

Abstract
Immature and nonnative proteins are retained in the endoplasmic reticulum (ER) by the quality control machinery. Folding-incompetent glycoproteins are eventually targeted for ER-associated protein degradation (ERAD). EDEM1 (ER degradation-enhancing α-mannosidase-like protein 1), a putative mannose-binding protein, targets misfolded glycoproteins for ERAD. We report that endogenous EDEM1 exists mainly as a soluble glycoprotein. By high-resolution immunolabeling and serial section analysis, we find that endogenous EDEM1 is sequestered in buds that form along cisternae of the rough ER at regions outside of the transitional ER. They give rise to ≈150-nm vesicles scattered throughout the cytoplasm that are lacking a recognizable COPII coat. About 87% of the immunogold labeling was over the vesicles and ≈11% over the ER lumen. Some of the EDEM1 vesicles also contain Derlin-2 and the misfolded Hong Kong variant of α-1-antitrypsin, a substrate for EDEM1 and ERAD. Our results demonstrate the existence of a vesicle budding transport pathway out of the rough ER that does not involve the canonical transitional ER exit sites and therefore represents a previously unrecognized passageway to remove potentially harmful misfolded luminal glycoproteins from the ER.
Keywords: electron microscopy, protein misfolding, protein quality control
The folding state of newly synthesized glycoproteins in the endoplasmic reticulum (ER) is monitored by a quality control machinery (1). Increased formation of misfolded proteins disturbs ER homeostasis resulting in protein degradation, as well as cell damage and death. This is the cause of many human diseases including cystic fibrosis, α-1-antitrypsin deficiency, renal diabetes insipidus, and congenital goiter (2, 3).
Orderly occurring processes can be distinguished during the life and death of a folding-incompetent glycoprotein. The first involves recognition by the quality control machinery (4–6). The lectin chaperones calnexin/calreticulin retain nonnative conformers with monoglucosylated glycans (5, 7). Mannose-trimmed glycans generated by ER-mannosidases appear to represent a quality control tag for routing misfolded glycoproteins to the ER-associated degradation (ERAD) (8–12). The current view is that misfolded, mannose-trimmed glycoproteins are then retrotranslocated to the cytoplasm, where they are ubiquitinated and deglycosylated before proteasomal degradation (13).
EDEM1 (ER degradation-enhancing α-mannosidase-like protein 1) and its yeast ortholog Htm1p/Mnl1p are putative mannose-binding proteins (14–18) that are transcriptionally induced by ER stress (14, 17, 18). They are believed to target misfolded glycoproteins for proteasomal degradation by removing them from the calnexin/calreticulin cycle (14–17). The overexpression of EDEM1 results in the accelerated proteasomal degradation of ERAD substrates such as the Hong Kong variant of α-1-antitrypsin (HK A1AT) and a luminal variant of beta-secretase, whereas the deletion or knockdown of EDEM1/Htm1p reduces the degradation rates of the ERAD substrates (14–17). The EDEM family also includes two soluble paralogues termed EDEM2 and EDEM3 (19, 20). Both have been shown to assist in the degradation of HK A1AT.
In the present study, we aimed to obtain further insight into how EDEM1 assists in the degradation of misfolded glycoproteins by characterizing the cellular properties of EDEM1 and establishing its subcellular distribution.
Results
To detect EDEM1, an antibody against its C-terminal sequence, which is unique to EDEM1 and conserved in both mice and human (19), was generated. Human EDEM1 was in vitro translated in the absence and presence of rough ER-derived canine microsomes to inspect the ability of the anti-peptide antibody to immunoprecipitate EDEM1. In the absence of microsomes, a single EDEM1 species was detected (Fig. 1A, EDEM1UT), whereas the addition of microsomes produced an additional slower migrating protein doublet (Fig. 1A, EDEM1). All EDEM1 bands were immunoprecipitated with an affinity-purified anti-EDEM1 antibody but not its corresponding preimmune serum. The decrease in mobility of EDEM1 caused by ER translocation was due to the addition of four or five N-linked glycans (Fig. 1B).
Fig. 1.

EDEM1 is a heterogeneously glycosylated, soluble protein that comigrates with restricted membrane fractions on Optiprep gradients. (A) EDEM1 was translated in rabbit reticulocyte lysate in the absence or presence of rough ER microsomes (MS). The total lysates were either analyzed directly (IP-) or after immunoprecipitation with either preimmune serum (P) or purified anti-EDEM1 antibodies (E). (B) EDEM1 translated in the presence of rough ER microsomes and subjected to deglycosylation using the indicated concentrations of PNGase F. The number of glycans is designated. (C and D) Isolated canine pancreas rough ER microsomes (C) or membranes from HepG2 cells (D) were alkaline extracted. The total membranes (TM) along with the membrane pellet (P) and soluble supernatant (S) were deglycosylated using PNGase F or Endo H where indicated, resolved on reducing SDS/PAGE, and immunoblotted with EDEM1 antibody (α-EDEM1). (E) Postnuclear cellular membranes from HepG2 cells were loaded onto a discontinuous Optiprep gradient. Fractions collected in the order of increasing density and resolved on a reducing SDS/PAGE were probed for EDEM1 (SI Fig. 6 shows full size blot), the ER markers calnexin and Sec61β, the Golgi marker GM130 and the ERAD proteins Derlin-1 and Derlin-2. P denotes the pellet. (F) Thin section from an Optiprep fraction corresponding to fraction 10 in E shows vesicles and cisternal membrane profiles. (G) Vesicles are positive for EDEM1 by immunoperoxidase electron microscopy. (Scale bars, 500 nm in F and 200 nm in G.)
To characterize the topology of EDEM1, rough ER microsomes were alkaline-extracted and membrane integrated proteins were separated from soluble proteins. By immunoblotting, three EDEM1 species were observed with the slowest band localized to the membrane pellet, and the fastest band to the soluble fraction (Fig. 1C, lanes 1–3). The middle band was also largely found in the soluble fraction. PNGase F digestion removed differences caused by glycosylation, to show that the soluble protein migrated faster than the membrane-associated protein, likely due to cleavage of its N-terminal signal sequence (21). The effectiveness of the alkaline extraction was demonstrated by the separation of the soluble ER enzyme glucosidase II and the integral ER membrane protein calnexin (Fig. 1C, lanes 2 and 3). Similar results were obtained with membranes from HepG2 cells (Fig. 1D). All together, these results indicated that EDEM1 exists as both a soluble and membrane-associated glycoprotein.
Initially to determine the distribution of endogenous EDEM1, total nuclei-free homogenates from human hepatoma HepG2 cells were separated by centrifugation in Optiprep density gradients and fractions were immunoblotted for EDEM1, the ER proteins calnexin and Sec61β, the ERAD proteins Derlin-1 and Derlin-2 and the Golgi protein GM130. Interestingly, EDEM1 immunoreactivity was restricted to the densest membrane fractions along the gradient although calnexin, Sec61β, Derlin-1 and Derlin-2 showed a broader distribution in the density profile [Fig. 1E and supporting information (SI) Fig. 6]. No EDEM1-immunoreactivity was detected in the GM130-immunoreactive fractions. These results indicated a restricted distribution of endogenous EDEM1 in cellular membranes. Electron microscopic analysis of fraction 10 of the Optiprep gradient revealed cisternal profiles and vesicles as main components (Fig. 1F). Some of the vesicles were positive for EDEM1 by immunoperoxidase labeling (Fig. 1G).
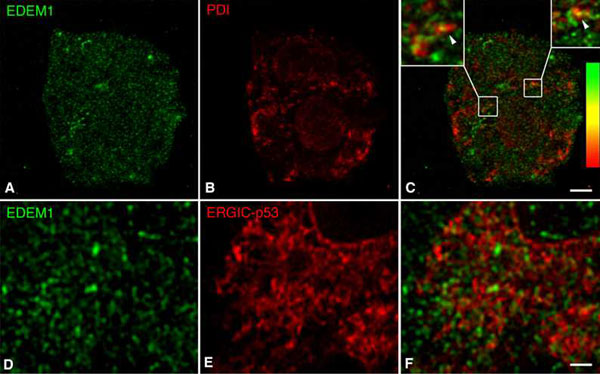
To extend these findings to the in situ subcellular distribution of endogenous EDEM1, we applied immunocytochemistry to a variety of mammalian cell types (HepG2, HEK293, CHO, and clone9 cells as well as MRC5 fibroblasts) including rat hepatoma clone9 cells stably expressing a known ERAD substrate, HK A1AT (17). When purified anti-EDEM1 antibodies were used for confocal immunofluorescence of endogenous EDEM1, an unusual pattern of well distributed punctate structures along with some localized finger-like structures was detected in HepG2 cells (Fig. 2A) and the other studied cell types (Fig. 3E). Under the various conditions of controls (see SI Text), immunolabeling for EDEM1 became undetectable. The labeling pattern for EDEM1 was not reminiscent of a typical ER pattern as it was observed for calnexin (Fig. 2B). Infrequently, overlap of immunofluorescence for EDEM1 and calnexin in punctate structures associated with the ER and regularly in EDEM1-reactive finger-like structures was noted (Fig. 2C). Occasional codistribution was observed with Sec61β, Derlin-2, and PDI (SI Figs. 7–9). Previous immunofluorescence investigations on EDEM1 have studied overexpressed tagged forms of the protein and found an ER pattern (11, 16, 17), which is in contrast to our localization of endogenous EDEM1. To confirm our findings, we compared the distribution of endogenous EDEM1 and of overexpressed EDEM1-FLAG. On Optiprep gradients, EDEM1-FLAG exhibited a broad density equilibrium distribution as calnexin in stably overexpressing CHO cells or transiently overexpressing HepG2 cells (Fig. 3 A–C). By confocal immunofluorescence, overexpressed EDEM1-FLAG in CHO cells showed a typical ER pattern (Fig. 3D), in contrast to the punctate pattern of endogenous EDEM1 in CHO cells (Fig. 3E). This finding must be considered in the interpretation of studies involving overexpressed EDEM1.
Fig. 2.

EDEM1 vesicles are formed outside of the transitional ER. (A–C) Confocal immunofluorescence, HepG2 hepatoma cells, staining for EDEM1 (A), calnexin (B), and overlay (C, codistribution is indicated by different shades of yellow). EDEM1 staining is essentially punctate along with some elongated structures, whereas that for calnexin is reticular (B). (C) EDEM1 and calnexin staining partially overlaps in elongated structures. (D and E) Ultrathin frozen sections from HepG2 cells with immunogold labeling for EDEM1 in the ER lumen (arrowheads) and ER-associated smooth vesicles (arrows). Clathrin-coated vesicles (cv in D) are unlabeled. (F–I) Immunoperoxidase labeling for EDEM1 reveals staining in the lumen of rough ER (F, arrowhead), in ER buds (G), and vesicles pinching-off the ER (H, arrowhead) or close to the ER (I, arrowheads). (K) Four serial sections from a HepG2 cell with luminal EDEM1 labeling (black dots) in parts of ER cisternae. Arrowhead in K2 points to ER membrane-associated EDEM1 staining and arrow to cytoplasmic EDEM1 staining. cp in K 1–3: clathrin-coated pit. (Scale bars, 10 μm in A–C; 60 nm in D and E; 80 nm in F; 95 nm in G; 155 nm in H; 130 nm in I; and 400 nm in K.)
Fig. 3.

Overexpressed EDEM1-FLAG, unlike endogenous EDEM1, is present throughout the ER. Immunoblots of postnuclear cellular membrane fractions collected in the order of increasing density of Optiprep gradients. (A–C) CHO cells stably overexpressing EDEM1-FLAG (A and B) and HepG2 cells transiently overexpressing EDEM1-FLAG (C) were resolved by reducing SDS/PAGE and immunoblotted for calnexin (A) or the FLAG tag (B and C). Both calnexin and the EDEM1-FLAG exhibited similar distribution. Confocal immunofluorescence reveals an ER pattern for overexpressed EDEM1-FLAG in CHO cells (D), which is in contrast with the punctate pattern of endogenous EDEM1 in mock-transfected CHO cells (E). (Scale bars, 10 μm in D and E.)
The subcellular localization of EDEM1 by immunoelectron microscopy was established. Immunogold labeling for EDEM1 in ultrathin frozen sections of CHO, clone 9, and HepG2 (arrowheads in Fig. 2 D and E) cells was sparse in the ER lumen. However, intense gold particle labeling was detected over smooth surfaced membranous elements often closely associated with ER cisternae (arrows in Fig. 2 D and E) that most likely corresponded to the punctate staining observed by confocal immunofluorescence. Vesicles exhibiting a clathrin coat were unlabeled (cv in Fig. 2D). Quantitative evaluation of the gold particle labeling revealed that 10.3% (clone9 hepatocytes) and 10.6% (CHO cells) of the gold particles were over the ER and 87.6% (clone9 hepatocytes) and 87.3% (CHO cells) over vesicles. In addition, 0.5% of immunogold labeling was over the Golgi apparatus, 0.7% over mitochondria and 0.9% over nuclei in both cell types. No labeling was detected over coated vesicles and pre-Golgi intermediates. For a systematic serial section analysis, we used preembedding immunoelectron microscopy. To this end, cells were subjected to preembedding immunoperoxidase or silver-intensified nanogold labeling for EDEM1, embedded in situ in resin, and series of consecutive ultrathin sections were cut in the plane of the cell layer. We observed sparse ER luminal labeling (arrowhead in Fig. 2F) and intensely EDEM1-immunoreactive membrane buds along rough ER cisternae (Fig. 2 G and H) that gave rise to ≈150-nm diameter smooth vesicles (arrowheads in Fig. 2 H and I). By tilting the sections, membrane continuities of the buds with the ER membrane were obvious. By serial section analysis, the vesicular nature of the EDEM1-positive structures was confirmed because they could be detected only in two consecutive ≈80-nm thin sections (SI Fig. 10). Neither EDEM1-reactive buds nor the vesicles had a recognizable coat. This was not due to any inherent difficulty in preserving or visualizing coats, because plasma membrane clathrin-coated pits and clathrin-coated vesicles (Fig. 2 D and K 1 and 2) as well as COPII-coated buds at transitional ER (Fig. 4D 2 and 3 and SI Figs. 11 and 12) were preserved in the same cells. Serial section analysis also revealed the nature of the finger-like EDEM1-reactive structures observed by confocal immunofluorescence. They corresponded to a few rough ER cisternae in a given cell with limited regions showing labeling (Fig. 2K). The EDEM1-immunolabeling in the ER appeared to be mainly luminal, which is in agreement with our biochemical data (Fig. 1). Together, these findings suggest that EDEM1 is sequestered into ER-budded vesicles of ≈150 nm in diameter lacking a recognizable coat.
Fig. 4.

EDEM1 does not localize to COPII component and to ER exit sites. (A–C) Confocal immunofluorescence for EDEM1 (A) or Sec23p (B) in CHO cells and overlay (C) are presented. Lack of codistribution is indicated by absence of yellow color best seen at higher magnification in C Inset. (D) Serial sections of HepG2 cells with immunoperoxidase staining for EDEM1. An ER cisterna forms an exit site with coated buds (arrowheads in D 2 and 3) and a vesiculotubular cluster (VTC in D 3 and 4). EDEM1 labeling is evident in the ER lumen (black spots) but undetectable in the coated buds of the transitional ER and the VTC. Arrow in D points to cytoplasmic EDEM1 labeling. For peroxisomes (PO), note the absence of DAB reaction product. (Scale bars, 8 μm in A–C and 400 nm in D.)
To evaluate a possible spatial relation of the EDEM1-containing buds and vesicles to transitional ER and ER-to-Golgi cargo vesicular carriers, double confocal immunofluorescence was performed for the COPII component Sec23p (22) and ERGIC-53 for pre-Golgi intermediates (23). Neither Sec23p (Fig. 4 A–C) nor ERGIC-53 (SI Fig. 9) exhibited colocalization with EDEM1. These findings were confirmed and extended by electron microscopy. Fig. 4D shows four consecutive serial sections (see also SI Fig. 11) with a transitional ER element exhibiting typical (COPII) coated buds and an associated vesiculotubular cluster (24, 25). Although EDEM1 labeling was present in parts of the lumen of the ER cisterna, the coated buds (arrowheads in Fig. 4D 2 and 3) and the vesiculotubular cluster (VTC in Fig. 4D 3 and 4) of the transitional ER domain exhibited no labeling for EDEM1 (see also SI Fig. 11). The same results were obtained when Golgi-associated ER exit sites were analyzed (SI Fig. 12). These results indicate an ER-budding pathway different from the COPII pathway used by cargo proteins destined for transport to the Golgi apparatus.
We next studied the subcellular distribution of an EDEM1-accelerated ERAD substrate, the misfolded HK A1AT (26), in relation to EDEM1. By confocal immunofluorescence, evidence for codistribution of EDEM1 and HK A1AT in punctate structures (arrows in Fig. 5 A and B) and in finger-like profiles was obtained. As expected, HK A1AT was also detectable throughout the ER (Fig. 5 A and B). By immunoelectron microscopy, labeling for HK A1AT was detected throughout the rough ER, in ER buds and vesicles with the latter showing a more intense immunostaining reaction than the ER cisternae (Fig. 5 D and E).
Fig. 5.

EDEM1 vesicles contain the HK A1AT. Rat clone9 cells stably expressing HK A1AT were studied. (A and B) Confocal double immunofluorescence for EDEM1 (red), A1AT (green) and overlays (codistribution indicated by yellow-orange) are shown. Enlarged boxed fields in A and B reveal partial codistribution of EDEM1 and HK A1AT in punctate structures (some labeled with arrows). (C–E) By immunoperoxidase labeling, EDEM1 (C) and HK A1AT (D and E) are detectable in ER buds and vesicles. (Scale bars, 1 μm in A and B, 270 nm in C, 285 nm in D, and 255 nm in E.)
Discussion
We report high-resolution data on the subcellular distribution of endogenous EDEM1. Our results suggest the existence of an ER vesicle budding pathway through which the putative glycoprotein quality receptor EDEM1 and the ERAD substrate HK A1AT become sequestered from the early secretory pathway. This finding is indicative of a role of vesicular trafficking in the clearance of misfolded luminal proteins from the ER.
Previous studies have analyzed the subcellular site(s) of accumulation of misfolded proteins and shown that they may be stored throughout the ER (27) and may cause local dilatation or proliferation of the ER or induce proliferation of pre-Golgi intermediates (28–33). However, the possible involvement of these induced structures in retrotranslocation of misfolded proteins remains enigmatic. Lederkremer and coworkers (34) identified a quality control compartment unrelated to the bulk of the ER and ERGIC-53 positive pre-Golgi intermediates that contains the ER chaperones calnexin and calreticulin but not BiP, PDI, or the glucosyltransferase. It was suggested to be a microtubule-dependent and brefeldin A-insensitive subcompartment of the ER and proposed to be the site for concentration and retrotranslocation of ERAD substrates. This ER quality control compartment differs from the EDEM1-containing vesicles identified in this study because of its juxtanuclear location adjacent to the centrosome, Golgi apparatus and pre-Golgi intermediates.
The ΔF508 variant of the cystic fibrosis transmembrane conductance regulator (CFTR), which is an ERAD substrate, does not appear to require COPII transport for its degradation in mammalian systems (35). However, when expressed in yeast, the role of COPII in ΔF508 degradation is controversial. Fu and Sztul (36) have found that CFTR degradation requires COPII transport and the segregation of CFTR to an ER subdomain containing the ER hsp70 family member Kar2p/BiP. In sharp contrast, others have found that CFTR degradation in yeast does not require COPII but does require Htmlp/EDEM1 (37, 38). Multiple sequentially acting quality control checkpoints appear to be in place in yeast that operate to sort misfolded proteins with damaged cytosolic domains for retention in the ER or proteins with damage in the luminal domain for COPII-mediated transport to the Golgi apparatus and subsequent retrieval via retrograde transport (39–41). In the end, both pathways meet in the ER where ERAD takes place (40, 41). In mammalian cells, the quality control machinery proteins glucosidase II, glucosyltransferase and calreticulin have been shown to be present beyond the ER in pre-Golgi intermediates (42–44), indicating their function as post-ER quality control checkpoints.
Our results suggest the existence of an ER vesicle budding pathway for aberrant luminal glycoproteins. Constitutive cargo transport from the ER to the Golgi apparatus is mediated by 60- to 70-nm vesicles with an easily discernible COPII coat (45). The exit of COPII vesicles occurs at specialized sites in the ER, termed the transitional ER (24, 25, 46). We observe that EDEM1 vesicles, some of which contain Derlin-2 and misfolded HK A1AT, are of ≈150 nm and lack a recognizable cytosolic coat. Single EDEM1-containing vesicles are formed at multiple sites that are independent of the canonical transitional ER. COPII-coated vesicles are engaged in ER-to-Golgi cargo transport, whereas the EDEM1-containing vesicles provide a path for the removal of potentially harmful misfolded luminal glycoproteins from the ER. Further studies are warranted to investigate how this pathway links up with the cellular degradation machinery.
Materials and Methods
See SI Text for details of EDEM1 antibody generation and affinity-purification, reagents, details of procedures, and SI Figs. 6–12.
Antibody Preparation and Affinity Purification.
A rabbit polyclonal anti-peptide antibody against the C-terminal 15 aa (N-KSIYMRQIDQMVGLI-C) of human EDEM1 protein that is conserved in mouse was generated and affinity-purified by using a soluble EDEM1 lacking its N-terminal hydrophobic domain, which was produced in Escherichia coli.
Cell Culture and Transfection.
Human HepG2, HEK293, and MRC5 fibroblasts, rat clone9 hepatoma cells, and CHO cells were obtained from American Type Culture Collection (Manassas, VA), and culture media were used according to the recommendations of the supplier. Full-length EDEM1 cDNA was subcloned into the p3xFLAG-CMV-14 vector and used for transfection of CHO and HepG2 cells.
Transcription, Translation, and Translocation.
Messenger RNA for EDEM1 was prepared by in vitro transcribing NheI linearized EDEM1-containing plasmid using T7 RNA polymerase. 35S-labeled EDEM1 was translated for 1 h at 27°C in the absence or presence of canine rough ER-derived microsomes.
Immunoprecipitation.
35S-labeled EDEM1 was resuspended in MNT lysis buffer (20 mM 2-morpholinoethanesulfonic acid/100 mM NaCl/30 mM Tris, pH 7.5/0.5% Triton X-100). Samples were precleared with Protein A-Sepharose for 1 h at 4°C, lysates were centrifuged, and supernatants were incubated with anti-EDEM1 or preimmune serum and 1% Protein-A Sepharose. The isolated immune complexes were resuspended in reducing sample buffer.
Alkaline Extraction and Deglycosylation.
RER microsomes were alkaline extracted, and membrane-bound and soluble fractions were separated by ultracentrifugation. Soluble proteins of the supernatant were concentrated by trichloroacetic acid precipitation. Both the concentrated soluble fraction and the membrane-bound pelleted fraction were dissolved in denaturing buffer (0.5% SDS/1% BME, sodium phosphate buffer, pH 7.5). Samples were then denatured, adjusted to 1% Nonidet P-40 and deglycosylated with 10 units/μl PNGase F or Endo H overnight at 37°C. For limited PNGase F digestion of in vitro translated EDEM1, the denaturing buffer was added directly to the lysates and treated with the PNGase F.
Preparation of Cellular Membranes, Optiprep Density Sedimentation, and Immunoblotting.
Cellular membranes from human HepG2 hepatoma cells and HepG2 cells transiently overexpressing EDEM1-FLAG or CHO cells stably overexpressing EDEM1-FLAG as well as the Optiprep gradients were prepared according to the manufacturer's instruction. Concentrated fractions were dissolved in reducing sample buffer, resolved on 7.5% SDS/PAGE, transferred to PVDF, and immunoblotted.
Confocal Immunofluorescence and Immunoelectron Microscopy.
Cells were grown on glass coverslips or in Petri dishes and fed with fresh medium 16 h before fixation. Details of fixation and immunolabeling conditions including specificity controls and quantification of immunogold labeling are given in SI Text.
Supplementary Material
Acknowledgments
We thank Dr. M. Ziak (University of Zurich) for rat clone9 hepatoma cells stably expressing the Hong Kong variant of α-1-antitrypsin; Drs. H.-P. Hauri (University of Basel, Basel, Switzerland), B. Dobberstein (University of Heidelberg, Heidelberg, Germany) and H. Ploegh (Whitehead Institute for Biomedical Research, Cambridge, MA) for the antibodies; and Drs. M. Aebi, C. Hirschberg, W. J. Lennarz and D. Schnell for critical reading of the manuscript. This work was supported by the Swiss National Science Foundation (to J.R.), the Canton of Zurich, and U. S. Public Health Service Grant CA79864 (to D.N.H.).
Abbreviations
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0700154104/DC1.
References
- 1.Ellgaard L, Helenius A. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 2.Petrucelli L, Dawson TM. Ann Med. 2004;36:315–320. doi: 10.1080/07853890410031948. [DOI] [PubMed] [Google Scholar]
- 3.Aridor M, Hannan LA. Traffic. 2002;3:781–790. doi: 10.1034/j.1600-0854.2002.31103.x. [DOI] [PubMed] [Google Scholar]
- 4.Molinari M, Helenius A. Science. 2000;288:331–333. doi: 10.1126/science.288.5464.331. [DOI] [PubMed] [Google Scholar]
- 5.Trombetta ES, Parodi AJ. Annu Rev Cell Dev Biol. 2003;19:649–676. doi: 10.1146/annurev.cellbio.19.110701.153949. [DOI] [PubMed] [Google Scholar]
- 6.Hebert DN, Garman SC, Molinari M. Trends Cell Biol. 2005;15:364–370. doi: 10.1016/j.tcb.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Bedard K, Szabo E, Michalak M, Opas M. Int Rev Cytol. 2005;245:91–121. doi: 10.1016/S0074-7696(05)45004-4. [DOI] [PubMed] [Google Scholar]
- 8.Su K, Stoller T, Rocco J, Zemsky J, Green R. J Biol Chem. 1993;268:14301–14309. [PubMed] [Google Scholar]
- 9.Jakob CA, Burda P, Roth J, Aebi M. J Cell Biol. 1998;142:1223–1233. doi: 10.1083/jcb.142.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Choudhury P, Cabral CM, Sifers RN. J Biol Chem. 1999;274:5861–5867. doi: 10.1074/jbc.274.9.5861. [DOI] [PubMed] [Google Scholar]
- 11.Hosokawa N, Tremblay LO, You Z, Herscovics A, Wada I, Nagata K. J Biol Chem. 2003;278:26287–26294. doi: 10.1074/jbc.M303395200. [DOI] [PubMed] [Google Scholar]
- 12.Lederkremer GZ, Glickman MH. Trends Biochem Sci. 2005;30:297–303. doi: 10.1016/j.tibs.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 13.Meusser B, Hirsch C, Jarosch E, Sommer T. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 14.Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A, Nagata K. EMBO Rep. 2001;2:415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakob CA, Bodmer D, Spirig U, Battig P, Marcil A, Dignard D, Bergeron JJ, Thomas DY, Aebi M. EMBO Rep. 2001;2:423–430. doi: 10.1093/embo-reports/kve089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molinari M, Calanca V, Galli C, Lucca P, Paganetti P. Science. 2003;299:1397–1400. doi: 10.1126/science.1079474. [DOI] [PubMed] [Google Scholar]
- 17.Oda Y, Hosokawa N, Wada I, Nagata K. Science. 2003;299:1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. Dev Cell. 2003;4:265–271. doi: 10.1016/s1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 19.Mast SW, Diekman K, Karaveg K, Davis A, Sifers RN, Moremen KW. Glycobiology. 2005;15:421–436. doi: 10.1093/glycob/cwi014. [DOI] [PubMed] [Google Scholar]
- 20.Hirao K, Natsuka Y, Tamura T, Wada I, Morito D, Natsuka S, Romero P, Sleno B, Tremblay LO, Herscovics A, et al. J Biol Chem. 2006;281:9650–9658. doi: 10.1074/jbc.M512191200. [DOI] [PubMed] [Google Scholar]
- 21.Olivari S, Galli C, Alanen H, Ruddock L, Molinari M. J Biol Chem. 2005;280:2424–2428. doi: 10.1074/jbc.C400534200. [DOI] [PubMed] [Google Scholar]
- 22.Orci L, Ravazzola M, Meda P, Holcomb C, Moore HP, Hicke L, Schekman R. Proc Natl Acad Sci USA. 1991;88:8611–8615. doi: 10.1073/pnas.88.19.8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schweizer A, Fransen J, Bächi T, Ginsel L, Hauri H. J Cell Biol. 1988;107:1643–1653. doi: 10.1083/jcb.107.5.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palade G. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- 25.Bannykh SI, Balch WE. J Cell Biol. 1997;138:1–4. doi: 10.1083/jcb.138.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torossi T, Fan JY, Sauter-Etter K, Roth J, Ziak M. Cell Mol Life Sci. 2006;63:1923–1932. doi: 10.1007/s00018-006-6175-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim PS, Arvan P. Endocrine Rev. 1998;19:173–202. doi: 10.1210/edrv.19.2.0327. [DOI] [PubMed] [Google Scholar]
- 28.Alanen A, Pira U, Lassila O, Roth J, Franklin R. Eur J Immunol. 1985;15:235–242. doi: 10.1002/eji.1830150306. [DOI] [PubMed] [Google Scholar]
- 29.Raposo G, van Santen HM, Leijendekker R, Geuze HJ, Ploegh HL. J Cell Biol. 1995;131:1403–1419. doi: 10.1083/jcb.131.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilbert A, Jadot M, Leontieva E, Wattiaux DCS, Wattiaux R. Exp Cell Res. 1998;242:144–152. doi: 10.1006/excr.1998.4101. [DOI] [PubMed] [Google Scholar]
- 31.Huyer G, Longsworth GL, Mason DL, Mallampalli MP, McCaffery JM, Wright RL, Michaelis S. Mol Biol Cell. 2004;15:908–921. doi: 10.1091/mbc.E03-07-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuber C, Fan JY, Guhl B, Roth J. FASEB J. 2004;18:917–919. doi: 10.1096/fj.03-1210fje. [DOI] [PubMed] [Google Scholar]
- 33.Yam GH-F, Bosshard N, Zuber C, Steinmann B, Roth J. Am J Physiol. 2006;290:C1076–C1082. doi: 10.1152/ajpcell.00426.2005. [DOI] [PubMed] [Google Scholar]
- 34.Kamhi-Nesher S, Shenkman M, Tolchinsky S, Fromm SV, Ehrlich R, Lederkremer GZ. Mol Biol Cell. 2001;12:1711–1723. doi: 10.1091/mbc.12.6.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Matteson J, An Y, Moyer B, Yoo JS, Bannykh S, Wilson IA, Riordan JR, Balch WE. J Cell Biol. 2004;167:65–74. doi: 10.1083/jcb.200401035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu L, Sztul E. J Cell Biol. 2003;160:157–163. doi: 10.1083/jcb.200210086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiser GL, Gentzsch M, Kloser AK, Balzi E, Wolf DH, Goffeau A, Riordan JR. Arch Biochem Biophys. 2001;390:195–205. doi: 10.1006/abbi.2001.2385. [DOI] [PubMed] [Google Scholar]
- 38.Gnann A, Riordan JR, Wolf DH. Mol Biol Cell. 2004;15:4125–4135. doi: 10.1091/mbc.E04-01-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vashist S, Kim W, Belden WJ, Spear ED, Barlowe C, Ng DT. J Cell Biol. 2001;155:355–368. doi: 10.1083/jcb.200106123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vashist S, Ng DT. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caldwell SR, Hill KJ, Cooper AA. J Biol Chem. 2001;276:23296–23303. doi: 10.1074/jbc.M102962200. [DOI] [PubMed] [Google Scholar]
- 42.Lucocq JM, Brada D, Roth J. J Cell Biol. 1986;102:2137–2146. doi: 10.1083/jcb.102.6.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuber C, Fan JY, Guhl B, Parodi A, Fessler JH, Parker C, Roth J. Proc Natl Acad Sci USA. 2001;98:10710–10715. doi: 10.1073/pnas.191359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuber C, Spiro MJ, Guhl B, Spiro RG, Roth J. Mol Biol Cell. 2000;11:4227–4240. doi: 10.1091/mbc.11.12.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Annu Rev Cell Dev Biol. 2004;20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307. [DOI] [PubMed] [Google Scholar]
- 46.Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}