

Abstract
Background
We present the results of EGASP, a community experiment to assess the state-of-the-art in genome annotation within the ENCODE regions, which span 1% of the human genome sequence. The experiment had two major goals: the assessment of the accuracy of computational methods to predict protein coding genes; and the overall assessment of the completeness of the current human genome annotations as represented in the ENCODE regions. For the computational prediction assessment, eighteen groups contributed gene predictions. We evaluated these submissions against each other based on a 'reference set' of annotations generated as part of the GENCODE project. These annotations were not available to the prediction groups prior to the submission deadline, so that their predictions were blind and an external advisory committee could perform a fair assessment.
Results
The best methods had at least one gene transcript correctly predicted for close to 70% of the annotated genes. Nevertheless, the multiple transcript accuracy, taking into account alternative splicing, reached only approximately 40% to 50% accuracy. At the coding nucleotide level, the best programs reached an accuracy of 90% in both sensitivity and specificity. Programs relying on mRNA and protein sequences were the most accurate in reproducing the manually curated annotations. Experimental validation shows that only a very small percentage (3.2%) of the selected 221 computationally predicted exons outside of the existing annotation could be verified.
Conclusion
This is the first such experiment in human DNA, and we have followed the standards established in a similar experiment, GASP1, in Drosophila melanogaster. We believe the results presented here contribute to the value of ongoing large-scale annotation projects and should guide further experimental methods when being scaled up to the entire human genome sequence.
Background
During the first decade of the 21st century the sequencing of whole genomes has become a routine biological practice. The list of chordates with assembled genome sequences now numbers nearly two dozen, while the total number of sequenced bacteria, archea, and eukaryota is approaching 2,000. The genome sequence is said to be an organism's blueprint: the set of instructions dictating its biological traits. In higher eukaryotic organisms, however, these traits are apparently encoded by only a small fraction of the genome sequence that is functional (possibly less than 5% in the case of the human genome). The genes are a major component of this functional sequence. While there is growing evidence for many functional non-protein coding RNA genes, such as miRNAs and snoRNAs, the largest and best studied subset of the human genes comprise the protein coding genes, genes specifying the amino acid sequence of the proteins. Thus, locating the genes in a newly sequenced genome is a first, essential step toward understanding how the organism translates its genome sequence into biological function. This paper focuses on the identification of protein coding genes, if not otherwise noted.
Maybe to the surprise of many, five years after the first drafts of the human genome sequence became available [1,2], and nearly three years after the announcement of the completion of the sequencing [3], a complete set of protein coding genes encoded in the human genome does not exist. One reason for the lack of a complete gene set is that an appropriately rigorous standard has been set for the human genome: every gene, exactly correct. And as shown in this paper, only very few of the human genes seem to be missing from the computational predictions, but the exact genomic structure of these genes is estimated to be correct for only 50% of the predicted genes. In other words, only very few protein coding genes appear to have been totally missed today. Nevertheless, getting the entire genomic structure of a protein coding gene right is still a very difficult task, compounded by the large amount of alternative splicing characterizing human genes. Our assessment here tries to quantify the status of these differences in the current human genome annotations and computational prediction programs.
Automatic genome annotation methods
To date, accurate automatic annotation of the human genome (and of other genomes with significant cDNA libraries) strongly relies on an elaborate mapping of these known gene sequences onto the genome sequence. This method of genome annotation requires high quality and a nearly complete set of cDNA sequences. Datasets trying to achieve this goal, but are still works in progress, are the RefSeq database [4] and those currently being produced by the Mammalian Gene Collection (MGC) [5]. As the MGC project - and similar efforts to deepen the coverage of the fraction of the human genome being transcribed - continues, cDNA mapping based gene identification methods are becoming increasingly accurate. While few organisms will have the rich cDNA libraries that are currently being developed for the human genome, the availability of protein sequence data from evolutionarily close relatives has been effectively used in addition to cDNA data for automatic gene prediction across many of the currently sequenced mammals. The most commonly used annotation pipelines are the ENSEMBL pipeline [6], the UCSC genome browser's [7] Known Genes (KG II) pipeline, and the Gnomon pipeline at the NCBI [8]. It remains unclear, however, what fraction of the low and specifically expressed transcripts and of alternatively spliced isoforms can be effectively recovered from cDNA libraries. Additionally, orthologous proteins from other species may not align genes that are rapidly evolving. For these reasons, current cDNA and protein-based methods are likely to provide an incomplete picture of the protein coding gene content of the human genome. These methods will be less accurate for genomes with fewer expressed sequences and comparative options.
For automatic annotation of genomes without deep expressed sequence libraries, any available cDNA or expressed sequence tag (EST) based annotation is often complemented by dual (or multiple) genome comparative predictions. These predictions are obtained by means of the analysis of the patterns of sequence conservation between genome sequences of evolutionarily related organisms. As examples, programs such SGP2 [9], SLAM [10,11] and TWINSCAN [12,13] have contributed efficiently to the annotation of a number of vertebrate genomes, including mouse [14], rat [15], and chicken [16]. This type of comparative-based automatic gene prediction can produce highly accurate gene sets when the sequence of related species is available, but few ESTs have been sequenced, such as the case with the fungus Cryptococcus neoformans [17].
Occasionally, the so-called single genome ab initio predictors - programs that use statistical sequence patterns, such as the coding reading frame, codon usage or splice site consensus sequences, for gene identification - are also used to complement cDNA and comparative based methods. When no genome exists at the appropriate phylogenetic distance, and the cDNA or EST coverage of the transcriptome is shallow, single genome ab initio predictions play an important role in genome annotation, such as those obtained, for example, by the programs GENSCAN [18] and GENEID [19] in the initial annotation of the genome of the fish Tetraodon nigroviridis [20].
In summary, despite substantial progress in the past decade and the existence of highly accurate gene sets in a number of organisms, current gene identification methods are, as yet, not able to produce a complete catalogue of the set of protein coding genes in higher eukaryotic genomes (see [21] for a recent review).
Assessing the accuracy of automatic genome annotation
Over the past quarter century, a large number of automated gene prediction algorithms have been introduced, which can be loosely grouped based on the general strategies described above. These methods vary widely in the details of their implementation and in the number and location of predicted protein coding genes. Thus, the issue of evaluating the accuracy of the predictive methods has been recurrent within the field of computational gene prediction. The early work of Burset and Guigó [22], and the subsequent analysis of Bajic [23], Baldi et al. [24], Guigó et al. [25], Rogic et al. [26] and others, provide a framework - a set of metrics and a protocol - to consistently evaluate gene prediction methods. Essentially, a set of well-annotated sequences are used as a test set. The gene prediction programs are run on these sequences, and the predictions obtained are compared with the annotations. A number of measures are computed to evaluate how well the predictions reproduce the annotation. Typically, predictions are evaluated at nucleotide, exon and gene levels. At all three levels, two basic measures are computed: sensitivity, the proportion of annotated features (nucleotide, exon, gene) that have been predicted; and specificity, the proportion of predicted features that is annotated. One problem with this approach is that, until recently, very few large genomic sequences were well annotated and only the coordinates of the coding exons within a gene could be considered. Moreover, because methods did not exist to predict alternative splicing, the test sets used to evaluate computational gene predictions consisted of a few hundred short sequences encoding single genes from which alternatively spliced isoforms had been removed. This led to an oversimplification of the problem and, in turn, to an overestimation of the real accuracy of the programs [25]. Furthermore, many programs were developed in-house and were, therefore, not accessible for independent evaluation.
To address the problem of independent, objective assessment of the state-of-the-art in automated tools and techniques for annotating large contiguous genomic DNA regions and eventually complete genomes, a first Genome Annotation Assessment Project (GASP1) was organized in 1999 [27]. In many ways, GASP1 was set up similarly to CASP (Critical Assessment of Techniques for Protein Structure Prediction) [28]. In short, at GASP1, a genomic region in Drosophila melanogaster, including auxiliary training data, was provided to the community and gene finding experts were invited to send the annotation files they had generated to the organizers before a fixed deadline. Then, a set of standards were developed to evaluate submissions against the later published annotations [29], which had been withheld until after the submission stage. Next, the evaluation results were assessed by an independent advisory team and publicly presented at a workshop at the Intelligent Systems in Molecular Biology (ISMB) 1999 meeting. This community experiment was then published as a collection of methods and evaluation papers in Genome Research [27].
The ENCODE Genome Annotation Assessment Project
Inspired by GASP1, and within the context of the ENCyclopedia Of DNA Elements (ENCODE) project, we organized the ENCODE GASP (EGASP) community experiment, which followed closely the model of its predecessor, GASP1 [27]. The ENCODE project was launched two years ago by the National Human Genome Research Institute (NHGRI) with the aim of identifying all functional elements in the genome sequence through the collaborative effort of computational and laboratory-based scientists [30]. The pilot phase of the project is focused on a selected 30 Mb of sequence within 44 selected regions (Table 1) across the human genome, which represents approximately 1% of the genome sequence.
Table 1.
The 44 selected sequences within the ENCODE region
| Random picks Mouse homology | |||||
| Sequence Set | Manual picks | Low | Medium | High | Gene density |
| Training | ENm006 | ENr132 | ENr231 | ENr333 | High |
| ENr232 | ENr334 | ||||
| ENm004 | - | ENr222 | ENr323 | Medium | |
| ENr223 | ENr324 | ||||
| - | ENr111 | - | - | Low | |
| ENr114 | |||||
| Test | ENm002 | ENr131 | ENr233 | ENr331 | High |
| ENm005 | ENr133 | ENr332 | |||
| ENm007 | |||||
| ENm008 | |||||
| ENm009 | |||||
| ENm010 | |||||
| ENm011 | |||||
| ENm001 | ENr121 | ENr221 | ENr321 | Medium | |
| ENm003 | ENr122 | ENr322 | |||
| ENm012 | ENr123 | ||||
| ENm013 | |||||
| ENm014 | |||||
| - | ENr112 | ENr211 | ENr311 | Low | |
| ENr113 | ENr212 | ENr312 | |||
| ENr213 | ENr313 | ||||
ENCODE sequences were assigned to either the training or the test set based on annotation data availability (see the section 'The EGASP experiment'). For the performance evaluation, only the test set sequences were used. The numeric code for the randomly picked sequence names correspond to the non-exonic conservation with the mouse genome, the density of previously identified genes, and the sequence number, respectively; numbers vary from 1 (low), to 3 (high). Manually selected sequences range in size from 500 kbp to 2 Mbp, while random regions are 500 kbp. The selection and stratification criteria for all the sequences is described at the ENCODE project web site [34].
Within the ENCODE project, the GENCODE consortium [31] was set up. This group, in collaboration with the HAVANA team [32] at the Sanger Institute, has produced a high quality annotation of the gene content of the ENCODE regions through a combined manual, computational and experimental strategy [33]. The EGASP experiment was organized with the main goal of evaluating how well automatic methods are able to reproduce this annotation produced by GENCODE. A second goal of EGASP was to assess the completeness of the GENCODE annotation and, in this regard, EGASP was designed such that, in a follow-up step, a number of computational gene predictions not included in GENCODE were tested experimentally.
In what follows, we first describe the organization and structure of the EGASP experiment. We then present the results of the evaluation of the submitted predictions against the GENCODE annotation, and finally we present the results of the experimental verification of the novel predictions.
The EGASP experiment
Data: the benchmark sequence of 44 selected ENCODE regions
Description of the sequence
The 44 ENCODE regions represent 30 Mb (approximately 1%) of the human genome [30]. Approximately half of the sequence corresponds to a set of 14 manually selected regions including well-studied genes and for which a significant amount of prior comparative sequence data was available. The remaining 30 genomic regions were chosen based on a stratified random sampling based on two measures: gene density (from previous annotations) and non-exonic conservation with the mouse genome sequence. Briefly, each portion of the human genome sequence was classified as high, medium, or low if it fell in the top 20%, the middle 30%, or the bottom 50%, respectively, of the above two measures. Several 500 kb sequences were chosen from each of the nine classifications created by this stratification procedure.
Table 1 lists the 44 selected sequences within the ENCODE region and classifies them based on random/manual selection, previously known gene density and non-exonic conservation to the mouse genome. It also describes the size differences between the sequences. Information about the criteria used to select the regions and their characteristics can be found on the ENCODE website [34]. The sequences of the ENCODE regions (as well as multiple functional annotations) can be downloaded from the UCSC ENCODE browser [35].
We defined the sequences used for the EGASP experimental evaluation by taking advantage of the prior work of the HAVANA team [32], which had previously comprehensively annotated and released annotation for several human chromosomes [36-42]. Updated annotation for the 13 ENCODE regions on these chromosomes was released in January 2005 as a 'training' set for the EGASP experiment. The manual annotation of the other 31 ENCODE regions was held back from release until after the automated gene predictions had been received. The 31 EGASP test regions represent a total of 21.6 million base-pairs (bp) of sequence. Further information is available at the GENCODE website [31].
The reference gene set: the GENCODE annotations
The ENCODE regions had been subjected to an exhaustive annotation strategy prior to EGASP by the HAVANA team. In short, the annotators initially build coding transcripts manually based on alignments of known mRNA, EST and protein sequences to the human genome. The initial gene map delineated in this way was then experimentally refined through reverse transcription (RT)-PCR and rapid amplification of cDNA ends (RACE), which essentially confirmed the existence of the mRNA sequences of the hypothesized genes. Finally, the initial annotation was refined by the annotators based on these experimental results. While the initial annotation by the HAVANA team is augmented by some experimentally verified ab initio and dual-genome gene predictions without a priori transcript sequence support, these constitute a marginal fraction of the entire GENCODE annotation set. The strategy is described in detail elsewhere in this issue [33]. We used this final annotation as the reference set for EGASP, and refer to it as the GENCODE annotation.
The protein coding GENCODE annotation for all 44 ENCODE regions consists of 2,471 total transcripts representing 434 unique protein coding gene loci. There are 1,097 coding transcripts that code for 993 unique proteins. The annotation identifies 5.7 total transcripts per locus, with an average of 2.52 coding transcripts. Of the 434 coding loci, 393 contain multi-exon transcripts. In line with earlier estimates [43], 86% of the multi-exon loci exhibit alternative splicing in either the coding or non-coding transcripts. Sixty percent of multi-exon loci have alternative coding transcripts. See [33] in this issue for additional details.
Incomplete annotation
The GENCODE annotation includes incomplete genes and transcripts. These are caused both by the truncation of some features at the end of the ENCODE regions and by transcript annotations that may be incomplete due to lack of evidence. In the rare case that an exon crossed an ENCODE region boundary, the exon was truncated at the ENCODE region boundary in both the annotations and the predictions to ensure that the nucleotide level evaluation statistics were computed correctly (see Materials and methods).
EGASP: a community experiment
To determine an automatic method's ability to reproduce the GENCODE annotation, we organized EGASP in the following way: In January 2005, the GENCODE annotation for 13 of the 44 ENCODE regions (the 'training regions' defined above) was publicly released. With the release of this annotation, EGASP was officially announced: gene and other DNA feature prediction groups world-wide were asked to submit genome annotations on the remaining 31 ENCODE regions, for which the GENCODE annotations would not be released until the deadline for submission expired. Participating groups had access to the annotation of the 13 training regions, as well as to the sequences and all additional publicly available data for all 44 ENCODE regions. No other pre-defined and pre-selected auxiliary data, such as cDNA databases, EST sequences or other genome alignments, were given to the submitters. However, many of the 31 test regions had been previously and extensively annotated by other groups. For example, ENm001, the greater cystic fibrosis transmembrane receptor (CFTR) region, has been extensively studied [44].
Participants were asked to submit their genome annotations on the 31 ENCODE test regions, using whatever methods and data were available to them. To be able to better compare different DNA feature prediction methods, we predefined the following prediction categories and asked the submitters to indicate in which category they were submitting: methods using any type of available information; single-genome ab initio methods; EST-, mRNA-, and protein-based methods; dual- or multiple-genome based methods; methods predicting unusual genes (non-canonical splicing, short intron-less genes, and so on); and exon-only predictions.
Finally, we allowed an extra category (category 7) for methods predicting other annotation features, including pseudogenes and promoters. Bajic et al. [45] have conducted a comprehensive evaluation of the promoter predictions and see Zheng and Gerstein [46] for a paper on pseudogenes.
A web server (Figure 1) [47] was set up to collect all the submissions and each group was able to submit predictions for more than one category. The submitted predictions, as well as the GENCODE annotations for the test sequence set, were kept confidential until the submission deadline on 15 April 2005. The format for submissions was the Gene Transfer Format (GTF) [48]. An advisory committee (Table 2) was formed to oversee the submission and evaluation processes and provide advice for the evaluation.
Figure 1.

A screenshot of the EGASP submission server [47]. The server was user-authenticated in order to keep the submitted predictions in private before the EGASP workshop. Initially, there were eight suggested submission categories. However, after the workshop, category 5 was not used at all and removed. Promoter and pseudogene predictions from category 8 were then kept as a new category 7, which is not analyzed in this paper (see [45] instead).
Table 2.
EGASP organizing and advisory committees
| Organizers | Advisory board |
| Jennifer Ashurst (Wellcome Trust Sanger Institute) | Michael Ashburner (Cambridge University) |
| Ewan Birney (European Bionformatics Institute) | Vladimir B Bajic (Institute for Infocomm Research) |
| Peter Good (National Human Genome Research Institute) | Tom Gingeras (Affymetrix, Inc.) |
| Roderic Guigó (Institut Municipal d'Investigació Mèdica) | Suzanna Lewis (Berkeley) |
| Tim Hubbard (Wellcome Trust Sanger Institute) | Martin Reese (Omicia, Inc.) |
By the submission deadline on 15 April 2005, 18 groups had submitted 30 prediction sets (Table 3). All the submitted predictions together with the annotations are available through the GencodeDB Genome Browser (Figure 2) [49], as well as through the UCSC Genome Browser ('EGASP' tracks). They can also be downloaded from the ftp server as plain text GTF files [50].
Table 3.
Summary of programs used to determine predictions submitted for each EGASP category
| Submission category | Program | Affiliation | Reference |
| 1 (AUGUSTUS-any) | AUGUSTUS | Georg-August-Universität, Göttingen | [58] |
| 2 (AUGUSTUS-abinit) | |||
| 3 (AUGUSTUS-EST) | |||
| 4 (AUGUSTUS-dual) | |||
| 1 | FGENESH++ | Softberry Inc. | [56] |
| 1 | JIGSAW | The Institute for Genomic Research (TIGR) | [59] |
| 1 (PAIRAGON-any) | PAIRAGON and NSCAN_EST | Washington University, Saint Louis (WUSTL) | [57] |
| 3 (PAIRAGON+NSCAN_EST) | |||
| 2 | GENEMARK.hmm | Georgia Institute of Technology | [60] |
| 2 | GENEZILLA | TIGR | [81] |
| 3 | ACEVIEW | National Center for Biotechnology Information (NCBI) | [52] |
| 3 | ENSEMBL | The Wellcome Trust Sanger Institute (WTSI) and | [64] |
| European Bioinformatics Institute (EBI) | |||
| 3 | EXOGEAN | Ecole Normale Superieure, Paris | [62] |
| 3 | EXONHUNTER | University of Waterloo | [63] |
| 4 | ACESCAN* | Salk Institute | [82] |
| 4 | DOGFISH-C | WTSI | [67] |
| 4 | NSCAN | WUSTL | [57] |
| 4 | SAGA | University of California at Berkeley | [66] |
| 4 | MARS | WUSTL - EBI | [65] |
| 5 | GENEID-U12 | Institut Municipal d'Investigació | - |
| 5 | SGP2-U12 | Mèdica, Barcelona | |
| 6 | ASPIC† | Università degli Studi di Milano | [83] |
| 6 (AUGUSTUS-exon) | AUGUSTUS | Georg-August-Universität, Göttingen | [58] |
| 6 | CSTMINER‡ | Università degli Studi di Milano | [84] |
| 6 | DOGFISH-C-E§ | WTSI | [67] |
| 6 | SPIDA | EBI | [85] |
| 6 | UNCOVER§ | Duke University | [86] |
| 1 | CCDSGene | UCSC tracks [7] | [55] |
| 1 | KNOWNGene | [54] | |
| 1 | REFSEQ (REFGene) | [4] | |
| 2 | GENEID | [19] | |
| 2 | GENSCAN | [18] | |
| 3 | ACEMBLY | [52] | |
| 3 | ECGene | [53] | |
| 3 | ENSEMBL (ENSGene) | [6] | |
| 3 | MGCGene | [5] | |
| 4 | SGP2 | [9] | |
| 4 | TWINSCAN | [12,13] | |
| - | CODING 20050607 | GENCODE annotation | [33] |
| - | GENES 20050607 |
A complete listing of the number of features for each sequence obtained by each method is available at the Supplementary material web page [51]. *The ACESCAN group submitted results only for the training set and, therefore, has not been evaluated. †ASPIC only provided results for the training regions and, therefore, has not been evaluated. Moreover, ASPIC submitted only intron annotations and should be considered in category 6. ‡CSTMINER predicts coding regions but does not provide strand information. §DOGFISH-C-E and UNCOVER predict only novel exons; this makes it difficult to compare these methods with the others in the same category.
Figure 2.

The GencodeDB Genome Browser. A screenshot of the GencodeDB Genome Browser [49], displaying the annotation features on 100 Kbp from the ENm001 region (chr7: 116,074,892-116,174,891). The annotations along with the predicted genes by each submitted method were made publicly available together with further experimental evidence, such as TARs/transfrags.
Predictions were compared with the reference set GENCODE annotations and assessed by members of the advisory and organizing committees (Table 2), all selected as independent experts in this field. The results of this assessment were presented at a workshop that took place at the Wellcome Trust Genome Campus in Hinxton, UK, on 6 and 7 May 2005. The advisory and organizing committees met on 4 May for a pre-evaluation of the predictions, and to determine a number of summary statistics. Each of the submitting groups was invited to present their methods and submissions at the workshop with a focus on what went right and what went wrong. In total, 16 groups were represented at the workshop. The final prediction evaluation results from the workshop are discussed in the next section.
Results
The evaluation of the predictions against the annotation
The protocol to evaluate the predictions
The main goal of the EGASP experiment was to evaluate the ability of automatic methods of genome annotation to reproduce the manual and experimental annotation of the ENCODE regions described above. By this standard, a perfect prediction strategy would produce annotation completely consistent with the GENCODE annotation.
For the purposes of evaluating the submitted predictions, we considered only the results for the 31 test ENCODE regions, which were the 'blinded' regions for which no GENCODE annotations were available during the submission phase. Potential biases introduced by this restriction will be addressed below. The statistics reported are computed globally for the test region, which means that the total number of prediction successes and failures for all 31 regions are compared directly to the total number of annotated exons, transcripts and genes for all 31 regions.
We evaluated each set of submitted predictions at four distinct levels: nucleotide accuracy, exon accuracy, transcript accuracy, and gene accuracy. At the earlier GASP1 workshop, transcript accuracy levels were not assessed due to the limited transcript information and the lower levels of alternatively spliced transcripts in Drosophila melanogaster [27]. For this study we also made a distinction between the statistics calculated for the coding portions of the mRNA transcripts (coding sequence (CDS) evaluations) and the mRNA transcripts as a whole (mRNA evaluations).
For each of the four levels, we calculated the sensitivity and specificity of the predictions as defined below. In some cases, we have also computed other standard measures previously used in the gene finding literature (see [22-27]). Many additional measures of accuracy have been computed on the EGASP predictions, and they are available through the Supplementary Material web page [51].
Non-EGASP entries
To compare the EGASP results to existing community standards, we also evaluated the performance of 11 gene annotation tracks published in the UCSC Browser [7] just before the start of the EGASP workshop. These tracks included two single genome ab initio prediction methods (GENSCAN [18] and GENEID [19]) and two dual-genome prediction methods (TWINSCAN [12,13] and SGP2 [9]). We also considered four methods we classified as using expressed sequence (ENSGENE [6], ACEMBLY [52], MGCGENES [5], and ECGENE [53]) and three we classified as using any information (UCSC 'KNOWN' genes [54], REFSEQ genes [4], and CCDSGENES [55]).
Measures used for evaluating predictions: definitions
Nucleotide level accuracy is a comparison of the annotated nucleotides with the predicted nucleotides. Individual nucleotides appearing in more than one transcript in either the annotation or the predictions are considered only once for the nucleotide level statistics (Figure 3a). Nucleotide predictions must be on the same strand as the annotations to be counted as correct. At the nucleotide level, sensitivity (Sn) is the proportion of annotated nucleotides (as being coding or part of an mRNA molecule) that is correctly predicted, and specificity (Sp) the proportion of predicted nucleotides (as being coding or part of an mRNA molecule) that is so annotated. As a summary measure, we have computed either the simple average of these two measures, or the correlation coefficient between the annotated and the predicted nucleotides (see [22-27]).
Figure 3.
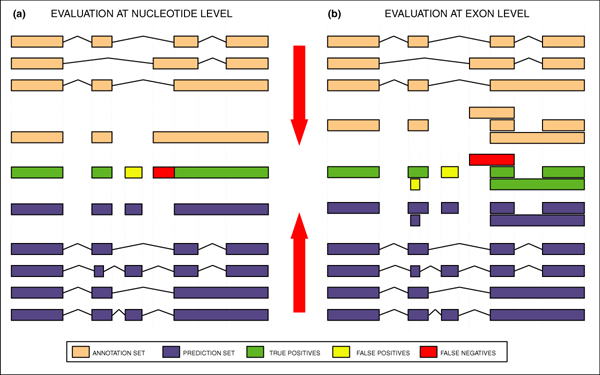
Gene Feature Projection for evaluation. The process of projecting genic features into unique nucleotide and exon coordinates in order to compute the accuracy values (see text for details).
The exon level accuracy is calculated with the requirement that an exon in the prediction must have identical start and end coordinates as an exon in the annotation to be counted correct. Only the unique exons in each set are considered (see Figure 3b for a graphical example of how unique exons are collected from both the annotation and prediction sets; also see [22-27] for more details on these definitions). At the exon level, sensitivity is computed as the proportion of annotated exons correctly predicted, and specificity as the proportion of predicted exons that is annotated. As a summary measure, we have computed the average of these two measures. In addition, we have computed 'missing exons' (MEs), the proportion of annotated exons totally missed by the predictions (that is, there is no overlap by a predicted exon by at least 1 bp), and 'wrong exons' (WEs), the proportion of predicted exons not overlapping annotated exons by at least 1 bp. A subset of predicted exons falling in regions annotated as intergenic have been tested experimentally (see the section 'The experimental test of unannotated predictions' below for details). Nucleotide and exon level accuracy are calculated for the CDS evaluation and for the mRNA evaluation. Comparison of the results of these evaluation strategies highlights the differences for those programs that attempt to predict untranslated regions (UTRs) of genes.
The transcript and gene level accuracy measures are more stringent. We consider a transcript accurately predicted for the CDS evaluation if the beginning and end of translation are correctly annotated and each of the 5' and 3' splice sites for the coding exons are correct. Similarly, for the mRNA evaluation, a transcript is counted correct if all of the exons from the start of transcription to the end of transcription are correctly predicted. Thus, at the transcript level, sensitivity is the proportion of annotated transcripts that is correctly predicted, and specificity is the proportion of predicted transcripts that is correct. A gene is counted correct if at least one transcript in the locus is correct as defined above, and sensitivity and specificity are defined accordingly. Using these definitions, transcript accuracy is the most stringent measure for both the CDS evaluation and for the mRNA evaluation (Figure 4).
Figure 4.
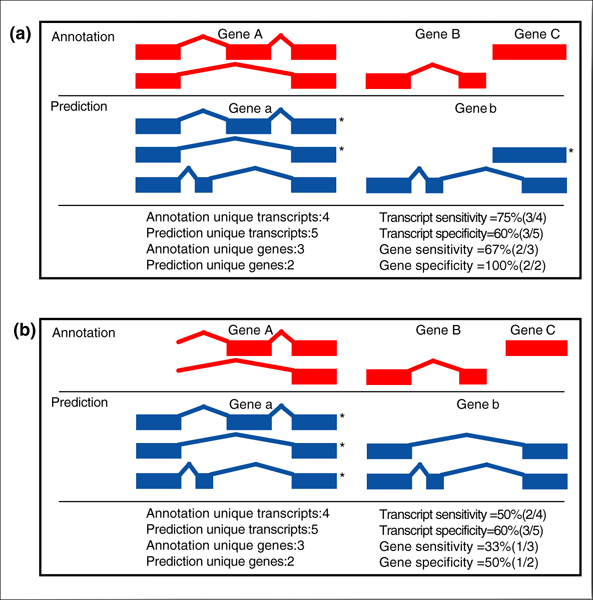
Gene transcript evaluation. Computing sensitivity and specificity at transcript level: (a) complete transcript annotation; (b) incomplete transcript annotation. Transcripts marked with an asterisk are considered 'consistent with the annotation' and will be scored as correct.
The accuracy of the prediction methods must be considered in the context of the annotation, which contains a significant fraction of incomplete transcripts. In the case of an incomplete transcript, we made the distinction that if a prediction is completely consistent with the annotation, it will be counted correct. For example, if the annotation contains an incomplete transcript with three exons and a prediction method includes a transcript with these exons plus an additional exon, we consider the prediction to be completely consistent with the annotation and count it as a correct prediction. For the CDS evaluation, if the annotation contains a complete coding transcript, it must be predicted correctly and no additional exons are allowed (Figure 4).
Global results and trends
The evaluation statistics discussed above for the CDS evaluation are provided in Tables 4 and 5 and for the mRNA evaluation in Table 6, which only lists methods that predict full mRNA transcripts. Figures 5, 6, 7, 8 display the results for the CDS evaluation at the nucleotide, exon, transcript and gene levels. Values are given for programs in categories 1 to 4 (see previous section and Table 3), which constitute the bulk of the submitted predictions. The accuracies of the programs in other categories are often not strictly comparable and, therefore, not shown in these figures. They are, however, given in the Supplementary material [51]. The top panel in Figures 5, 6, 7, 8 is a dotplot of sensitivity versus specificity, where each dot represents the performance of one program. The bottom panel includes a boxplot for each program displaying the average of sensitivity and specificity (that is, (Sn + Sp)/2) for the given program on each of 27 test sequences (see Materials and methods). Four test sequences (ENr112, ENr113, ENr311, ENr313) were removed from the original set of 31 because they did not contain any annotated protein coding genes and, therefore, sensitivity and specificity could not be computed for them. The dotplot intends to capture the global balance between sensitivity and specificity for each program, while the boxplots provide the dispersion of the accuracy of each program predictions across test sequences. At similar average accuracies, programs providing more consistent predictions across sequences may be preferable since their behavior can be better anticipated.
Table 4.
CDS assessment: summary of accuracy measures for CDS features at the nucleotide and exon levels
| Nucleotide | Exon | ||||||
| NSn | NSp | N CC | ESn | ESp | ME | WE | |
| Category 1 | |||||||
| AUGUSTUS-any | 94.42% | 82.43% | 0.88 | 74.67% | 76.76% | 8.25% | 16.29% |
| FGENESH++ | 91.09% | 76.89% | 0.83 | 75.18% | 69.31% | 9.73% | 24.64% |
| JIGSAW | 94.56% | 92.19% | 0.93 | 80.61% | 89.33% | 6.22% | 7.78% |
| PAIRAGON-any | 87.77% | 92.78% | 0.90 | 76.85% | 88.91% | 11.18% | 6.82% |
| Category 2 | |||||||
| AUGUSTUS-abinit | 78.65% | 75.29% | 0.76 | 52.39% | 62.93% | 29.09% | 24.82% |
| GENEMARK.hmm-A | 78.43% | 37.97% | 0.53 | 50.58% | 29.01% | 27.86% | 63.27% |
| GENEMARK.hmm-B | 76.09% | 62.94% | 0.69 | 48.15% | 47.25% | 31.77% | 40.68% |
| GENEZILLA | 87.56% | 50.93% | 0.66 | 62.08% | 50.25% | 19.14% | 41.93% |
| Category 3 | |||||||
| ACEVIEW | 90.94% | 79.14% | 0.84 | 85.75% | 56.98% | 4.38% | 16.69% |
| AUGUSTUS-EST | 92.62% | 83.45% | 0.88 | 74.10% | 77.40% | 9.01% | 15.61% |
| ENSEMBL | 90.18% | 92.02% | 0.91 | 77.53% | 82.65% | 9.99% | 9.22% |
| EXOGEAN | 84.18% | 94.33% | 0.89 | 79.34% | 83.45% | 9.88% | 5.06% |
| EXONHUNTER | 90.46% | 59.67% | 0.73 | 64.44% | 41.77% | 14.29% | 50.94% |
| PAIRAGON+NSCAN_EST | 87.56% | 92.77% | 0.90 | 76.63% | 88.95% | 11.51% | 6.85% |
| Category 4 | |||||||
| AUGUSTUS-dual | 88.86% | 80.15% | 0.84 | 63.06% | 69.14% | 16.82% | 19.60% |
| DOGFISH | 64.81% | 88.24% | 0.74 | 53.11% | 77.34% | 32.67% | 11.70% |
| MARS | 84.25% | 74.13% | 0.78 | 65.56% | 61.65% | 20.26% | 26.10% |
| NSCAN | 85.38% | 89.02% | 0.87 | 67.66% | 82.05% | 17.11% | 10.93% |
| SAGA | 52.54% | 81.39% | 0.65 | 38.82% | 50.73% | 40.48% | 27.85% |
| UCSC Tracks | |||||||
| ACEMBLY | 96.43% | 58.47% | 0.74 | 84.66% | 38.32% | 2.71% | 28.55% |
| CCDSgene | 56.87% | 99.52% | 0.75 | 51.95% | 97.75% | 40.38% | 0.27% |
| ECgene | 96.36% | 47.30% | 0.66 | 86.22% | 35.08% | 2.64% | 45.92% |
| ENSgene | 91.39% | 91.92% | 0.92 | 77.71% | 82.39% | 9.80% | 9.21% |
| GENEID | 76.77% | 76.48% | 0.76 | 53.84% | 61.08% | 27.86% | 27.26% |
| GENSCAN | 84.17% | 60.60% | 0.71 | 58.65% | 46.37% | 19.50% | 42.91% |
| KNOWNgene | 89.10% | 93.61% | 0.91 | 78.11% | 82.28% | 10.27% | 4.30% |
| MGCgene | 44.06% | 97.56% | 0.65 | 42.95% | 93.61% | 49.28% | 2.68% |
| REFgene | 85.34% | 98.50% | 0.92 | 73.23% | 94.67% | 15.38% | 1.22% |
| SGPgene | 82.81% | 82.20% | 0.82 | 60.56% | 65.16% | 19.36% | 22.85% |
| TWINSCAN | 78.16% | 84.59% | 0.81 | 58.43% | 73.11% | 24.64% | 16.30% |
CC, correlation coefficient.
Table 5.
CDS assessment at the transcript and gene levels
| Transcript | Gene | ||||
| TSn | TSp | GSn | GSp | Ratio CDS/UTR | |
| Category 1 | |||||
| AUGUSTUS-any | 22.65% | 35.59% | 47.97% | 35.59% | 100.00% |
| FGENESH++ | 36.21% | 41.61% | 69.93% | 42.09% | 78.25% |
| JIGSAW | 34.05% | 65.95% | 72.64% | 65.95% | 100.00% |
| PAIRAGON-any | 39.29% | 60.34% | 69.59% | 61.32% | 62.92% |
| Category 2 | |||||
| AUGUSTUS-abinit | 11.09% | 17.22% | 24.32% | 17.22% | 100.00% |
| GENEMARK.hmm-A | 6.93% | 3.24% | 15.20% | 3.24% | 100.00% |
| GENEMARK.hmm-B | 7.70% | 7.91% | 16.89% | 7.91% | 100.00% |
| GENEZILLA | 9.09% | 8.84% | 19.59% | 8.84% | 100.00% |
| Category 3 | |||||
| ACEVIEW | 44.68% | 19.31% | 63.51% | 48.65% | 49.15% |
| AUGUSTUS-EST | 22.50% | 37.01% | 47.64% | 37.01% | 100.00% |
| ENSEMBL | 39.75% | 54.64% | 71.62% | 67.32% | 65.77% |
| EXOGEAN | 42.53% | 52.44% | 63.18% | 80.82% | 59.60% |
| EXONHUNTER | 10.48% | 6.33% | 21.96% | 6.33% | 100.00% |
| PAIRAGON+NSCAN_EST | 39.29% | 60.64% | 69.59% | 61.71% | 62.89% |
| Category 4 | |||||
| AUGUSTUS-dual | 12.33% | 18.64% | 26.01% | 18.64% | 100.00% |
| DOGFISH | 5.08% | 14.61% | 10.81% | 14.61% | 100.00% |
| MARS | 15.87% | 15.11% | 33.45% | 24.94% | 100.00% |
| NSCAN | 16.95% | 36.71% | 35.47% | 36.71% | 79.80% |
| SAGA | 2.16% | 3.44% | 4.39% | 3.44% | 100.00% |
| UCSC Tracks | |||||
| ACEMBLY | 33.90% | 7.96% | 54.39% | 21.24% | 48.56% |
| CCDSgene | 28.97% | 85.58% | 55.41% | 89.39% | 100.00% |
| ECgene | 56.86% | 8.84% | 79.05% | 12.42% | 46.11% |
| ENSgene | 40.52% | 54.09% | 73.99% | 68.30% | 65.62% |
| GENEID | 4.78% | 8.78% | 10.47% | 8.78% | 100.00% |
| GENSCAN | 7.40% | 10.13% | 15.54% | 10.13% | 100.00% |
| KNOWNgene | 43.45% | 46.93% | 77.03% | 72.79% | 60.03% |
| MGCgene | 23.73% | 78.24% | 49.32% | 82.56% | 63.43% |
| REFgene | 41.91% | 75.21% | 77.03% | 82.76% | 61.82% |
| SGPgene | 8.17% | 12.59% | 17.57% | 12.59% | 100.00% |
| TWINSCAN | 10.63% | 20.25% | 22.30% | 20.25% | 100.00% |
The ratio CDS/UTR was obtained by summing up all the coding exons' lengths and dividing by the sum of all the exons' lengths. The ratio CDS/UTR for the annotations is 36.78%.
Table 6.
mRNA assessment: summary of accuracy measures of mRNA features at the nucleotide and exon levels
| Nucleotide | Exon | ||||||
| NSn | NSp | N CC | ESn | ESp | ME | WE | |
| Category 1 | |||||||
| FGENESH++ | 48.87% | 81.16% | 0.62 | 35.84% | 58.41% | 19.20% | 22.84% |
| PAIRAGON-any | 56.31% | 89.36% | 0.70 | 41.23% | 74.93% | 15.83% | 7.95% |
| Category 3 | |||||||
| ACEVIEW | 88.08% | 79.47% | 0.83 | 64.16% | 61.18% | 3.60% | 10.41% |
| ENSEMBL | 61.61% | 95.26% | 0.76 | 41.61% | 73.41% | 12.84% | 7.09% |
| EXOGEAN | 60.58% | 94.73% | 0.75 | 48.87% | 76.29% | 10.38% | 4.16% |
| PAIRAGON+NSCAN_EST | 56.22% | 89.35% | 0.70 | 41.11% | 74.98% | 16.06% | 7.98% |
| Category 4 | |||||||
| NSCAN | 39.55% | 78.69% | 0.55 | 32.41% | 65.25% | 26.10% | 14.69% |
| UCSC Tracks | |||||||
| ACEMBLY | 91.94% | 53.98% | 0.70 | 65.51% | 44.28% | 2.15% | 18.26% |
| ECgene | 93.00% | 38.68% | 0.59 | 58.17% | 34.83% | 1.81% | 34.31% |
| ENSgene | 62.43% | 95.27% | 0.77 | 41.71% | 72.65% | 12.62% | 7.10% |
| KNOWNgene | 65.74% | 91.82% | 0.77 | 43.84% | 74.57% | 13.58% | 2.74% |
| MGCgene | 29.17% | 96.73% | 0.53 | 21.21% | 74.10% | 47.16% | 2.22% |
| REFgene | 57.51% | 97.07% | 0.74 | 38.35% | 83.51% | 19.28% | 0.91% |
Only programs that submitted 5' or 3' UTR exon annotations besides the CDS parts of exons are shown. CC, correlation coefficient.
Figure 5.
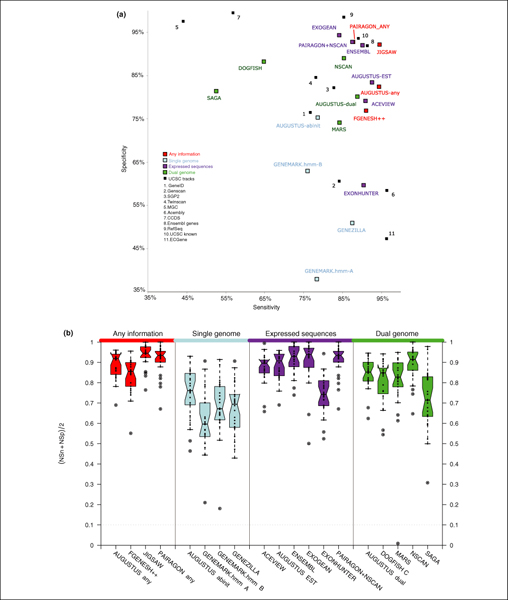
Gene Prediction Accuracy at the nucleotide level: Sensitivity versus specificity. Top panel: dotplot for sensitivity versus specificity at the nucleotide level for CDS evaluation. Each dot represents the overall value for each program on the 31 test sequences. Bottom panel: boxplots of the average sensitivity and specificity ((Sn + Sp)/2) for each program. Each dot corresponds to the average in each of the test sequences for which a GENCODE annotation existed (27 out of 31 sequences).
Figure 6.
Gene Prediction Accuracy at the exon level: Sensitivity versus specificity. Top panel: dotplot for sensitivity versus specificity at the exon level for CDS evaluation. Each dot represents the overall value for each program on the 31 test sequences. Bottom panel: boxplots of the average sensitivity and specificity for each program. Each dot corresponds to the average in each of the test sequences for which GENCODE annotation existed.
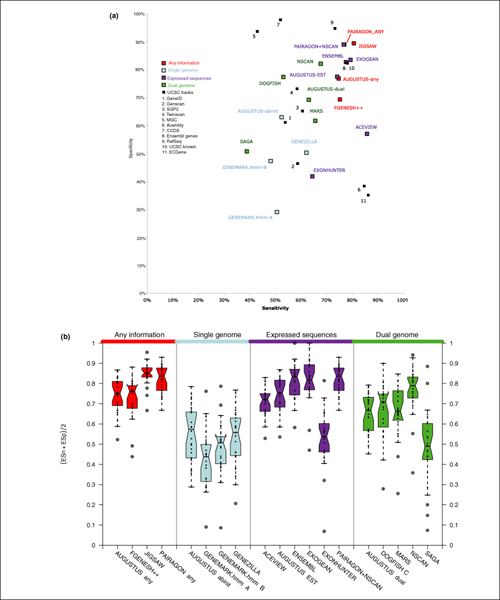
Figure 7.
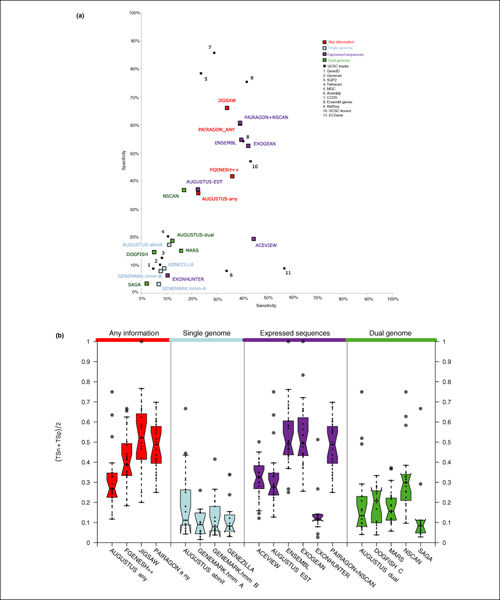
Gene Prediction Accuracy at the transcript level: Sensitivity versus specificity. Top panel: dotplot for sensitivity versus specificity at the transcript level for CDS evaluation. Each dot represents the overall value for each program on the 31 test sequences. Bottom panel: boxplots of the average sensitivity and specificity for each program. Each dot corresponds to the average in each of the test sequences for which GENCODE annotation existed.
Figure 8.
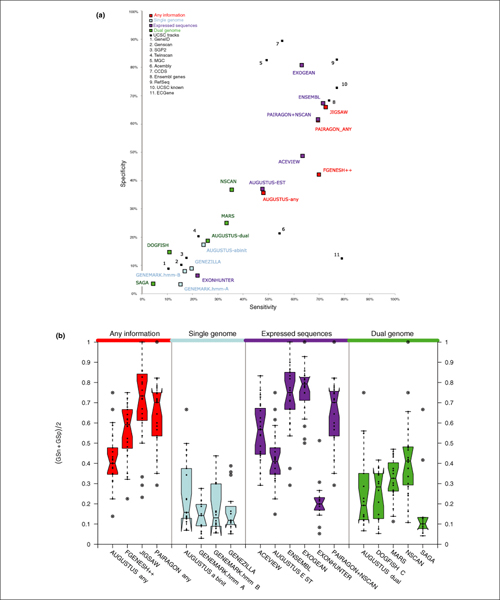
Gene Prediction Accuracy at the gene level: Sensitivity versus specificity. Top panel: dotplot for sensitivity versus specificity at the gene level for CDS evaluation. Each dot represents the overall value for each program on the 31 test sequences. Bottom panel: boxplots of the average sensitivity and specificity for each program. Each dot corresponds to the average in each of the test sequences for which GENCODE annotation existed.
No annotation strategy produced perfect predictions, but several clear trends emerged from the evaluations and are summarized here.
The prediction methods that used expressed sequence information (category 3) and those that used any information (category 1 prediction methods often used expressed sequence information) were generally the most accurate for all measures.
The three best category 4 dual-genome methods (NSCAN, MARS, and AUGUSTUS-dual) were more accurate than the category 2 single genome ab initio prediction methods.
At the nucleotide level, JIGSAW and ENSEMBL both achieved greater than 90% for both sensitivity and specificity for the CDS evaluation, while several other methods scored greater than 80% for both sensitivity and specificity on the same measure, including the NSCAN and AUGUSTUS dual-genome methods (Figure 5). For the mRNA evaluation, ACEVIEW reached 88% sensitivity at 79% specificity, while ENSEMBL and EXOGEAN were more specific with 95% and 94%, respectively, but at much lower sensitivities of 61% and 60%, respectively.
At the exon level, the most accurate predictor of coding exons was JIGSAW with greater than 80% sensitivity while maintaining nearly 90% specificity. ACEVIEW was the most sensitive prediction method for all exons (coding and non-coding) with greater than 85% (CDS) and 64% (mRNA) exon sensitivity while still being reasonably specific (Figure 6).
At the transcript level, no prediction method correctly identified greater than 45% of the coding transcripts exactly (see sensitivity in Figure 7).
At the gene level, using the measure of averaged sensitivity and specificity, the most accurate gene level predictions in the CDS evaluation were produced by EXOGEAN followed by JIGSAW and ENSEMBL. JIGSAW and ENSEMBL were the only two methods with greater than 70% gene level sensitivity. Of the two, JIGSAW was slightly more sensitive, while ENSEMBL was slightly more specific. EXOGEAN's specificity was higher than 80%, which is more than 13% higher than any other program (Figure 8; Table 5).
Relatively few prediction methods are able to predict multiple transcripts per gene locus. These include four expressed sequence methods from category 3 (PAIRAGON+NSCAN_EST, EXOGEAN, ACEVIEW, and ENSEMBL), FGENESH++ and PAIRAGON-any from category 1, and MARS from category 4.
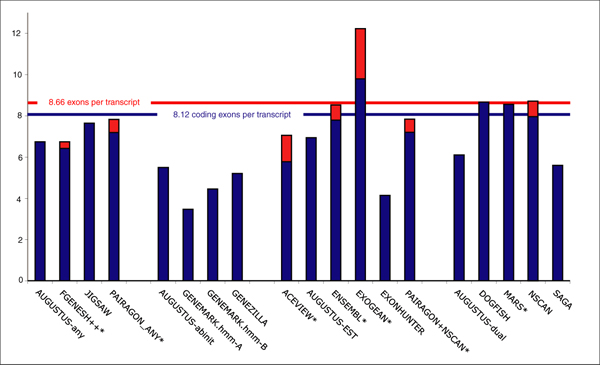
Most of the methods predict genes that, on average, have fewer coding exons per gene than the GENCODE annotation (Figure 9). The only exceptions to this observation are EXOGEAN, DOGFISH, and MARS, which all predict more coding exons than the annotation.
Figure 9.
Exon counts per gene transcript. A comparison of the number of exons per transcript and coding exons per transcript in the GENCODE annotation of the 31 test regions and in the predictions. Blue bars show the average number of coding exons per coding transcript for each of the programs in categories 1, 2, 3, and 4; the blue line shows this for the GENCODE annotation. The number of all exons per transcript in the GENCODE annotation is shown with a red line. Those programs that predict non-coding exons are noted with red bars. Programs marked with an asterisk predict multiple transcripts per gene locus.
Prediction tracks from the UCSC browser were generally clustered near the EGASP entries for similar categories. At the transcript level, the BLAT aligned REFSEQ mRNAs ('REFgene') were both more sensitive than all of the prediction methods except EXOGEAN and ACEVIEW, and approximately 8% more specific than the best EGASP entries. The MGC transcripts ('MGCGene') and the CCDS transcripts ('CCDSgene') were 10% and 18% more specific at the transcript level, but had significantly lower sensitivity than the best EGASP method due to the incomplete nature of these sets at the time of the workshop.
In general, the accuracy of the programs varied substantially across test sequences, but some programs appear to behave more consistently than others (as is reflected in the boxplots in Figures 5, 6, 7, 8).
Programs performed in general better in the training than in the test sequences, the two exceptions being ACEVIEW from category 3 and FGENESH++ from category 1 (Figure 10).
Figure 10.
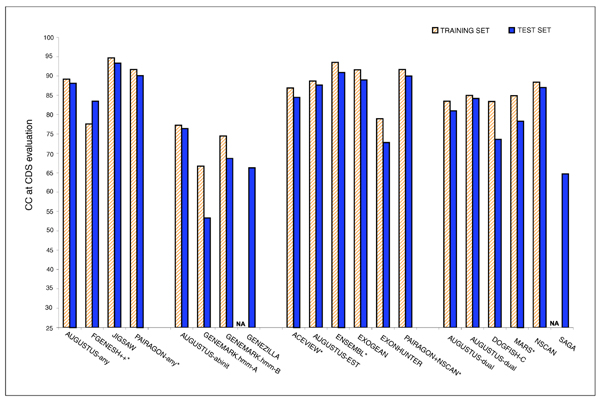
Correlation Coefficient Accuracy for Training and Test Sequences. The correlation coefficient (CC) at the nucleotide level for CDS evaluation for sequences EN_TRN13 and EN_PRD31 for training and test set sequences. NA, not available; because the submitters did not send their results for the training set.
No overall trend was observed when comparing performance between manually placed ENCODE regions and the random ones (Figure 11). Even though, programs in category 4 performed consistently better in the random picks.
Figure 11.
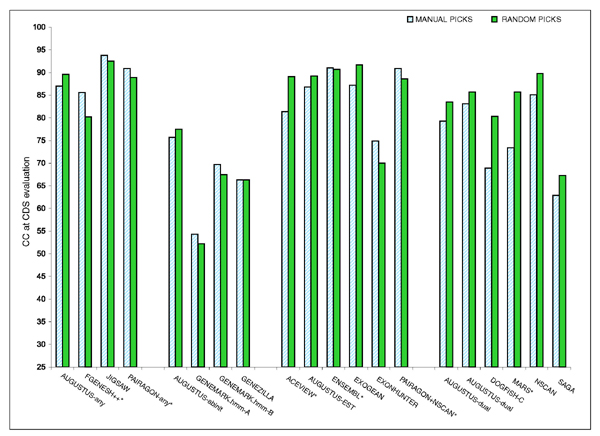
Correlation Coefficient Accuracy for manually and randomly selected Sequences. The correlation coefficient (CC) at the nucleotide level for CDS evaluation for EN_MNLp12 and EN_RNDp19 for manually and randomly selected sequences within the test set.
Programs performed clearly better in medium or high gene dense regions than in regions poor in genes (Figure 12). Only the category 6 method SPIDA had higher accuracy in regions of low gene density.
Figure 12.
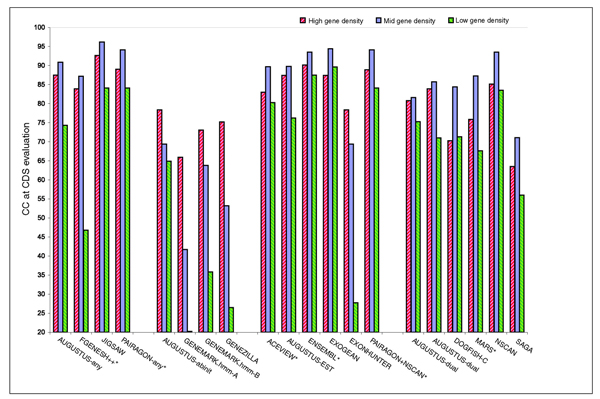
Correlation Coefficient Accuracy in relation to gene density. The correlation coefficient (CC) at the nucleotide level for sequences EN_PGH12, EN_PGM11 and EN_PGL8 for high, mid and low gene density sequence sets within the test set.
The accuracy of the programs was also related to the level of conservation of the genomic sequence in the mouse genome, with programs performing generally better in the test sequences showing stronger conservation in the mouse genome, but the trend was not as strong as with gene density (Figure 13).
Figure 13.
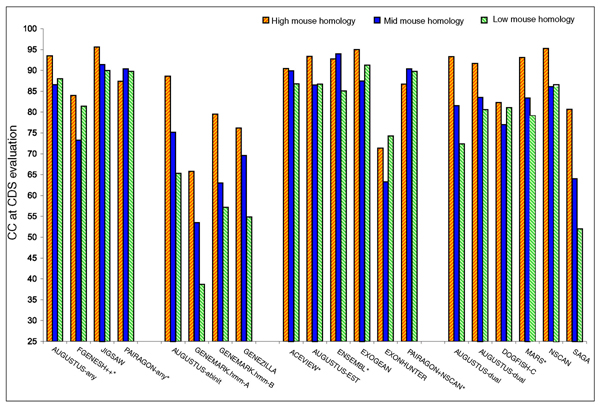
Correlation Coefficient Accuracy in relation to sequence conservation. The correlation coefficient (CC) at the nucleotide level for sequences EN_PMH7, EN_PMM5 and EN_PML7 for high, mid and low conservation with mouse sequences only for the randomly selected sequences in the test set.
Results by category for the CDS evaluation
Category 1: methods using any type of available information
Four prediction methods were considered in EGASP category 1. Of these the FGENESH++ pipeline [56], the PAIRAGON-any pipeline [57], and AUGUSTUS-any [58] are conceptually similar. Each of these approaches uses information from both expressed sequences and from ab initio or de novo gene prediction strategies.
FGENESH++ and PAIRAGON-any consist of an alignment step followed by de novo prediction in the regions where there are not alignments. The sensitivity of these two methods is similar for all levels of the evaluation, but PAIRAGON-any is significantly more specific. AUGUSTUS-any uses both the 'hints' discovered in its expressed sequence (category 3) strategy and those discovered in its dual-genome (category 4) strategy.
Both the AUGUSTUS and the PAIRAGON groups submitted predictions in categories 1 and 3, allowing us to judge the value of the additional information that each of the programs used in producing the category 1 predictions. Neither program shows a significant increase in predictive performance in this category over their respective category 3 predictions (see below). For AUGUSTUS-any, this suggests that its models get very little additional information from the inclusion of the dual-genome prediction information. For PAIRAGON-any, the category 1 prediction set included only two transcripts not included in the category 3 prediction set (PAIRAGON+NSCAN).
JIGSAW [59] is unlike the other three methods. It uses a statistical combination of several sources of evidence to create the best consensus prediction. Considering all the evaluation measures, JIGSAW is the most accurate category 1 prediction method, although both PAIRAGON-any and FGENESH++ are more sensitive than JIGSAW at the transcript level. FGENESH++ and PAIRAGON-any predict multiple transcripts per gene locus.
Category 2: single-genome ab initio methods
Three ab initio prediction methods use only the information found in the human genome sequence. All three methods only predict coding transcripts and are thus only considered by the CDS evaluation. Of the three, GENEZILLA is the most sensitive at the nucleotide and exon levels, while AUGUSTUS-abinit is the most specific. AUGUSTUS-abinit is consistently better than the other two at finding the start and end of translation and is thus both more sensitive and more specific at both the transcript and the gene level.
There are two variants of the predictions made by (the human genome version) of the GENEMARK.hmm program [60]. Data marked GENEMARK.hmm-A were produced and submitted prior to the deadline and inadvertently used unmasked genomic sequence (communication at the workshop by M Borodovsky). This is also the case for the GENEZILLA predictions in the single genome category, which were also created using unmasked sequence. Therefore, we caution the direct comparison of GENEMARK.hmm-A and the GENEZILLA results to the results of the other programs, which in general used masked genomic sequence. It is well known that gene finding programs do worse on unmasked sequences due to the high 'protein-coding-like' content of repetitive elements, resulting in an increase of the number of false positive predictions [61]. Data marked GENEMARK.hmm-B were produced by the same human genome version of the GENEMARK.hmm algorithm run on the masked sequence (communication by M Borodovsky), although this was a post-deadline submission. It is clearly seen that the specificity values for GENEMARK.hmm are higher when run on masked sequence due to the significant decrease in the false positive rate.
Category 3: EST-, mRNA-, and protein-based methods
More submissions were received for category 3 than for any of the other categories and the type of expressed sequence information (EST, mRNA, protein sequence) varied among the methods, as did the strategy for incorporating the information. As such, it is not surprising that the methods have various strengths and weaknesses depending on the details of the method. For example, ACEVIEW [52] has the highest transcript sensitivity and predicts an average of 4.05 coding transcripts per gene locus. This is nearly twice as many transcripts per gene compared to EXOGEAN [62], which is nearly as sensitive (44.7% and 42.5%, respectively) and predicts only 2.34 coding transcripts per gene locus. ACEVIEW also has the highest coding exon sensitivity, but its high sensitivity comes at a cost of a relatively low specificity.
For the CDS evaluation at the nucleotide level, AUGUSTUS-EST [58] is the most sensitive program and EXOGEAN is the most specific. There is little distinction at the nucleotide level among most of the category 3 programs with the exception of EXONHUNTER [63], which seems to get less information from expressed sequences and scores significantly lower than the other programs.
At the coding exon level, the best programs (EXOGEAN, PAIRAGON+NSCAN_EST, and ENSEMBL) predict more than 75% of the exons correctly, while maintaining specificity greater than 80%. Of these three, EXOGEAN is the most sensitive, and PAIRAGON+NSCAN_EST is the most specific. A similar story exists at the transcript level, where each of these 3 programs predicts more than 39% of the coding transcripts correctly, with specificity greater than 50%. Again EXOGEAN is the most sensitive (42.5% compared with 39.3% for PAIRAGON+NSCAN_EST and 39.8% for ENSEMBL) and PAIRAGON+NSCAN_EST is the most specific.
At the gene level, ENSEMBL [64] is more sensitive than PAIRAGON+NSCAN_EST (71.6% versus 69.6%) and more specific. EXOGEAN is the most specific program at the gene level at a specificity of 80.8% with a sensitivity of 63.2%.
Category 4: dual- or multiple-genome based methods
Six groups submitted gene structure predictions that were assigned to the dual-genome category. ACESCAN, however, submitted predictions only on the 13 training regions and was, therefore, not evaluated.
Of the dual-genome prediction programs, NSCAN [57] is generally the most sensitive and the most specific for all evaluation levels. The only exception is at the nucleotide level, where AUGUSTUS-dual [58] is more sensitive (88.9% versus 85.4%) at a cost of being less specific than NSCAN (80.2% versus 89.0%). All of the dual-genome predictors except MARS [65] are limited to predicting one transcript per gene locus.
NSCAN is one of the most conservative of the dual-genome gene predictors, which partly explains its high transcript and gene specificity. It predicts approximately 90 fewer genes than SAGA [66], approximately 110 fewer than MARS, and almost 130 fewer than AUGUSTUS-dual. Only DOGFISH [67], which predicts 219 genes, is more conservative.
Other predictions
Two programs submitted predictions on the test regions for category 5 (methods predicting unusual genes, non-canonical splicing, short intronless genes, and so on). Both GENEID-U12 and SGP2-U12 (T. Alioto, unpublished) are optimized to find genes that contain U12 introns (see Patel and Steitz [68] for an in-depth review on U12 splicing).
Six programs submitted predictions that were included in category 6 (exon only predictions). ASPIC predicted only introns for the training regions, CSTMINER predicted coding regions, but did not provide strand information or splice site boundaries, DOGFISH-C-E and UNCOVER predicted only novel exons, and AUGUSTUS-exon and SPIDA predicted exons but they did not attempt to link them into transcript structures.
The programs in categories 5 and 6 have very specialized and diverse goals and cannot easily be compared to each other or to the predictions in other categories. Their accuracy values, however, have been computed when possible, and they are provided in the Supplementary material.
Results for the mRNA evaluation
In the computational gene finding literature, gene predictions have traditionally been evaluated using coding transcripts only. That is, only the exonic structure of the coding fraction of the gene or transcript is taken into account both in the prediction and in the annotation. One reason for this has been the difficulty of experimentally determining 'full length' cDNAs, which represent a full mRNA transcript. While it is difficult to accurately clone and sequence the 3' UTRs of cDNA clones, it is even harder to obtain and sequence the 5' UTRs of a gene transcript. Besides the limitation of existing experimental data, very little signal information exists in the sequence of 5' and 3' UTRs of genes that can be statistically modeled. Therefore, most of the computational gene finders have historically made no attempt to predict UTRs, and instead predicted genes from the start codon to the stop codon.
Apparently, encouraged by the announcement to explicitly try to "replicate the GENCODE annotations", which included many full mRNA transcript annotations in the training set, several programs submitted predictions of the entire exonic structure of the mRNA molecules. FGENESH++, PAIRAGON+NSCAN_EST, ACEVIEW, ENSEMBL, EXOGEAN, and NSCAN programs all submitted full transcript predictions, including coding and untranslated (UTR) exons. We have compared these predictions with the annotated exonic structure of the mRNA transcripts within the GENCODE annotation. Accuracy results for the mRNA evaluation of these programs are given in Table 6.
In general, programs performed worse when predicting the exonic structure of the entire transcript than when predicting only the coding exons. This is consistent with the fact that the UTR sequences are less constrained than regions coding for amino acid sequences. Note, however, that the 3' and 5' end of the genes are particularly difficult to delineate experimentally. Therefore, a metric that emphasizes prediction of exact exon boundaries will lead to an underestimation of the accuracy of the predictions. Evaluation of the predictions at the intron level, instead of exon level, could partially address this limitation. In any case, given these limitations, ACEVIEW exhibits the highest accuracy of mRNA evaluations and has similar accuracy, at least at the nucleotide level, when considering either the entire mRNA or the CDS. In contrast to other programs, ACEVIEW is more specific in the entire mRNA than on the CDS. It also has the highest sensitivity, although ENSEMBL, EXOGEAN and PAIRAGON+NSCAN_EST are more specific.
Interpreting the results
The 44 ENCODE regions represent 30 Mb (approximately 1%) of the human genome. The 31 EGASP test regions include 21.6 Mb and represent an even smaller fraction of the human genome. Although this is the largest region ever used for benchmarking automatic genome annotation, it is not a random selection of the human genome, and, therefore, results obtained in them should only be extrapolated to the whole genome with appropriate caution. The stratification of the ENCODE regions into 'manually' versus 'randomly' selected and according to gene density and conservation with mouse (Table 1) allows for an investigation into how these factors affect the accuracy of gene predictions.
Figure 14a displays the accuracy of each program (average sensitivity and specificity at the nucleotide level) in the form of boxplots for each individual sequence in which genes were annotated (27 of the 31 EGASP regions) and for the collections of sequences discussed next (random versus manual, training versus test, low, medium and high gene density, and low, medium and high conservation with the mouse genome). The accuracy of the programs varied substantially across sequences (with a median value raging from values below 0.7 (ENm011) to above 0.95 (ENr332). Figure 14b shows similar results but on the exon level. In what follows, we describe potential biases in the evaluation results that can be explained by the characteristics of the raw sequences, instead of the behavior of the prediction methods.
Figure 14.
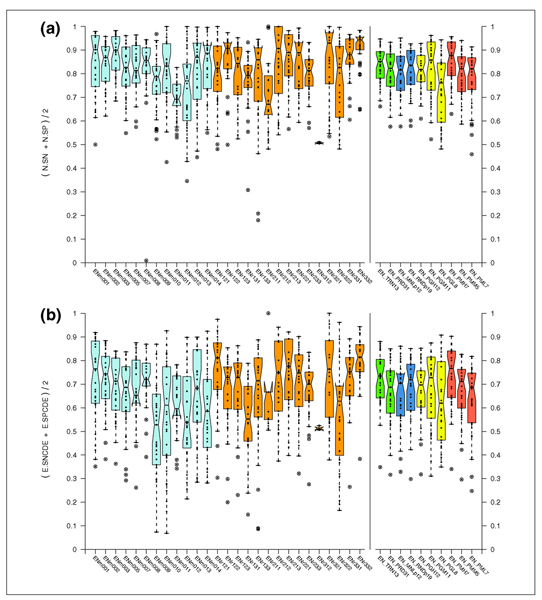
Gene Prediction Accuracy for each ENCODE sequence at the nucleotide and exon levels. Boxplots showing the average sensitivity and specificity at the (a) nucleotide level and (b) exon level for CDS evaluation of each program on every sequence of the test set. Sequences are displayed across the x-axes. Manual picks are shown in in light blue; random picks are shown in orange. Boxplots corresponding to the overall average sensitivity and specificity at the nucleotide level for CDS evaluation in different subsets of the ENCODE sequences are shown at the right of the graph. EN_TRN13, the set of 13 training regions, and EN_PRD31, the set of 31 test regions, are shown in green. EN_MNLp12, the 12 manual picks in the test set, and EN_RNDp19, the 19 random picks in the test set are shown in dark blue. EN_PGH12/EN_PGM11/EN_PGL8, the subsets of 12 high, 11 medium and 8 low gene dense sequences from the set of test sequences, are shown in yellow. EN_PMH7/EN_PMM5/EN_PML7, the subsets of seven regions with high sequence conservation with mouse, five regions with medium conservation, and seven regions with low conservation from random picks in the test set, are shown in red.
Training versus testing regions
We compared the predictive accuracy of each of the programs on the set of 13 training regions to their performance on the 31 test regions. Most of the gene prediction programs were more accurate on the training set compared to the test set (Figure 10). This can be partially explained by the training set being enriched in gene dense regions (see the section Gene rich versus gene poor regions below; Table 1). Indeed, 11 of the 13 training regions (85%) had a high or medium gene density, compared with 23 out of the 31 test regions (74%).
Random versus manual regions
Within the test set, we compared the performance of each of the gene prediction programs on the set of 12 manually placed ENCODE regions to their performance on the set of 19 ENCODE regions chosen randomly (Figure 11). Some programs performed better in the manual regions, while others did on the random ones, but no overall trend could be observed. Only programs in category 4 (dual- or multiple-genome predictors) performed consistently better in the random than in the manual picks. One possible explanation for this might be that the GENCODE annotation is more exhaustive in the regions selected manually. These regions contain genes of interest, and some of them have been extensively investigated. Therefore, the coverage by cDNAs -on which the GENCODE annotation is based - is likely to be higher in the manual than in the randomly chosen regions, which might explain the difference in class performance.
Gene rich versus gene poor regions
We compared the performance of the prediction programs based on the stratification of high (12 sequences), medium (11 sequences) and low (8 sequences) gene dense regions (see the data description in the section Description of the sequence above). In general, all programs performed better in regions with medium or high gene density than in regions with low gene density (Figure 12). This reflects the low specificity resulting from a higher rate of false positive predictions. Interestingly, single genome ab initio gene finders (category 2) performed the best in very gene rich regions, while the programs in all other categories performed the best in regions with an intermediate density of genes. This is possibly due to the training of the programs tuned to balance over- and under-prediction.
High versus low conservation with mouse regions
We also compared the performance of the prediction programs in the randomly selected sequences with high (seven sequences), medium (five sequences) and low (seven sequences) conservation with mouse (see the data description in the section Description of the sequence above). Programs performed generally better in sequences showing higher conservation with mouse, but the trend was not as strong as with gene density (Figure 13). As expected, dual genome predictors performed better in sequences with high mouse homology, but the trend was also observed for single genome predictors. It is possible to speculate that genes conserved across species are also likely to exhibit more typical sequence characteristics, in terms of codon bias and splice site signals, whereas fast evolving genes may have undergone changes toward unusual sequence biases. Single genome predictors are likely to perform better on genes exhibiting typical features.
The experimental test of unannotated predictions
The second major goal of EGASP was to assess the completeness of the GENCODE annotation. This annotation is based on available evidence, and we cannot immediately rule out the possibility that it misses a fraction of protein coding genes. Indeed, many predictions submitted for EGASP did not match any GENCODE annotated exon. Table 7 lists the total number of unique coding and non-coding exons predicted by each program, as well as the number of unannotated exons (that is, predicted exons not overlapped by a GENCODE annotated exon by 1 bp or more). Also listed is the number of unannotated exons predicted in intergenic regions. The unannotated are termed 'wrong exons' in the evaluation section above. It is unclear what fraction of these unannotated exons belong to annotated genes or are exons of novel, unannotated genes. We have carried out an initial investigation by comparing the unannotated exons with transcript data obtained from the hybridization of polyA+ cytosolic RNA onto Affymetrix high-density genome tiling microarrays covering the ENCODE regions. Details of the technology and applications have recently been published [69,70]. Briefly, positive hybridization probes are combined into discrete sites of transcription, which are usually known as TARs (transcriptionally active regions) or transfrags (transcribed fragments). It is important to note that identification of TARs/transfrags is based on selecting a threshold of detection that is derived from estimates of 5% false positive detection using a bacterial sequence spike in controls [71]. Raising or lowering the false positive rates can alter these thresholds. More or fewer detected regions of transcription will likely follow from these changes. TAR/transfrag maps corresponding to different cell lines and conditions have been downloaded from the ENCODE specific UCSC browser [35]. Table 7 lists the number of predicted unannotated exons that overlap TARs/transfrags by at least 1 bp. Overall, 44.7% of the EGASP predicted exons overlap TARs/transfrags. Importantly, while 71% of the annotated exons overlap TARs/transfrags, only 13% of the unannotated exons do. This difference suggests that many of the predicted, but not annotated, exons are false positives.
Table 7.
Exons predicted by the programs not overlapping GENCODE annotated exons, and supported by transfrag evidence from genome tiling microarrays
| Total number of unique exons | Number of exons overlapping TARs/transfrags (%) | Number of non-annotated exons | Number of non-annotated exons overlapping TARs/transfrags (%) | Number of intergenic exons | Number of Intergenic exons overlapping TARs/transfrags (%) | |
| Category 1 | ||||||
| AUGUSTUS-any | 4,160 | 2,718 (65.3%) | 484 | 74 (15.3%) | 281 | 38 (13.5%) |
| FGENESH++ | 4,784 | 2,766 (57.8%) | 1,071 | 146 (13.6%) | 885 | 123 (13.9%) |
| JIGSAW | 3,935 | 2,673 (67.9%) | 206 | 34 (16.5%) | 130 | 19 (14.6%) |
| PAIRAGON-any | 4,414 | 3,080 (69.8%) | 284 | 84 (29.6%) | 221 | 68 (30.8%) |
| Category 2 | ||||||
| AUGUSTUS-abinit | 3,699 | 2,336 (63.2%) | 776 | 175 (22.6%) | 482 | 111 (23.0%) |
| GENEMARK.hmm | 6,897 | 2,552 (37.0%) | 3,796 | 319 (8.4%) | 2,826 | 244 (8.6%) |
| GENEZILLA | 3,415 | 1,535 (44.9%) | 1,361 | 110 (8.1%) | 970 | 69 (7.1%) |
| Category 3 | ||||||
| ACEVIEW | 8,410 | 5,756 (68.4%) | 539 | 118 (21.9%) | 449 | 106 (23.6%) |
| AUGUSTUS-EST | 4,073 | 2,700 (66.3%) | 439 | 73 (16.6%) | 253 | 37 (14.6%) |
| ENSEMBL | 4,505 | 3,094 (68.7%) | 251 | 27 (10.8%) | 187 | 17 (9.1%) |
| EXOGEAN | 5,014 | 3,480 (69.4%) | 183 | 21 (11.5%) | 100 | 15 (15.0%) |
| EXONHUNTER | 6,376 | 2,843 (44.6%) | 2,782 | 257 (9.2%) | 2,055 | 187 (9.1%) |
| PAIRAGON+NSCAN_EST 4,404 | 3,073 (69.8%) | 284 | 84 (29.6%) | 221 | 68 (30.8%) | |
| Category 4 | ||||||
| AUGUSTUS-dual | 4,024 | 2,588 (64.3%) | 629 | 114 (18.1%) | 364 | 65 (17.9%) |
| DOGFISH | 3,194 | 2,290 (71.7%) | 267 | 115 (43.1%) | 225 | 99 (44.0%) |
| MARS | 4,623 | 2,801 (60.6%) | 948 | 161 (17.0%) | 528 | 89 (16.9%) |
| NSCAN | 3,996 | 2,686 (67.2%) | 500 | 133 (26.6%) | 342 | 92 (26.9%) |
| SAGA | 2,115 | 1,147 (54.2%) | 564 | 36 (6.4%) | 433 | 26 (6.0%) |
| All unique exons (18 progs) | 26,818 | 12,001 (44.7%) | 12,025 | 1,563 (13.0%) | 8,634 | 1,163 (13.5%) |
Support by TARs/transfrags, however, indicates only that the predicted exon appears to be transcribed and, possibly, processed into an RNA sequence. It does not allow us to infer that the predicted exons assemble into the predicted transcript structure, or that the transcript is a protein-coding RNA. Therefore, to better assess the likelihood of the predicted exonic structures, we selected a subset of the predicted but unannotated exon pairs to be tested experimentally by RT-PCR. We focused our verification efforts on the subset of 8,634 intergenic exons (Table 7), since these predictions could correspond to yet undetected, novel genes. We ranked the predictions based on the predictive specificity at the exon level for the given programs and then selected the top 200 ranking exons. We next identified all predicted introns (exon pairs) radiating from this set of exons. That is, we paired each of these exons with its immediate upstream and downstream neighbors within the same predicted transcript. Selection of those pairs not overlapping any GENCODE annotation resulted in 238 unique non-inclusive exon pairs (pairs in which one of the exons was included in an exon from another selected exon pair were discarded). Of these pairs, 221 could be tested by RT-PCR (see Materials and methods) in 24 tissues. All data files can be accessed through the Supplementary material web page. Of the assayed exon pairs, only seven (3.2%) produced a positive result, all with perfectly predicted exon boundaries. Of the seven validated exon pairs, three were intergenic, presumably representing new transcribed loci, while four extended existing gene annotations. Every positive case was expressed in only a single tissue out of the 24 tested. This result is comparable to that obtained for novel human genes identified using the chicken genome as reference (expressed on average in 3.3 tissues) [72] or for the recently described chimeric transcripts (expressed on average in 2.5 tissues) [73]. This result is significantly below the 7 to 8 average positive tissues out of 12 tested found for known mammalian genes [14,74], suggesting that the majority of yet unanno-tated genes have a restricted pattern of expression. This also suggests an explanation for why these transcripts have eluded identification by experimental means until now.
The number of exon pairs (introns) tested per program, and the number of positive verifications are given in Table 8 (see also the Supplementary material web page for information about the positive cases [51]). There appear to be differences in the success rate by program, but the numbers are too small to draw significant conclusions. Interestingly, the positive predictions tended to be classified high in our ranking based on the specificity of the programs: 3 out of the 7 positive predictions ranked among the top 50 ranked predictions, and 6 ranked among the 100 top ranking predictions. This suggests that combining multiple sources of evidence helps to identify the computational predictions that correspond to 'bona fide' genes. Consistent with these observations, TARs/transfrags overlap with about 20% of the exons classified among the 200 top ranking ones, but only with 13% of the intergenic predicted exons overall. Two of the positive predictions had already been included in later releases of the GENCODE annotation, but were unknown at the time of experimental verification. Two are extending 'putative' GENCODE loci, and three could correspond to novel genes - one being antisense to an annotated GENCODE locus. In the GENCODE annotation, the sequence of the RT-PCR products is passed back into the GENCODE pipeline, where it is used as another source of transcript sequence evidence. Future versions of GENCODE will incorporate the validated computational predictions.
Table 8.
Number of exon pairs (introns) tested per program, and the number of positive verifications
| Number of tested exon pairs | Number of positive RT-PCR exon pairs and % over tested | Number of tested exon pairs suported by TARs/transfrags and % over tested pairs | Number of positive RT-PCR pairs supported by TARs/transfrags and % over supported pairs | % of positive RT-PCR pairs supported by TARs/transfrags over positive | |
| Category 1 | |||||
| AUGUSTUS-any | 31 | 2 (6.5%) | 3 (9.7%) | 1 (33.3%) | 50.0% |
| FGENESH++ | 119 | 3 (2.5%) | 3 (2.5%) | 1 (33.3%) | 33.3% |
| JIGSAW | 19 | 2 (10.5%) | 3 (15.8%) | 1 (33.3%) | 50.0% |
| PAIRAGON-any | 1 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0.0% |
| Category 2 | |||||
| AUGUSTUS-abinit | 29 | 2 (6.9%) | 2 (6.9%) | 1 (50.0%) | 50.0% |
| GENEMARK.hmm | 99 | 1 (1.0%) | 3 (3.0%) | 1 (33.3%) | 100.0% |
| GENEZILLA | 34 | 2 (5.9%) | 1 (2.9%) | 0 (0.0%) | 0.0% |
| Category 3 | |||||
| ACEVIEW | 13 | 4 (30.8%) | 2 (15.4%) | 2 (100.0%) | 50.0% |
| AUGUSTUS-EST | 31 | 2 (6.5%) | 3 (9.7%) | 1 (33.3%) | 50.0% |
| ENSEMBL | 10 | 1 (10.0%) | 2 (20.0%) | 1 (50.0%) | 100.0% |
| EXOGEAN | 18 | 1 (5.6%) | 3 (16.7%) | 1 (33.3%) | 100.0% |
| EXONHUNTER | 23 | 1 (4.3%) | 2 (8.7%) | 0 (0.0%) | 0.0% |
| PAIRAGON+NSCAN_EST | 1 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0.0% |
| Category 4 | |||||
| AUGUSTUS-dual | 26 | 1 (3.8%) | 3 (11.5%) | 1 (33.3%) | 100.0% |
| DOGFISH | 11 | 2 (18.2%) | 1 (9.1%) | 1 (100.0%) | 50.0% |
| MARS | 47 | 7 (14.9%) | 5 (10.6%) | 3 (60.0%) | 42.9% |
| NSCAN | 26 | 0 (0.0%) | 2 (7.7%) | 0 (0.0%) | 0.0% |
| SAGA | 9 | 0 (0.0%) | 1 (11.1%) | 0 (0.0%) | 0.0% |
| All unique exon pairs | 238 | 7 (2.9%) | 7 (2.9%) | 3 (42.9%) | 42.9% |
The percentage of success has been computed in the table on the 238 selected exon pairs. For technical reasons, only 221 of them could be tested by RT-PCR. In the text the percentages are given with respect to this number.
Since TAR/transfrag support has not been used to prioritize predicted exons for experimental verification, it is possible to investigate whether the results of the RT-PCR experiments are consistent with the TAR/transfrag data, and whether these data can be used to prioritize verification experiments. For example, one would expect the likelihood of RT-PCR success to be higher when the two predicted exons to be tested are both supported by TARs/transfrags from the same cell line and condition, suggesting that the two exons are connected into the same RNA sequence. In only seven of the 221 exon pairs tested were the two exons supported by TARs/transfrags from the same cell line. Interestingly, three of these cases were positive by RT-PCR. This is a success rate of 43%, compared with 4 successful RT-PCRs out of 214 exons not having consistent transfrag support (less than 2% success rate). While the numbers are too small for significant conclusions, the trend is quite striking: consistent transfrag support of computational predictions is strongly indicative of RT-PCR success. Conversely, the reasons why exon pairs fail RT-PCR verification when supported by consistent transcription evidence from the same cell line and condition are multiple. Depending on the primers chosen, for instance, wrong prediction of the exon boundaries, even by a small offset, may lead to failed RT-PCR amplification. Moreover, TAR/transfrag maps have been obtained from cell lines different from the tissues used for RT-PCR. Given the extremely restricted expression pattern that these novel transcripts appear to show, transcripts expressed in one given cell line may not be expressed in any of the 24 tissues analyzed. In this regard, it is interesting to note that the four negative RT-PCR exon pairs cluster into a single locus, and even share some sequence (see the Supplementary material web page), and, therefore, may represent the same transcript, while the three other transfrag-supported positive RT-PCR exon pairs correspond to three distinct loci mapped to three different ENCODE regions.
Discussion
The unfolding of the instructions encoded in the DNA sequence is initiated by the transcription of DNA to RNA, and the subsequent processing of the primary transcript to functional RNA sequences. According to the central dogma, most of these processed RNAs correspond to mRNAs that are eventually translated to proteins. Despite the fact that the identification of the protein-coding mRNAs (or genes) is essential for our understanding of how the genome sequence translates into biological phenomena, uncertainty still remains with respect to the set of human genes. The lack of an accurate and complete gene catalogue undermines the impact of the genome sequence on human biology and biomedical research. Experimental determination of expressed mRNA sequences and computational mapping of this sequence onto the sequence of the genome constitutes the most reliable approach to identify the exonic structure, and chromosomal location, of protein-coding genes. However, this approach has limitations. First, it is unclear what fraction of low and specifically expressed transcripts can be effectively sequenced, and high throughput mRNA sequencing often leads to only partial sequences. Second, computational mapping of mRNA to genomic sequences is not trivial, and it is complicated by fragmentary mRNA sequences, sequencing errors, sequence polymorphism, and the highly repetitive nature of the human genome. Moreover, the high pseudogene content of the human genome, and the presence of small exons, leads to uncertain or incorrect mapping of exon boundaries. Therefore, substantial manual intervention is required to delineate an accurate protein coding gene map from the available mRNA sequence data.
We organized EGASP as a community experiment with the goal of assessing the ability of computational methods to automatically reproduce the accurate protein-coding gene map produced by a team of expert human curators. Such a map [33], subsequently verified experimentally, has been obtained for only 1% of the human genome selected by the ENCODE project [30]. Scaling the map to the entire human genome will require substantial additional resources, and it will enormously benefit from improved computational strategies for gene finding. With its focus on this 1% of the human genome, EGASP has indeed demonstrated progress in the performance of newly developed computational gene finding pipelines, with accuracies of about 80% at the coding exon level for both sensitivity and specificity, and of nearly 90% at the coding nucleotide level (Table 4). However, the success of these metrics is significantly tempered by the relatively low numbers of coding transcripts that are predicted correctly. Programs relying on mRNA and protein sequences were the most accurate in reproducing the manually curated annotation. This is not unexpected, and, to some extent, circular, since the manually curated annotation relies on mRNA and protein sequences as well. Notably, however, programs based on sequence comparisons across two or more genomes - which do not use information from known mRNA or protein sequences - also exhibited impressive accuracy at the nucleotide and exon levels (Table 6). Dual genome prediction programs, however, were significantly less accurate at finding complete genes than the expressed sequence based methods. Finally, with few exceptions, all of the methods struggled to predict correctly the non-coding exons of transcripts. Indeed, UTRs are often predicted as mere extensions of first and terminal exons, if predicted at all. Thus, while the computational methods are quite reliable in predicting the protein coding components of transcripts, they have difficulties in linking them into transcript structures. Indeed, the most accurate programs were only able to correctly predict about 40% of the annotated transcripts, meaning the correct prediction of all of the exons constituting a transcript (Table 5). The results of coding gene predictions were more encouraging. For up to 80% of human genes the exact structure of the coding part, including all the splice junctions and start/stop codons, could be predicted correctly in at least one transcript.
Contributing to the difficulty is the unexpected complexity of the protein coding loci in higher eukaryotic genomes. Indeed, as revealed in the GENCODE annotation, most protein coding loci appear to encode a mixture of coding and non-coding transcripts, sharing part of their sequence. Additional transcriptional activity, including chimeric, overlapping and antisense transcripts, transcripts within introns, and other transcriptional phenomena, appear to be less exceptional than had been previously suspected. Thus, the model of a eukaryotic gene currently implicit in most computational methods is too simple to capture this complexity, leading to relatively poor prediction performance.
The second goal of EGASP was to assess the completeness of the manual/computational/experimental GENCODE annotation. This annotation is based on available evidence, and thus may miss some protein coding genes and exons. Indeed, in EGASP, computational methods predict many exons and transcripts that are not included in the GENCODE annotation (Table 7), a trend accentuated in ab initio and comparative gene finders, which do not rely on available evidence from transcript sequences. While we were not able to confirm experimentally the bulk of these predictions and they are likely to be false positives, some might be real.
To assess what fraction of the predicted exons unannotated in GENCODE could correspond to novel genes, we prioritized - based on the reliability of the programs predicting them - a subset of intergenic predicted exon pairs, and attempted to experimentally verify them by RT-PCR in 24 human tissues. Only 3.2% of these pairs tested positive, a result consistent with most of the computational predictions outside of GENCODE being false positives. All verified cases tested positive in only one tissue among the 24 tested, emphasizing the extremely restricted expression patterns of these novel, unannotated exons. Since many more tissues and cell lines exist, it cannot be ruled out that some other predictions could also be positive in other tissues. Support for a larger fraction of predictions corresponding to real exons comes from the observation that 13% of these predictions overlap sites of transcription (or TARs/transfrags) as detected by genome tiling experiments. Interestingly, the success rate of RT-PCR was much higher (at least 40%) for those few tested exon pairs that both overlapped TARs/transfrags and were detected in the same cell line and condition. Thus, consistent TAR/transfrag support is strongly indicative of an underlying transcript, including exons predicted to be connected. In total, about 100 unannotated predicted exons in EGASP are consistently supported by TARs/transfrags, and are, therefore, likely to belong to transcribed RNAs. In summary, a non-negligible fraction of unannotated exons predicted in EGASP have some evidence of transcription (not necessarily associated with protein coding), but only a small fraction of the predicted structures connecting exons could be verified experimentally here.
In this regard, the EGASP experiment seems to indicate that the GENCODE annotation of protein coding genes is quite complete, although it is still unclear what fraction of all the alternative transcript diversity of gene loci is captured by GENCODE. EGASP was also useful in helping to identify the software tools that can contribute to reduce the amount of human intervention required to delineate the GENCODE annotation. Programs accelerating and improving the mapping of cDNA sequences (partial or complete) into the genome sequence could be particularly useful towards that end.
Overall, we believe that the EGASP project has given a fair assessment of the state-of-the-art of gene prediction in human DNA. This will allow biologists to interpret better the annotations presented to them in public genome databases such as GenBank, the UCSC browser, ENSEMBL and others. It has also clearly shown that we are still far from being able to computationally predict human gene structures with total accuracy from the DNA sequence alone. Furthermore, while we believe the experiment has shown that only very few protein-coding human genes seem to missing from the annotations, the exact protein sequences are annotated for roughly over 50% of the sequences. Getting a complete protein sequence correct is also made difficult by the existence of many splice forms, mis-assembled cDNAs and additional contamination in cDNA/EST sequences in the public databases. Each can lead to various spurious protein sequence annotations. Unfortunately, there are very few processes in place to remove erroneous sequences and annotations from the public databases, so it will still take some time to get a better picture of exact gene structures. It has to be noted that the human genome and its annotation for protein coding genes are still works in progress.
Another class of genes, non-protein coding transcripts, which were not generally considered by EGASP, are thought to be especially difficult to predict. These genes, such as those that encode miRNAs and snoRNAs, were not addressed in this experiment; nevertheless, they seem to play a very important role in physiological processes such as development and disease.
One of the most difficult problems in gene prediction accuracy assessment is the definition of a reference set against which to evaluate. Ultimately, this reference set should be 'unknown' to the prediction teams. In EGASP, the delayed publication of the GENCODE annotations partially achieved this goal, although a significant amount of the annotation information was known from previously submitted cDNA and EST sequences to public databases such as ENSEMBL or Genbank. This is slightly different to GASP1 [27], where novel cDNA sequences had been withheld before the experiment. Additionally, it may be optimal if each group used the same auxiliary data for their predictions. One suggestion would be to 'freeze' databases of auxiliary data and allow only the inclusion in the predictions of these frozen databases, so that progress in these assessment experiments can be measured independently of growing experimental data.
Furthermore, while our assessments have started to evaluate gene annotations on the transcript level, better and additional evaluation methods for evaluating UTRs are needed. One suggestion would be to evaluate the transcript performance at the intron level (similar to the exon evaluation above). This measure would exclude the beginning and end of a gene, two coordinates that are considered the most difficult to obtain experimentally, but would include non-coding introns that are determined by their splice sites.
One of the major benefits of this kind of experiment is that it allows prediction teams to measure their programs and methods against each other, to learn from their failures, and, as a community, to identify the open and difficult questions in this area of research.
Materials and methods
Submitted predictions
Files submitted to the EGASP server were validated to conform to the GTF specifications [33] and the use of standard annotation features such as 'exon', 'CDS', 'stop codon' and 'start codon'. Submissions not conforming to this format were rejected, although the participants were allowed to fix prediction files accordingly and to resubmit to the server (Figure 1) [47]. Submissions were clipped to the ENCODE region sequence boundaries. The clipping criteria were the following: (feature_start < 1 and feature_end >= 1 then feature_start == 1) and (feature_end > sequence_end and feature_start <= sequence_end, then feature_end == sequence_end); while those records where (feature_end < 1 or feature_start > sequence_end) were removed from the GENCODE annotation and the submitted predictions before performing the evaluations.
Evaluations
We used different programs to obtain accuracy values at nucleotide, exon, gene/transcript and clustered transcripts. These programs included software developed by the authors at IMIM and at the EBI. We also used the Eval package [75]. We confirmed the results obtained with the evaluation programs by comparison. The programs can be downloaded, along with a small description on how to use them, from the Supplementary material web page [51].
When comparing annotations against the predictions for each individual sequence in the test sets for the boxplots, sequences that contained no feature annotations either in the annotations or in the predictions were excluded from the analysis for the boxplots. This did not happen when the numbers were computed globally for the sequences. We considered the following sequence sets build up by concatenating (without overlap) the coordinates of different sequence annotation and prediction sets: EN_TRN13, all training set sequences (13 sequences); EN_PRD31, all evaluation set sequences (31 sequences); EN_MNLp12, evaluation set sequences, manual picks (12 sequences); EN_RNDp19, evaluation set sequences, random picks (19 sequences); EN_PGH12/EN_PGM11/EN_PGL8, all the sequences of the test set were collected into three sequence sets based on their gene density into three sequence sets, for high, medium and low densities (12, 11 and 8 sequences, respectively); EN_PMH7/EN_PMM5/ENPML7, in this case, the random sequences from the evaluation set were considered, depending on their sequence conservation with mouse, into three sequences, for high, medium and low conservation (7, 5 and 7 sequences, respectively). See the Supplementary materials web page for the complete set of results on all sequences and sequence sets [51].
The box-and-whisker plots [76] (simply 'boxplots') describe graphically how the data being analyzed are distributed. The horizontal line within the box shows the median value of the data set, while the top and the bottom of the box correspond to the third and first quartiles, respectively; therefore, the box represents the interquartile range (IQR). The whiskers represent the range of the data and show a maximum and a minimum, which are based on 1.5 times the length of the IQR. The notches centered on the median correspond to the 5% interval of confidence for this median (median ± 1.57·IQR/√n, as defined in R [77]).
RT-PCR
Primers mapping in the two predicted exons spanning the exon junction to be tested were designed using Primer3 [78] with the following parameters: 18 ≤ primer size ≤ 27, optimal size = 20, 57°C ≤ primer Tm ≤ 63°C, optimal Tm = 60°C, 20% ≤ primer GC percentage ≤ 80%. Similar amounts of 24 human cDNAs (brain, heart, kidney, spleen, liver, colon, small intestine, muscle, lung, stomach, testis, placenta, skin, peripheral blood lymphocytes, bone marrow, fetal brain, fetal liver, fetal kidney, fetal heart, fetal lung, thymus, pancreas, mammary glands, prostate, final dilution 1,000·) were mixed with JumpStart REDTaq ReadyMix (Sigma-Aldrich, St. Louis, MO, USA) and 4 ng/μl primers (Sigma-Genosys, Cambridge, U.K.) with a BioMek 2000 robot (Beckman, Fullerton, CA, USA) as described and modified [14,79,80]. The 10 first cycles of PCR amplification were performed with a touchdown annealing temperature decreasing from 60°C to 50°C; the annealing temperature of the next 30 cycles was 50°C. Amplimers were separated on 'Ready to Run' precast gels (Amersham Pharmacia, Sunnyvale, CA, USA) and sequenced. We tested 221 exon pairs out of the 238 exon pairs with an exon ranked in the top 200. The remaining 17 exon pairs were not experimentally evaluated because either the targeted amplimer was too small (8 cases) or one of the exons was too short to allow us to design a primer (9 cases).
Acknowledgments
Acknowledgements
We would like to thank Peter Good from the National Human Genome Research Institute (NHGRI) for motivating EGASP and, together with Elise Feingold and Mark Guyer, also from NHGRI, for their continuous support. We thank all participants for their enthusiasm, commitment and efforts to meet all the EGASP deadlines. We also acknowledge Evan Keibler for providing support for the Eval software package. We are particularly thankful to the National Human Genome Research Institute for inspiring and providing funding for EGASP. We also thank the Wellcome Trust Conference staff for their assistance during the workshop. RG is supported by grants from the NHGRI ENCODE Project, the European Biosapiens Project, and from the Spanish Ministry of Education and Science. PF is supported by EMBL. AR acknowledges the Swiss National Science Foundation for financial support. MR is partially supported by the NHGRI.
This article has been published as part of Genome Biology Volume 7, Supplement 1, 2006: EGASP '05. The full contents of the supplement are available online at http://genomebiology.com/supplements/7/S1.
Contributor Information
Roderic Guigó, Email: rguigo@imim.es.
Martin G Reese, Email: mreese@omicia.com.
References
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;(33 Database):D501–504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard DS, Wagner L, Feingold EA, Shenmen CM, Grouse LH, Schuler G, Klein SL, Old S, Rasooly R, Good P, et al. The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC). Genome Res. 2004;14:2121–2127. doi: 10.1101/gr.2596504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard T, Andrews D, Caccamo M, Cameron G, Chen Y, Clamp M, Clarke L, Coates G, Cox T, Cunningham F, et al. Ensembl 2005. Nucleic Acids Res. 2005;(33 Database):D447–453. doi: 10.1093/nar/gki138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2005;(33 Database):D54–58. doi: 10.1093/nar/gki031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Agarwal P, Abril JF, Wiehe T, Fickett JW, Guigo R. Comparative gene prediction in human and mouse. Genome Res. 2003;13:108–117. doi: 10.1101/gr.871403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawley S, Pachter L, Alexandersson M. SLAM web server for comparative gene finding and alignment. Nucleic Acids Res. 2003;31:3507–3509. doi: 10.1093/nar/gkg583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandersson M, Cawley S, Pachter L. SLAM: cross-species gene finding and alignment with a generalized pair hidden Markov model. Genome Res. 2003;13:496–502. doi: 10.1101/gr.424203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I, Flicek P, Duan D, Brent MR. Integrating genomic homology into gene structure prediction. Bioinformatics. 2001;17(Suppl 1):S140–148. doi: 10.1093/bioinformatics/17.suppl_1.s140. [DOI] [PubMed] [Google Scholar]
- Flicek P, Keibler E, Hu P, Korf I, Brent MR. Leveraging the mouse genome for gene prediction in human: from whole-genome shotgun reads to a global synteny map. Genome Res. 2003;13:46–54. doi: 10.1101/gr.830003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigo R, Dermitzakis ET, Agarwal P, Ponting CP, Parra G, Reymond A, Abril JF, Keibler E, Lyle R, Ucla C, et al. Comparison of mouse and human genomes followed by experimental verification yields an estimated 1,019 additional genes. Proc Natl Acad Sci USA. 2003;100:1140–1145. doi: 10.1073/pnas.0337561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Shteynberg D, Arumugam M, Gibbs RA, Brent MR. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 2004;14:665–671. doi: 10.1101/gr.1959604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyras E, Reymond A, Castelo R, Bye JM, Camara F, Flicek P, Huckle EJ, Parra G, Shteynberg DD, Wyss C, et al. Gene finding in the chicken genome. BMC Bioinformatics. 2005;6:131. doi: 10.1186/1471-2105-6-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenney AE, Brown RH, Vaske C, Lodge JK, Doering TL, Brent MR. Gene prediction and verification in a compact genome with numerous small introns. Genome Res. 2004;14:2330–2335. doi: 10.1101/gr.2816704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Parra G, Blanco E, Guigo R. GeneID in Drosophila. Genome Res. 2000;10:511–515. doi: 10.1101/gr.10.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004;431:946–957. doi: 10.1038/nature03025. [DOI] [PubMed] [Google Scholar]
- Brent MR, Guigo R. Recent advances in gene structure prediction. Curr Opin Struct Biol. 2004;14:264–272. doi: 10.1016/j.sbi.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Burset M, Guigo R. Evaluation of gene structure prediction programs. Genomics. 1996;34:353–367. doi: 10.1006/geno.1996.0298. [DOI] [PubMed] [Google Scholar]
- Bajic VB. Comparing the success of different prediction software in sequence analysis: a review. Brief Bioinform. 2000;1:214–228. doi: 10.1093/bib/1.3.214. [DOI] [PubMed] [Google Scholar]
- Baldi P, Brunak S, Chauvin Y, Andersen CA, Nielsen H. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000;16:412–424. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
- Guigo R, Agarwal P, Abril JF, Burset M, Fickett JW. An assessment of gene prediction accuracy in large DNA sequences. Genome Res. 2000;10:1631–1642. doi: 10.1101/gr.122800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogic S, Mackworth AK, Ouellette FB. Evaluation of gene-finding programs on mammalian sequences. Genome Res. 2001;11:817–832. doi: 10.1101/gr.147901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SE. Genome annotation assessment in Drosophila melano-gaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack RL, Jr, Gerloff DL, Bower M, Chen X, Lichtarge O, Cohen FE. Meeting review: the Second meeting on the Critical Assessment of Techniques for Protein Structure Prediction (CASP2), Asilomar, California, December 13-16, 1996. Fold Des. 1997;2:R27–42. doi: 10.1016/S1359-0278(97)00011-4. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Misra S, Roote J, Lewis SE, Blazej R, Davis T, Doyle C, Galle R, George R, Harris N, et al. An exploration of the sequence of a 2.9-Mb region of the genome of Drosophila melanogaster: the Adh region. Genetics. 1999;153:179–219. doi: 10.1093/genetics/153.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- The GENCODE Project http://genome.imim.es/gencode/
- The HAVANA Team http://www.sanger.ac.uk/HGP/havana/
- Harrow J, Denoeud F, Frankish A, Reymond A, Chen C-K, Chrast J, Lagarde J, Gilbert JGR, Storey R, Swarbreck D, et al. GENCODE: Producing a reference annotation for ENCODE. Genome Biology. 2006;7(Suppl 1):S4. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Target Selection Process and Target Regions http://genome.gov/10506161
- ENCODE Project at UCSC Genome Browser http://genome.cse.ucsc.edu/ENCODE/
- Mungall AJ, Palmer SA, Sims SK, Edwards CA, Ashurst JL, Wilming L, Jones MC, Horton R, Hunt SE, Scott CE, et al. The DNA sequence and analysis of human chromosome 6. Nature. 2003;425:805–811. doi: 10.1038/nature02055. [DOI] [PubMed] [Google Scholar]
- Humphray SJ, Oliver K, Hunt AR, Plumb RW, Loveland JE, Howe KL, Andrews TD, Searle S, Hunt SE, Scott CE, et al. DNA sequence and analysis of human chromosome 9. Nature. 2004;429:369–374. doi: 10.1038/nature02465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deloukas P, Earthrowl ME, Grafham DV, Rubenfield M, French L, Steward CA, Sims SK, Jones MC, Searle S, Scott C, et al. The DNA sequence and comparative analysis of human chromosome 10. Nature. 2004;429:375–381. doi: 10.1038/nature02462. [DOI] [PubMed] [Google Scholar]
- Dunham A, Matthews LH, Burton J, Ashurst JL, Howe KL, Ashcroft KJ, Beare DM, Burford DC, Hunt SE, Griffiths-Jones S, et al. The DNA sequence and analysis of human chromosome 13. Nature. 2004;428:522–528. doi: 10.1038/nature02379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deloukas P, Matthews LH, Ashurst J, Burton J, Gilbert JG, Jones M, Stavrides G, Almeida JP, Babbage AK, Bagguley CL, et al. The DNA sequence and comparative analysis of human chromosome 20. Nature. 2001;414:865–871. doi: 10.1038/414865a. [DOI] [PubMed] [Google Scholar]
- Collins JE, Goward ME, Cole CG, Smink LJ, Huckle EJ, Knowles S, Bye JM, Beare DM, Dunham I. Reevaluating human gene annotation: a second-generation analysis of chromosome 22. Genome Res. 2003;13:27–36. doi: 10.1101/gr.695703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M, Howell GR, Burrows C, Bird CP, et al. The DNA sequence of the human X chromosome. Nature. 2005;434:325–337. doi: 10.1038/nature03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Rouchka EC, Gish WR, States DJ. Gene structure prediction and alternative splicing analysis using genomically aligned ESTs. Genome Res. 2001;11:889–900. doi: 10.1101/gr.155001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth RE, Jamison DC, Touchman JW, Chissoe SL, Braden Maduro VV, Bouffard GG, Dietrich NL, Beckstrom-Sternberg SM, Iyer LM, Weintraub LA, et al. Comparative genomic sequence analysis of the human and mouse cystic fibrosis transmem-brane conductance regulator genes. Proc Natl Acad Sci USA. 2000;97:1172–1177. doi: 10.1073/pnas.97.3.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic H, Brent MR, Brown RH, Frankish A, Harrow J, Ohler U, Solovyev VV, Tan SL. Performance assessment of promoter predictions on ENCODE regions in the EGASP experiment. Genome Biology. 2006;7(Suppl 1):S3. doi: 10.1186/gb-2006-7-s1-s3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Gerstein M. A computational approach for identifying pseudogenes in the ENCODE regions. Genome Biol. 2006;7(Suppl 1):S13. doi: 10.1186/gb-2006-7-s1-s13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The EGASP Submission Server for Predictions http://genome.imim.es/cgi-bin/EGASP2005/submission
- Gene Transfer Format Specifications http://genes.cs.wustl.edu/GTF2.html
- GencodeDB Genome Browser http://genome.imim.es/cgi-bin/gbrowse/encode
- EGASP ftp Server ftp://genome.imim.es/pub/projects/gencode/data/egasp05/submitted_predictions/
- Supplementary Material http://genome.imim.es/datasets/egasp2005/
- AceView http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/
- Kim N, Shin S, Lee S. ECgene: genome-based EST clustering and gene modeling for alternative splicing. Genome Res. 2005;15:566–576. doi: 10.1101/gr.3030405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UCSC Known Genes Track http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=63708116&c=chr7&g=knownGene [DOI] [PubMed]
- The Consensus CDS (CCDS) Project http://www.ncbi.nlm.nih.gov/CCDS/
- Solovyev V, Kosarev P, Seledsov I, Vorobyev D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biology. 2006;7(Suppl 1):S10. doi: 10.1186/gb-2006-7-s1-s10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam M, Wei C, Brown RH, Brent MR. Pairagon+N-SCAN_EST: a model-based gene annotation pipeline. Genome Biology. 2006;7(Suppl 1):S5. doi: 10.1186/gb-2006-7-s1-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Tzvetkova A, Morgenstern B. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biology. 2006;7(Suppl 1):S11. doi: 10.1186/gb-2006-7-s1-s11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JE, Majoros WH, Pertea M, Salzberg SL. JIGSAW, GeneZilla and GlimmerHMM: puzzling out the features of human genes in the ENCODE regions. Genome Biology. 2006;7(Suppl 1):S9. doi: 10.1186/gb-2006-7-s1-s9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005;(33 Web Server):W451–454. doi: 10.1093/nar/gki487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell JA, Korf I, Gish W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics. 2000;16:1040–1041. doi: 10.1093/bioinformatics/16.11.1040. [DOI] [PubMed] [Google Scholar]
- Djebali S, Delaplace F, Roest Crollius H. Exogean: a framework for annotating protein-coding genes in eukaryotic genomic DNA. Genome Biology. 2006;7(Suppl 1):S7. doi: 10.1186/gb-2006-7-s1-s7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejova B, Brown DG, Li M, Vinar T. ExonHunter: a comprehensive approach to gene finding. Bioinformatics. 2005;21(Suppl 1):i57–i65. doi: 10.1093/bioinformatics/bti1040. [DOI] [PubMed] [Google Scholar]
- Curwen V, Eyras E, Andrews TD, Clarke L, Mongin E, Searle SM, Clamp M. The Ensembl automatic gene annotation system. Genome Res. 2004;14:942–950. doi: 10.1101/gr.1858004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Brent MR. Using several pair-wise informant sequences for de novo prediction of alternatively spliced transcripts. Genome Biology. 2006;7(Suppl 1):S8. doi: 10.1186/gb-2006-7-s1-s8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterji S, Pachter L. Large multiple organism gene finding by collapsed Gibbs sampling. J Comput Biol. 2005;12(6):599–608. doi: 10.1089/cmb.2005.12.599. [DOI] [PubMed] [Google Scholar]
- Carter D, Durbin R. Vertebrate gene finding from multiple-species alignments using a two-level strategy. Genome Biology. 2006;7(Suppl 1):S6. doi: 10.1186/gb-2006-7-s1-s6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AA, Steitz JA. Splicing double: insights from the second spliceosome. Nat Rev Mol Cell Biol. 2003;4:960–970. doi: 10.1038/nrm1259. [DOI] [PubMed] [Google Scholar]
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–919. doi: 10.1126/science.1068597. [DOI] [PubMed] [Google Scholar]
- Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, Drenkow J, Piccolboni A, Bekiranov S, Helt G, et al. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004;14:331–342. doi: 10.1101/gr.2094104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo R, Reymond A, Wyss C, Camara F, Parra G, Antonarakis SE, Guigo R, Eyras E. Comparative gene finding in chicken indicates that we are closing in on the set of multi-exonic widely expressed human genes. Nucleic Acids Res. 2005;33:1935–1939. doi: 10.1093/nar/gki328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Reymond A, Dabbouseh N, Dermitzakis ET, Castelo R, Thomson TM, Antonarakis SE, Guigo R. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res. 2006;16:37–44. doi: 10.1101/gr.4145906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond A, Marigo V, Yaylaoglu MB, Leoni A, Ucla C, Scamuffa N, Caccioppoli C, Dermitzakis ET, Lyle R, Banfi S, et al. Human chromosome 21 gene expression atlas in the mouse. Nature. 2002;420:582–586. doi: 10.1038/nature01178. [DOI] [PubMed] [Google Scholar]
- Keibler E, Brent MR. Eval: A software package for analysis of genome annotations. BMC Bioinformatics. 2003;4:50. doi: 10.1186/1471-2105-4-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukey JW. Exploratory Data Analysis. Reading, MA: Addison-Wesley; 1977. [Google Scholar]
- R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2005. [Google Scholar]
- Primer3 http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi
- Reymond A, Friedli M, Henrichsen CN, Chapot F, Deutsch S, Ucla C, Rossier C, Lyle R, Guipponi M, Antonarakis SE. From PREDs and open reading frames to cDNA isolation: revisiting the human chromosome 21 transcription map. Genomics. 2001;78:46–54. doi: 10.1006/geno.2001.6640. [DOI] [PubMed] [Google Scholar]
- Reymond A, Camargo AA, Deutsch S, Stevenson BJ, Parmigiani RB, Ucla C, Bettoni F, Rossier C, Lyle R, Guipponi M, et al. Nineteen additional unpredicted transcripts from human chromosome 21. Genomics. 2002;79:824–832. doi: 10.1006/geno.2002.6781. [DOI] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Salzberg SL. TigrScan and Glim-merHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- Yeo GW, Van Nostrand E, Holste D, Poggio T, Burge CB. Identification and analysis of alternative splicing events conserved in human and mouse. Proc Natl Acad Sci USA. 2005;102:2850–2855. doi: 10.1073/pnas.0409742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzoni P, Rizzi R, Pesole G. ASPIC: a novel method to predict the exon-intron structure of a gene that is optimally compatible to a set of transcript sequences. BMC Bioinformatics. 2005;6:244. doi: 10.1186/1471-2105-6-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrignano T, Canali A, Grillo G, Liuni S, Mignone F, Pesole G. CSTminer: a web tool for the identification of coding and noncoding conserved sequence tags through cross-species genome comparison. Nucleic Acids Res. 2004;(32 Web Server):W624–627. doi: 10.1093/nar/gkh486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe D. SPIDA: Substitution Periodicity Index and Domain Analysis. ftp://ftp.ebi.ac.uk/pub/databases/ensembl/encode/egasp05
- Ohler U, Shomron N, Burge CB. Recognition of unknown conserved alternatively spliced exons. PLoS Comput Biol. 2005;1:113–122. doi: 10.1371/journal.pcbi.0010015. [DOI] [PMC free article] [PubMed] [Google Scholar]