antibody (R&D Systems Inc.) [14]. In a separate set of experiments, PBL (2 × 105) of the patients were incubated with the CDR-based peptides in the absence of the human anti-DNA 16/6Id MoAb. Supernatants were collected after 48 h and tested for TGF-β secretion as above.
Statistical analysis
Results presented as mean ± s.d. Chi-square, Wilcoxon and t-tests were employed for statistical analysis. P ≤ 0·05 was considered significant.
Results
Proliferative capacity and clinical characterization of SLE patients
It was of interest to find out whether the peptides, based on CDR1 and CDR3 of monoclonal anti-DNA 16/6 Id antibodies, are capable of inhibiting the specific proliferative responses of PBL of SLE patients to the human 16/6 Id. Furthermore, we wanted to compare the inhibitory capacity of the peptides based on the CDRs of the murine anti-DNA autoantibody to that of the newly synthesized peptides based on CDR1 and CDR3 of the human monoclonal anti-DNA 16/6 Id (Table 1). To this end, we first had to identify the patients whose PBL could be stimulated to proliferate by the human 16/6 Id. Therefore, PBL of 62 consecutive SLE patients were cultured in the presence of the human 16/6 Id and their proliferative responses and ability to secrete IL-2 were determined. PBL of 24 of the total of 62 (39%) and of 23 of 55 (42%) SLE patients tested responded (SI = 2, range 2–5·6) by proliferation and by IL-2 secretion (SI = 2, range 2–60), respectively. The frequency of responders in the group of SLE patients was lower than that observed in the group of healthy donors that was tested as control. Thus, PBL of 21 of a total of 36 (58%) healthy donors responded by proliferation to the 16/6 Id. These results are similar to those obtained in our previous studies [3,4]. The extent of proliferation (SI levels) was similar for the SLE patients and for the healthy controls who responded to the 16/6 Id. However, the optimal response to the 16/6 Id of PBL of the control donors was observed at higher concentrations of 16/6 Id as compared to the SLE patients (Fig. 1). It is shown in the Fig. 1 that whereas PBL of most SLE patients responded to the 16/6Id at a concentration of 1–10 µg/well, PBL of healthy controls responded mainly to concentrations of 5–20 µg/well (P = 0·016 for the 20 µg/well dose when numbers of responders out of the healthy donors and SLE patients were compared).
Fig. 1.

Concentrations of 16/6 Id required for optimal stimulation of PBL of SLE patients and of healthy controls. PBL were stimulated with various concentrations (0·1–40 µg/well) of the 16/6 Id. The concentration yielding the highest stimulation index was defined as optimal for triggering a proliferative response. ▪, SLE patients; □, healthy controls.
No differences could be demonstrated between gender and age of SLE patients that responded to the 16/6 Id and of the non-responder group of patients. However, the patients whose PBL proliferated in response to the 16/6 Id were sick for a shorter period of time (a mean of 9·78 ± 8·36 versus 11·73 ± 12·06 years for responders and non-responders, respectively; P = 0·036). Table 2 summarizes the clinical characterization of the 16/6 Id-specific responder and non-responder groups of SLE patients. As can be seen in the Table 2, both groups were similar in most SLE-related clinical manifestations. The disease activity score (SLEDAI) and the number of SLE diagnostic criteria were also similar in the two groups. Nevertheless, a higher frequency of neurological (both siezures and psychosis) and haematological involvement and a lower rate of renal involvement were noted in the responder group of patients in comparison to the group of non-responders. However, probably because of the low number of patients in the relevant subgroups, the above differences did not reach statistical significance. Moreover, relatively less responder patients were determined between those treated with either steroids or cytotoxic agents at the time of the study. It is noteworthy that significantly more patients who never received steroids responded to the 16/6 Id in comparison to the non responder group (54%versus 21%; P = 0·023).
Table 2.
Clinical and laboratory characterization of SLE patients
| All patients | Responders | Non-responders | |
|---|---|---|---|
| (a) Diagnostic criteria | |||
| No. of patients (%) | 62 (100) | 24 (39) | 38 (61) |
| Malar rash | 19/62 (30·1) | 8/24 (33·3) | 11/38 (29) |
| Discoid rash | 9/62 (15) | 3/24 (12·5) | 6/38 (16) |
| Photosensitivity | 21/62 (34) | 9/24 (37·5) | 12/38 (32) |
| Mucosal ulcers | 17/62 (27·4) | 8/24 (33·3) | 9/38 (23·7) |
| Arthritis | 46/62 (74·2) | 19/24 (79·2) | 27/38 (71) |
| Serositis | 14/62 (22·6) | 5/24 (20·8) | 9/38 (23·7) |
| Neurological disorders‡ | 5/62 (8·1) | 4/24 (16·7) | 1/38 (2·7) |
| Renal disorder‡ | 24/62 (38·8) | 7/24 (29·2) | 17/38 (44·8) |
| Haematological disorders‡ | 44/62 (71) | 19/24 (79·2) | 25/38 (65·8) |
| ANA | 61/62 (98·4) | 24/24 (100) | 37/38 (92·1) |
| α-dsDNA | 54/62 (87·1) | 19/24 (79·2) | 35/38 (92·1) |
| APLA | 35/62 (56·5) | 12/24 (50·0) | 23/38 (60·53) |
| (b) Disease activity | |||
| SLEDAI score | 6·65 ± 5·12 | 7·29 ± 1·06 | 6·24 ± 0·84 |
| Number of ACR diagnostic criteria | 5·44 ± 1·39 | 5·54 ± 0·33 | 5·34 ± 0·2 |
| (c) Current treatment† | |||
| NSAIDS | 17/62 (27·4) | 6/24 (25) | 11/38 (29) |
| Anti-malarial | 37/62 (59·7) | 15/24 (62·5) | 22/38 (57·9) |
| Steroids‡ | 33/62 (53·2) | 11/24 (45·8) | 22/38 (57·9) |
| Cytotoxic‡ | 10/62 (16·1) | 2/24 (8·3) | 8/38 (21) |
*Clinical involvement was defined according to the ACR revised criteria [16]. Anti-nuclear antibodies (ANA) and anti-dsDNA antibodies were determined by Hep2 cells and Crithidia luciliae, respectively. Anti-phospholipid antibodies (APLA) were defined as reactivity in one or more of the following assays: false positive VDRL, lupus anticoagulant (LAC) or ELISA for anticardiolipin antibodies.
†The antimalarial agent, hydroxychloroquine, was used at a dose of 200–400 mg/day; steroid treatment was defined as a daily dose≥5 mg of prednisone; cytotoxic agents used were cyclophosphamide (0·75–1·0 g/m2; monthly) or azathioprine (100–150 mg/day).
‡Parameters for which tendency was observed towards differences between the two groups of responder and non responder SLE patients.
In vitroinhibition of 16/6 Id-induced stimulation of PBL of SLE patients
The ability of the peptides, based on the CDRs of the murine (mCDR1 and mCDR3) and of the human (hCDR1 and hCDR3) autoantibodies, to inhibit the proliferative responses of PBL of SLE patients and of healthy controls to the human 16/6 Id was tested. Table 3 summarizes the inhibitory capacity of the peptides. Peptides mCDR1, hCDR1 and hCDR3 inhibited the proliferative response to the 16/6 Id of PBL of a similar number of SLE patients (15/19, 16/19 and 15/19, respectively). Peptide mCDR3 inhibited the proliferation of PBL of fewer patients (6/19) when added to cultures of PBL stimulated with the 16/6 Id. The mean maximum percentage inhibition was comparable for all four CDR based peptides tested (Table 3). When the ability of the peptides to inhibit the IL-2 secretion of PBL stimulated by the human 16/6 Id was tested, mCDR1 and hCDR1 inhibited the secretion by PBL of 23/23 and 21/23, respectively. Similarly, secretion of IL-2 by PBL of 19/23 tested individuals was inhibited by either mCDR3 or hCDR3 (Table 3b). Inhibition of proliferative responses correlated directly with IL-2 inhibition by the CDR-based peptides. Thus, inhibition of IL-2 secretion was observed in all cases where inhibition of proliferation were determined. It is noteworthy that the efficacy of the CDR-based peptides to inhibit the proliferative responses of PBL of healthy donors to the 16/6 Id was much lower than that observed for PBL of SLE patients. The proliferation of PBL of only four, 10, five and nine of the 18 healthy controls tested was inhibited by mCDR1, hCDR1, mCDR3 and hCDR3, respectively (Table 3c). The effect of the CDR-based peptides on IL-2 secretion by 16/6 Id-stimulated PBL of healthy controls was not tested.
Table 3.
Inhibition of 16/6 Id-induced stimulation of PBL by the CDR-based peptides
| Peptide | Inhibitory activity%* | Maximum inhibition%† |
|---|---|---|
| (a) SLE patients − proliferation | ||
| mCDR1 | 79 (15/19) | 64 ± 9 |
| hCDR1 | 84 (16/19) | 68 ± 10 |
| mCDR3 | 32 (6/19) | 75 ± 8 |
| hCDR3 | 79 (15/19) | 72 ± 8 |
| (b) SLE patients − IL-2 secretion | ||
| mCDR1 | 100 (23/23)‡ | 88 ± 16 |
| hCDR1 | 91 (21/23) | 84 ± 31 |
| mCDR3 | 83 19/23) | 58 ± 41 |
| hCDR3 | 83(19/23) | 78 ± 34 |
| (c) Healthy controls − proliferation | ||
| mCDR1 | 22 (4/18) | 75 ± 7 |
| hCDR1 | 56 (10/18) | 68 ± 10 |
| mCDR3 | 28 (5/18) | 58 ± 3 |
| hCDR3 | 50 (9/18) | 68 ± 8 |
The percentage of SLE patients or healthy controls in whom the responses of the PBL were inhibited by the CDR based peptides (above 50% inhibition, as in Methods). Parentheses represent the number of patients or controls out of the groups tested whose PBL were inhibited.
The mean ± s.d. of percentage maximum inhibition by the CDR-based peptides. Proliferative responses in the presence of the 16/6 Id without any CDR-based peptide were considered as 100%.
IL-2 secretion in the presence of 16/6 Id alone was considered as 100%. Inhibition of 50% or more was considered significant.
The inhibition of responses of PBL to the human 16/6 Id was shown to be specific, because two control peptides that were synthesized in the reversed order of mCDR1 and mCDR3 (Table 1) could not inhibit the 16/6 Id-specific proliferative responses. Figure 2 represents a typical experiment with PBL of one SLE patient. It can be seen that, whereas all four peptides based on CDR1 and CDR3 of the murine and human anti-DNA 16/6 Id inhibited efficiently the proliferative response to the 16/6 Id in this SLE patient, neither of the control reversed peptides could do so. The specificity of the inhibition by the CDR-based peptides was tested further. Thus, because PBL of all patients who responded to the 16/6 Id proliferated to PHA as well, the ability of the CDR-based peptides to inhibit the latter responses was tested. Figure 3 demonstrates results of a representative experiment. Neither peptide could inhibit the proliferative responses to the mitogen, confirming further the specificity of their inhibitory effects (Fig. 3).
Fig. 2.
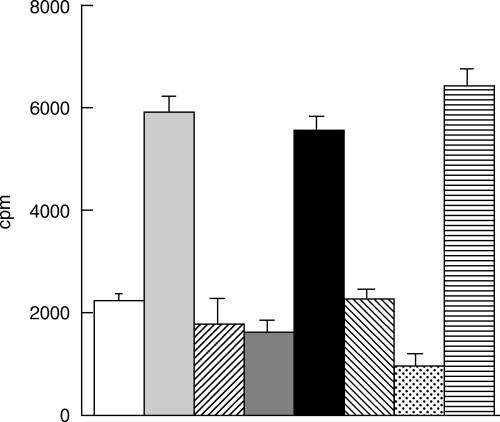
Inhibition of 16/6 Id-stimulated proliferation by peptides based on the human or murine anti-DNA autoantibodies. PBL of a representative SLE patient were cultured for 6 days in the presence of 16/6 Id with or without the various peptides based on the human (hCDR1, hCDR3) or murine (mCDR1, mCDR3) autoantibodies or with the control peptides (revmCDR1, revmCDR3). For the last 18 h 0·5 µCi of [3H]thymidine was added. Thereafter cells were harvested and radioactivity was counted. Results are expressed as mean cpm of triplicates ± s.d. □, Medium;  , 16/6Id;
, 16/6Id;  , 16/6Id + mCDR1;
, 16/6Id + mCDR1;  , 16/6Id + hCDR1; ▪, 16/6Id + revmCDR1;
, 16/6Id + hCDR1; ▪, 16/6Id + revmCDR1;  , 16/6Id + mCDR3;
, 16/6Id + mCDR3;  , 16/6Id + hCDR3;
, 16/6Id + hCDR3;  , 16/6Id + revmCDR3.
, 16/6Id + revmCDR3.
Fig. 3.

The CDR-based peptides do not affect the proliferative responses to PHA. PBL of a representative SLE patient were incubated in the presence of 2 µg/well of PHA and the various CDR-based peptides. Results are expressed as mean cpm of triplicates ± s.d. □, Medium;  , PHA; ▪, PHA + mCDR1;
, PHA; ▪, PHA + mCDR1;  , PHA + hCDR1;
, PHA + hCDR1;  , PHA + mCDR3;
, PHA + mCDR3;  , PHA + hCDR3.
, PHA + hCDR3.
Up-regulation of the secretion of TGF-β by the CDR-based peptides
Because we have shown that down-regulation of SLE manifestations by the CDR-based peptides in murine models is associated with up-regulated secretion of the immunosuppressive cytokine, TGF-β[14], it was of interest to find out whether incubation of PBL in the presence of the latter CDR-based peptides will stimulate the secretion of TGF-β. To this end, supernatants of PBL of SLE patients that were incubated in culture with the 16/6 Id and with either of the CDR-based peptides, were tested for the content of TGF-β. The results are summarized in Table 4. It can be seen that in most cases the CDR-based peptides up-regulated significantly the secretion of TGF-β by the PBL. The up-regulation of TGF-β correlated directly with the inhibition of proliferative responses and IL-2 secretion. The up-regulation of TGF-β secretion by the CDR-based peptides is specific because the reversed CDR-based peptides, used as controls, did not trigger the secretion of TGF-β to levels above those observed in the presence of the 16/6 Id alone (Fig. 4).
Table 4.
Up-regulation of TGF-β secretion of 16/6 Id-induced stimulation of PBL of SLE patients with CDR peptides
| Peptide | Up-regulation of TGF-β% | Maximum up-regulation % |
|---|---|---|
| mCDR1 | 84 (16/19) | 259 ± 240 |
| hCDR1 | 100 (19/19) | 305 ± 221 |
| mCDR3 | 89 (17/19) | 269 ± 170 |
| hCDR3 | 100 (19/19) | 338 ± 242 |
Secretion of TGF-β in the presence of 16/6 Id alone (mean 636 ± 25 pg/ml) was considered as 100%. Results are expressed as percentage secretion above that in the presence of 16/6Id alone.
Fig. 4.

Up-regulated secretion of TGF-β triggered by CDR-based peptides. PBL of a representative SLE patient were cultured with 16/6 Id with or without the various CDR-based peptides and the control peptides. After 48 h of incubation supernatants were collected and tested by ELISA for the content of TGF-β. Results are expressed as mean (pg/ml) of triplicates ± s.d. □, Medium;  , 16/6Id;
, 16/6Id;  , 16/6Id + mCDR1;
, 16/6Id + mCDR1;  , 16/6Id + hCDR1; ▪, 16/6Id + revmCDR1;
, 16/6Id + hCDR1; ▪, 16/6Id + revmCDR1;  , 16/6Id + mCDR3;
, 16/6Id + mCDR3;  , 16/6Id + hCDR3;
, 16/6Id + hCDR3;  , 16/6Id + revmCDR3.
, 16/6Id + revmCDR3.
Thus, the peptides based on the human and murine autoantibodies are capable of inhibiting the proliferative responses and IL-2 secretion of PBL of SLE patients that are stimulated by the human 16/6 Id. The latter correlates with an increased production of the immunosuppressive cytokine, TGF-β.
It was of interest to find out whether the various CDR-based peptides are capable of up-regulating the secretion of TGF-β by PBL of the various patients when incubated in the absence of the 16/6 Id. To this end PBL of a group of patients were incubated with the CDR-based peptides. Supernatants of the cultures were tested for the content of TGF-β. The results demonstrated that in the majority of the cases incubation of PBL with the peptides caused a moderate but significant increased secretion of 1·5–3·5-fold compared to the levels of TGF-β secreted by cells that were incubated in the presence of medium alone. Figure 5 demonstrates the results obtained with PBL of a representative SLE patient. It is noteworthy that the levels of TGF-β secreted by PBL incubated with the peptide only are lower than those measured in supernatants of PBL that were in culture with the 16/6 Id MoAb and the CDR-based peptides. Nevertheless, the results demonstrate that the peptides are capable of stimulating the secretion of the immunosuppressive cytokine TGF-β.
Fig. 5.
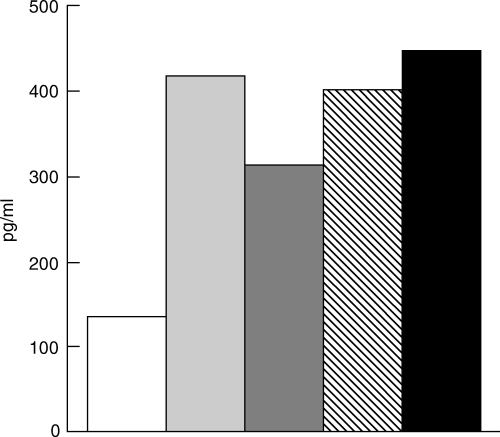
The CDR-based peptides trigger PBL of SLE patients to secrete elevated levels of TGF-β. PBL (2 × 105/well) of a representative SLE patient were cultured with 25 µg/well (total volume 200 µl/well) of the various CDR-based peptides. After 48 h of incubation supernatants were collected and tested by ELISA for the content of TGF-β. Results are expressed as pg/ml of secreted TGF-β. □, Medium;  , mCDR1;
, mCDR1;  , mCDR3;
, mCDR3;  , hCDR1; ▪, hCDR3.
, hCDR1; ▪, hCDR3.
Discussion
The main findings of the present study are that peptides based on the CDRs of human and murine pathogenic monoclonal anti-DNA 16/6 Id bearing autoantibodies, specifically inhibited the 16/6 Id-induced proliferation and IL-2 secretion by PBL obtained from SLE patients. This inhibition correlated with up-regulated secretion of the immunosuppressive cytokine, TGF-β. To our best knowledge this is the first study demonstrating the potential of CDR-based peptides to down-regulate in vitro SLE-related responses of human autoreactive T cells.
PBL obtained from 24 of 62 SLE patients responded (SI ≥ 2) to stimulation by the human anti-DNA 16/6 Id autoantibody. The rate of 16/6 Id-specific proliferative responses (39%) of PBL of SLE patients is similar to our previously reported data [3–5]. We have also reported [3–5], as shown in the present study, that the proliferative rate of PBL of healthy controls to stimulation by the 16/6 Id was higher (58%) than that of SLE patients. Low rates of proliferative responses to autoantibody variable region peptides in SLE patients were also observed by Williams et al. [23]. The decreased capacity of PBL of SLE patients to proliferate following in vitro stimulation with 16/6Id may be due in part to an in vivo excessive spontaneous response to the 16/6 Id-related network that leads to the exhaustion of the immune cells. Other mechanisms that may account for the low T cell-responsive rate in SLE patients are probably related to the dysbalance between Th1 and Th2 cell types observed in SLE during the disease course [24–26]. Analysis of the clinical characterization of the group of SLE patients (Table 2) suggest that renal involvement and immunosuppressive treatment may also contribute to the reduced ability of PBL of SLE patients to respond in vitro to the 16/6 Id.
Although PBL of less SLE patients than those of healthy controls responded to in vitro stimulation by the 16/6 Id it is likely that the former responses were more specific and of higher affinity, as the concentrations of 16/6 Id that triggered optimal proliferative responses were lower for most PBL of most SLE patients in comparison to those required for PBL of healthy controls (Fig. 1). Alternatively, the response to lower 16/6Id concentrations may be due to the hyperresponsiveness of SLE T cells to antigenic stimulation, as was shown recently by Vratsanos et al. [27].
Studies from our laboratory demonstrated significant beneficial effects of treatment with mCDR1 and mCDR3 in induced [10, 14, 15] as well as in spontaneous (NZB × NZW) F1[13] murine SLE. It was of interest, therefore, to investigate the ability of the peptides and of the newly synthesized peptides based on the CDRs of the human 16/6 Id (Table 1) to down-regulate the 16/6 Id-specific autoreactive responses of PBL of SLE patients. The human CDR-based peptides (hCDR1, hCDR3) inhibited efficiently the 16/6 Id-induced proliferation and IL-2 secretion of PBL of patients. Peptide mCDR1 demonstrated a similar inhibitory effect, whereas mCDR3 was less effective in its inhibitory capacity (Table 3a,b). Those inhibitions were specific because two control peptides, namely reversed mCDR1 and reversed mCDR3, had no inhibitory effects on the 16/6 Id-specific proliferative responses (Fig. 2) and there were no effects of any of the mouse or human CDR based peptides on PHA-stimulated PBL of SLE patients (Fig. 3). The modulating effects of the various CDR-based peptides on cytokine production (IL-2 and TGF-β) by the PBL of SLE patients is more prominent than their inhibitory effects on proliferation (Tables 3 and 4), reflecting the higher sensitivity of cytokine secretion in comparison to the proliferative responses.
In contrast to the high rate of inhibition observed for 16/6 Id-stimulated PBL of SLE patients, the effects of the various CDR-based peptides on PBL of healthy donors was less prominent (Table 3c). It is unlikely that the latter is due to insufficient amount of inhibitorory peptides because in all cultures (using PBL either of SLE patients or of healthy controls) the concentration of the CDR-based peptides was at least 10-fold higher than that of the 16/6 Id. In addition, for cases where optimal proliferations were observed with the same concentrations of the 16/6 Id, a much better inhibition was determined for stimulated PBL of SLE patients. Our inhibition experiments suggest that PBL of SLE patients and of healthy controls might recognize different determinants within the anti-DNA 16/6 Id autoantibody. Thus, T-cells of SLE patients recognize and react mainly to the CDR1 and CDR3 epitopes whereas T cells of healthy donors react probably to a variety of epitopes within the autoantibody macromolecule and therefore the inhibition of proliferation with the CDR-based peptides is less efficient in the controls.
The inhibitory effects of the CDR-based peptides, on 16/6 Id-stimulated PBL of SLE patients correlated with up-regulated secretion of TGF-β (Table 4, Fig. 3). The latter immunosuppressive cytokine was shown to inhibit IL-1 and IL-6 production [28] and to suppress IgG production [29]. Both constitutive and stimulated levels of TGF-β are low in SLE patients, especially during active disease and the high IgG levels seen in those patients is attributed, in part, to low levels of TGF-β[30]. In animal models, TGF-β knockout mice were shown to develop SLE-like disease [31], whereas up-regulation of TGF-β production in the lupus prone MRL/lpr/lpr mice decreased autoantibody production [32]. Our previous observations also demonstrated that the beneficial effects in prevention and treatment of SLE in various animal models, following administration of the CDR-based peptides, were associated with up-regulation of TGF-β secretion [14].
It is not likely that the observed inhibition of the 16/6 Id-induced proliferative responses and IL-2 secretion by PBL of SLE patients is due merely to MHC blocking, because the effect of the peptides was specific (Figs 2 and 3) and the control peptides that were shown to bind to MHC class II on APC of various SLE patients with a similar affinity (unpublished) to that of the CDR-based peptides [3] did not inhibit any of the above responses (Figs 2 and 3). Further, the CDR-based peptides stimulated specifically the secretion of the immunosuppressive cytokine TGF-β (Table 4, Fig. 3). These peptides might act as partial agonists stimulating a subset of PBL, such as CD4+CD25+ regulatory T cells that were shown to produce TGF-β[33] or by activating different signalling pathways aimed at the production of immunosuppressive cytokines (e.g. TGF-β) rather than the Th-1 type cytokines (e.g. IL-2). Indeed, in our previous studies the beneficial effects of the CDR-based peptides on two murine models of SLE were associated with a diminished secretion of the Th1-type cytokines and up-regulation of TGF-β production [13,14]. The fact that an elevated release of TGF-β could be triggered following incubation of PBL with the peptides themselves in the absence of the 16/6 Id-induced stimulation (Fig. 5) suggests that up-regulating the latter immunosupressive cytokine is a central step in the mechanism of action of the CDR-based peptides.
The peptides that are based on the CDR of the murine anti-DNA 16/6 Id autoantibody were shown previously by us to be capable of ameliorating experimental SLE in induced and spontaneous animal models [10,13–15]. Because of the observed high homology between various pathogenic anti-DNA antibodies [10] it is likely that our CDR-based peptides are capable of down-regulating other (non 16/6 Id) SLE-related aotoimmune responses. The amelioration of SLE-like disease in (NZB × NZW) F1 mice by treatment with the CDR-based peptides [13] strengthens the latter assumption. A limited number of studies by other investigators also demonstraed beneficial effects on SLE manifestations in animal models by peptides based on variable regions of autoantibodies [34–37] or by synthetic peptides that can bind and block the reactivity of anti-DNA autoantibodies [38]. Recently, Hahn et al. reported the amelioration of SLE-like disease in (NZB × NZW) F1 mice following treatment with a cosensus peptide based on amino acid sequences of murine anti-DNA MoAb probably via induction of tolerance in murine autoreactive T cells [39].
To conclude, we report here that mCDR1, to a lesser degree mCDR3, and the newly synthesized peptides based on the human anti-DNA 16/6 Id autoantibody (hCDR1, hCDR3), inhibited efficiently and specifically the in vitro 16/6 Id stimulation of PBL of SLE patients, apparently via a mechanism similar to that observed by us for the animal models. Thus, the above results suggest that these peptides might be novel potential candidates for specific treatment of SLE patients.
Acknowledgments
This work was supported by Teva Pharmaceutical Industries Limited, Israel. We would like to thank Ms Galia Shifroni, senior statistician and data manager at Teva Pharmaceutical Industries Limited, for the statistical analysis.
References
- 1.Winchester RJ. Systemic lupus erythematosus pathogenesis. In: Koopman WJ, editor. Arthritis and allied conditions. Birmingham, Alabama: Williams and Wilkins; 1996. pp. 1361–91. [Google Scholar]
- 2.Isenberg DA, Shoenfeld Y, Madaio MP, et al. Anti-DNA antibody idiotypes in systemic lupus erythematosus. Lancet. 1984;2:417–22. doi: 10.1016/s0140-6736(84)92904-0. [DOI] [PubMed] [Google Scholar]
- 3.Dayan M, Segal R, Waisman A, et al. Immune response of SLE patients to peptides based on the complementary determining regions (CDR) of a pathogenic anti-DNA monoclonal antibody. J Clin Immunol. 2000;20:187–94. doi: 10.1023/a:1006685413157. [DOI] [PubMed] [Google Scholar]
- 4.Mendlovic S, Shoenfeld Y, Bakimer R, Segal R, Dayan M, Mozes E. In vitro T-cell functions specific to an anti-DNA idiotype and serological markers in patients with systemic lupus erythematosus. J Clin Immunol. 1988;8:178–87. doi: 10.1007/BF00917564. [DOI] [PubMed] [Google Scholar]
- 5.Mendlovic S, Segal R, Shoenfeld Y, Mozes E. Anti-DNA idiotype and anti-idiotype-specific T cell responses in patients with systemic lupus erythematosus and their first-degree relatives. Clin Exp Immunol. 1990;82:504–8. doi: 10.1111/j.1365-2249.1990.tb05480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendlovic S, Brocke S, Shoenfeld Y, et al. Induction of a systemic lupus erythematosus-like disease in mice by a common human anti-DNA idiotype. Proc Natl Acad Sci USA. 1988;85:2260–4. doi: 10.1073/pnas.85.7.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waisman A, Mendelovic S, Ruiz JP, Zinger H, Meshorer A, Mozes E. The role of the 16/6 idiotype network in the induction and manifestation of systemic lupus erythematosus. Int Immunol. 1993;5:1293–300. doi: 10.1093/intimm/5.10.1293. [DOI] [PubMed] [Google Scholar]
- 8.Mendlovic S, Fricke H, Shoenfeld Y, Mozes E. The role of anti-idiotypic antibodies in the induction of experimental systemic lupus erythematosus in mice. Eur J Immunol. 1989;19:729–34. doi: 10.1002/eji.1830190424. [DOI] [PubMed] [Google Scholar]
- 9.Waisman A, Mozes E. Variable region sequences of autoantibodies from mice with experimental systemic lupus erythematosus. Eur J Immunol. 1993;23:1566–73. doi: 10.1002/eji.1830230726. [DOI] [PubMed] [Google Scholar]
- 10.Waisman A, Ruiz PJ, Israeli E, et al. Modulation of murine systemic lupus erythematosus with peptides based on complementarity determining regions of a pathogenic anti-DNA monoclonal antibody. Proc Natl Acad Sci USA. 1997;94:4620–5. doi: 10.1073/pnas.94.9.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brosh N, Eilat E, Zinger H, Mozes E. Characterization and role in experimental systemic lupus erythematosus of T-cell lines specific to peptides based on complementarity-determining region-1 and complementarity-determining region-3 of a pathogenic anti-DNA monoclonal antibody. Immunology. 2000;99:257–65. doi: 10.1046/j.1365-2567.2000.00957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brosh N, Dayan M, Fridkin M, Mozes E. A peptide based on the CDR3 of an anti-DNA antibody of experimental SLE origin is also a dominant T-cell epitope in (NZB × NZW) F1 lupus-prone mice. Immunol Lett. 2000;72:61–8. doi: 10.1016/s0165-2478(00)00161-9. [DOI] [PubMed] [Google Scholar]
- 13.Eilat E, Zinger H, Nyska A, Mozes E. Prevention of systemic lupus erythematosus-like disease in (NZB × NZW) F1 mice by treating with CDR1- and CDR3-based peptides of a pathogenic autoantibody. J Clin Immunol. 2000;20:268–78. doi: 10.1023/a:1006663519132. [DOI] [PubMed] [Google Scholar]
- 14.Eilat E, Dayan M, Zinger H, Mozes E. The mechanism by which a peptide based on the complemetarity determining region-1 of a pathogenic anti-DNA autoantibody ameliorates experimental SLE. Proc Natl Acad Sci USA. 2001;98:1148–53. doi: 10.1073/pnas.98.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brosh N, Zinger H, Fridkin M, Mozes E. A peptide based on the sequence of the CDR3 of a murine anti-DNA MoAb is a better modulator of experimental SLE than its single amino acid-substituted analogs. Cell Immunol. 2000;205:52–61. doi: 10.1006/cimm.2000.1711. [DOI] [PubMed] [Google Scholar]
- 16.Eilat E, Fridkin M, Mozes E. A peptide based on the CDR1 of a pathogenic anti-DNA antibody is more efficient than its analogs in inhibiting autoreactive T cells. Immunobiology. 2000;202:383–93. doi: 10.1016/S0171-2985(00)80041-8. [DOI] [PubMed] [Google Scholar]
- 17.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 18.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. Arthritis Rheum. 1992;35:630–40. doi: 10.1002/art.1780350606. .The Committee on Prognosis Studies in SLE. [DOI] [PubMed] [Google Scholar]
- 19.Shoenfeld Y, Hsu-Lin SC, Gabriels JE, et al. Production of autoantibodies by human human hybridomas. J Clin Invest. 1982;70:205–8. doi: 10.1172/JCI110595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schnolzer M, Alewood PF, Kent SBH. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int J Pept Protein Res. 1992;40:180–93. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 21.Sthoeger ZM, Chiorazzi N, Lahita RG. Regulation of the immune response by sex hormones. I. In vitro effects of estradiol and testosterone on pokeweed mitogen-induced human B cell differentiation. J Immunol. 1988;141:91–8. [PubMed] [Google Scholar]
- 22.Zisman E, Katz Levy Y, Dayan M, et al. Peptide analogs to pathogenic epitopes of the human acetylcholine receptor alpha subunit as potential modulators of myasthenia gravis. Proc Natl Acad Sci USA. 1996;93:4492–7. doi: 10.1073/pnas.93.9.4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams WM, Staines NA, Muller S, Isenberg DA. Human T cell responses to autoantibody variable region peptides. Lupus. 1995;4:464–71. doi: 10.1177/096120339500400608. [DOI] [PubMed] [Google Scholar]
- 24.Bermas BL, Petri M, Goldman D, et al. T helper cell dysfunction in systemic lupus erythematosus (SLE): relation to disease activity. J Clin Immunol. 1994;14:169–77. doi: 10.1007/BF01533366. [DOI] [PubMed] [Google Scholar]
- 25.Segal R, Bermas BL, Dayan M, Kalush F, Shearer GM, Mozes E. Kinetics of cytokine production in experimental systemic lupus erythematosus: involvement of T helper cell 1/T helper cell 2-type cytokines in disease. J Immunol. 1997;158:3009–16. [PubMed] [Google Scholar]
- 26.Funauchi M, Ikoma S, Enomoto H, et al. Decreased Th1-like and increased Th2-like cells in systemic lupus erythematosus. Scand J Rheumatol. 1998;27:219–24. doi: 10.1080/030097498440859. [DOI] [PubMed] [Google Scholar]
- 27.Vratsanos GS, Jung S, Park YM, Craft J. CD4+ T cells from lupus-prone mice are hyperresponsive to T cell receptor engagement with low and high affinity peptide antigens: a model to explain spontaneous T cell activation in lupus. J Exp Med. 2001;193:329–37. doi: 10.1084/jem.193.3.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitamura M, Suto T, Yokoo T, Shimizu F, Fine LG. Transforming growth factor-beta 1 is the predominant paracrine inhibitor of macrophage cytokine synthesis produced by glomerular mesangial cells. J Immunol. 1996;156:2964–71. [PubMed] [Google Scholar]
- 29.Horwitz DA, Gray JD, Ohtsuka K, Hirokawa M, Takahashi T. The immunoregulatory effects of NK cells: the role of TGF-beta and implications for autoimmunity. Immunol Today. 1997;18:538–42. doi: 10.1016/s0167-5699(97)01149-3. [DOI] [PubMed] [Google Scholar]
- 30.Ohtsuka K, Gray JD, Stimmler MM, Toro B, Horwitz DA. Decreased production of TGF-beta by lymphocytes from patients with systemic lupus erythematosus. J Immunol. 1998;160:2539–45. [PubMed] [Google Scholar]
- 31.Yaswen L, Kulkarni AB, Fredrickson T, et al. Autoimmune manifestations in the transforming growth factor-beta 1 knockout mouse. Blood. 1996;87:1439–45. [PubMed] [Google Scholar]
- 32.Raz E, Watanabe A, Baird SM, et al. Systemic immunological effects of cytokine genes injected into skeletal muscle. Proc Natl Acad Sci USA. 1993;90:4523–7. doi: 10.1073/pnas.90.10.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4 (+) CD25 (+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–44. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh RR, Ebling FM, Sercarz EE, Hahn BH. Immune tolerance to autoantibody-derived peptides delays development of autoimmunity in murine lupus. J Clin Invest. 1995;96:2990–6. doi: 10.1172/JCI118371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh RR, Kumar V, Ebling FM, et al. T cell determinants from autoantibodies to DNA can upregulate autoimmunity in murine systemic lupus erythematosus. J Exp Med. 1995;181:2017–27. doi: 10.1084/jem.181.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaliyaperumal A, Michaels MA, Datta SK. Antigen-specific therapy of murine lupus nephritis using nucleosomal peptides: tolerance spreading impairs pathogenic function of autoimmune T and B cells. J Immunol. 1999;162:5775–83. [PubMed] [Google Scholar]
- 37.Jouanne C, Avrameas S, Payelle-Brogard B. A peptide derived from a polyreactive monoclonal anti-DNA natural antibody can modulate lupus development in (NZB × NZW) F1 mice. Immunology. 1999;96:333–9. doi: 10.1046/j.1365-2567.1999.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaynor B, Putterman C, Valadon P, Spatz L, Scharff MD, Diamond B. Peptide inhibition of glomerular deposition of an anti-DNA antibody. Proc Natl Acad Sci USA. 1997;94:1955–60. doi: 10.1073/pnas.94.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hahn BH, Singh RR, Wong WK, Tsao BP, Bulpitt K, Ebling FM. Treatment with a consensus peptide based on amino acid sequences in autoantibodies prevents T cell activation by autoantigens and delays disease onset in murine lupus. Arthritis Rheum. 2001;44:432–41. doi: 10.1002/1529-0131(200102)44:2<432::AID-ANR62>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]