

Abstract
The yeast prion protein Sup35 is a translation termination factor, whose activity is modulated by sequestration into a self-perpetuating amyloid. The prion-determining domain, NM, consists of two distinct regions: an amyloidogenic N terminus domain (N) and a charged solubilizing middle region (M). To gain insight into prion conversion, we used single-molecule fluorescence resonance energy transfer (SM-FRET) and fluorescence correlation spectroscopy to investigate the structure and dynamics of monomeric NM. Low protein concentrations in these experiments prevented the formation of obligate on-pathway oligomers, allowing us to study early folding intermediates in isolation from higher-order species. SM-FRET experiments on a dual-labeled amyloid core variant (N21C/S121C, retaining wild-type prion behavior) indicated that the N region of NM adopts a collapsed form similar to “burst-phase” intermediates formed during the folding of many globular proteins, even though it lacks a typical hydrophobic core. The mean distance between residues 21 and 121 was ≈43 Å. This increased with denaturant in a noncooperative fashion to ≈63 Å, suggesting a multitude of interconverting species rather than a small number of discrete monomeric conformers. Fluorescence correlation spectroscopy analysis of singly labeled NM revealed fast conformational fluctuations on the 20- to 300-ns time scale. Quenching from proximal and distal tyrosines resulted in distinct fast and slower fluctuations. Our results indicate that native monomeric NM is composed of an ensemble of structures, having a collapsed and rapidly fluctuating N region juxtaposed with a more extended M region. The stability of such ensembles is likely to play a key role in prion conversion.
Keywords: amyloid, conformational fluctuation, single-molecule fluorescence, yeast prion
The prion hypothesis (1), involving self-replicating or infectious protein conformations, has attracted broad interest in recent times due to its role in the biology of debilitating neurodegenerative diseases (1, 2), protein-based inheritance of novel phenotypes in yeast (3–5), and (potentially) long-term memory (6, 7). The Saccharomyces cerevisiae translational termination factor, Sup35, is one such protein capable of switching to a self-perpetuating state. In the prion state [PSI+], the glutamine/asparagine (Q/N)-rich prion domain of Sup35 is sequestered into an amyloid conformer, reducing the efficiencies of translation termination (8). This switch causes ribosomes to read through stop codons at biologically significant rates, changing a multitude of phenotypes (9).
The NM segment (253 residues) of Sup35 determines the prion state and comprises two distinct regions. The N-terminal region (residues 1–123) is abundant in uncharged polar amino acids (glutamines, asparagines, and tyrosines), and forms the major part of the amyloid core that directs the protein into the [PSI+] prion state. The highly charged middle region M (residues 124–250) confers solubility in vitro and in vivo, allowing the protein to exist in the non-prion [psi−] state. In the prion state, the N region adopts a β-sheet-rich conformation, whereas the M region remains relatively unstructured (10).
Structural studies on NM amyloids have provided several insights into the molecular basis of prion nucleation (11–14). An early step is the establishment of an equilibrium between predominantly unstructured NM polypeptide monomers and molten oligomeric intermediates that are obligate on-pathway species (14–16). The structure and dynamics of early monomeric intermediates are of considerable interest in deciphering the molecular mechanism of amyloid formation (17, 18). However, the heterogeneity and transient nature of these partially folded intermediates impede structural characterization by steady-state bulk measurements, which provide ensemble-averaged information for monomers and oligomers.
In recent years, single-molecule measurements have proven very useful in studying such complex systems, which are expected to demonstrate static and/or dynamic conformational heterogeneity (19–25). These experiments permit direct observations and quantification of intermediates, their structural distributions and dynamics during assembly. Furthermore, these studies can be carried out at subnanomolar protein concentrations, minimizing oligomerization or aggregation.
Single-molecule FRET (SM-FRET) can be used to estimate intramolecular distances (r) of individual molecules, fluorescently labeled with donor and acceptor dyes, from the FRET-efficiency (E), by using Förster's equation
where R0 is the Förster's distance (at which E = 0.5) for a given dye pair (20, 21).
Although very powerful as commonly practiced, SM-FRET does not probe conformational dynamics occurring faster than ≈50 μs. Fluorescence correlation spectroscopy (FCS) is a closely related single-molecule or small-ensemble method that can be used to measure such faster fluctuations via an autocorrelation analysis of conformationally coupled fluorescence intensity fluctuations (26–28). In this work, we describe single-molecule studies on a monomeric amyloidogenic protein, employing both SM-FRET and FCS to provide insights into the structure and dynamics of monomeric NM.
Results
Ensemble FRET Measurements.
Our previous work indicates that the region from amino acids ≈20 to 120 encompasses the amyloid core in the prion state of NM fibers assembled at room temperature (13). Therefore, to probe the structure and dynamics of this region in the monomeric non-prion form, a double cysteine variant (N21C, S121C) was generated by site-directed mutagenesis to provide sites for attachment of fluorescent labels (Fig. 1a). This mutant was used to replace the NM portion of the wild-type SUP35 gene in vivo, resulting in a single, full length functional SUP35 gene. This variant retained its capacity to support both the prion [PSI+] and the non-prion [psi−] states. Next, the dual cysteine variant was expressed in and purified from Escherichia coli. This variant spontaneously assembled into amyloid at the same rate as wild-type NM under reducing conditions, and labeling with a variety of fluorescent probes had no effect on its assembly kinetics (data not shown). Having established that the cysteine variant behaves like the wild-type protein both in vivo and in vitro, we labeled it with Alexa Fluor 488 (donor) and Alexa Fluor 594 (acceptor) for FRET experiments (Fig. 1a). As a prelude to SM-FRET experiments, we performed ensemble FRET measurements on the protein. Steady-state fluorescence spectra were recorded using donor excitation at 488 nm under denaturing and native conditions during the lag phase, before amyloid assembly. Under denaturing conditions (6 M GdmCl), the acceptor emission was very weak (Fig. 1b), consistent with a low FRET efficiency expected for a dye-pair with a Förster distance of ≈54 Å, separated by 100 amino acid residues in a polypeptide chain. Immediately upon dilution into buffer, emission from the acceptor increased significantly with a concomitant decrease in donor signal, indicative of a significant increase in the FRET-efficiency and a decrease in the inter-dye distance in the native protein.
Fig. 1.

Amino acid sequence, ensemble, and single-molecule data for NM. (a) Sequence of the prion domain (NM) of Sup35 showing residues mutated to cysteine in green for single fluorescence labeling (Alexa Fluor 488). Underscores (21 and 121) indicate the dual cysteine mutation for donor (Alexa Fluor 488) and acceptor (Alexa Fluor 594) labeling for FRET studies. Tyrosines are shown in red. (b) Steady-state ensemble fluorescence spectra of double-labeled NM (0.1 μM) showing energy transfer under denatured (6 M GdmCl) conditions (blue) and under native condition (red) obtained by exciting the donor (λex 488 nm). (c) Two-color single-molecule fluorescence coincidence: Coincident bursts and stoichiometric factor histogram for dual-labeled DNA (non-FRET) standard sample (Upper) and for a mixture of two single-labeled NM (100 pM each) under native condition showing no intermolecular association under this condition (Lower). (d) Single-molecule FRET-efficiency histogram measured ratiometrically for NM under native conditions. Black curves are the best fits using Gaussian functions. See SI Text for details.
This observation was consistent with our previous investigation of assembly kinetics using steady-state fluorescence measurements, wherein acrylodan-labeled core mutants (amino acids ≈20–120) showed an immediate increase in fluorescence intensity and a concomitant blue shift in wavelength before any amyloid formation, suggestive of a collapsed intermediate (13). However, under both these experimental conditions (0.1 μM to low micromolar concentrations), NM undergoes partial conversion to oligomeric species that are on-pathway for assembly. Ensemble data cannot distinguish whether the blue shift in acrylodan fluorescence in our previous experiments or the increased FRET signal in our present experiments arise from intermolecular interactions (due to the formation of oligomers), or intramolecular energy transfer (due to protein compaction), or both. Furthermore, they provide no information about the possible existence of conformational subpopulations and the dynamics of their interconversion. Hence, we next used single-molecule techniques, which allow us to distinguish between these possibilities and to observe the conformational distributions of proteins at very low concentrations (50–200 pM). Under these conditions, most proteins are not expected to form any aggregates, or to form aggregates only very slowly.
Single-Molecule Fluorescence Coincidence: NM Is Monomeric at Low Concentration.
To determine whether aggregation of NM takes place at subnanomolar concentrations, single-molecule fluorescence coincidence experiments were performed (29, 30). In these experiments, we monitored fluorescence bursts from individual protein species, labeled with either one or two different dyes, as they freely diffused through a confocal volume. Overlapped two-color (blue and red) laser excitation and two-channel detection were used to simultaneously excite and detect fluorescence from the two dyes. In equimolar mixtures of proteins individually labeled with the two dyes, fluorescence bursts will be observed on either of the detection channels as these proteins diffuse through the confocal volume, but simultaneous bursts on the two channels will be absent if the protein remains monomeric. However, if the proteins oligomerise to form dimers or higher order species, a significant fraction of diffusing protein will contain both dyes, and hence two-channel (coincident) bursts will be observed. For data analysis, a stoichiometry factor (S) was calculated from the signals obtained in the two channels (blue-excited and red-excited fluorescence) using
Using calculated S values for individual bursts, S histograms can be plotted. In such a histogram, S can vary from 0 to 1, with S = 0 and 1 indicating the absence of coincidence (and hence no aggregation), whereas an S of 0.5 indicates maximum coincidence [corresponding to aggregates with a 1:1 stoichiometric detection of the dyes, using appropriate relative excitation intensities; see supporting information (SI) Text].
Coincidence experiments were first performed with a standard single-stranded DNA sample labeled with Alexa Fluor 488 and Cy5 positioned 40 bases apart, so that the dyes were beyond FRET range. A prominent coincidence peak (at 0.5 S) was observed, as expected for such a dual-dye sample (Fig. 1c Upper). In sharp contrast, under the same conditions, a mixture of two singly labeled NM proteins (Alexa Fluor 488- and Cy5-labeled NM), each at 100 pM concentration showed only peaks close to S = 0 and 1 (Fig. 1c Lower). The absence of a coincidence peak for NM demonstrates that the protein is predominantly monomeric under these conditions. Indeed, NM remained monomeric even at twice this concentration. We used a 100 pM protein concentration for all subsequent SM-FRET experiments.
SM-FRET: Native NM Adopts a Compact State.
Having established conditions under which NM polypeptides remain monomeric, SM-FRET was next used to monitor the conformational properties of dual-labeled NM. In these experiments, after donor excitation, fluorescence bursts were separated into donor (ID) and acceptor (IA) signals. We estimated the FRET efficiency for each molecule using the relationship
where γ is a factor correcting for differences in quantum yields of the dyes and the detection efficiencies of the two optical paths (see SI Text).
The FRET histogram showed a compact Gaussian distribution with a FRET-peak centered around 0.8 E, indicative of a uniform population of molecules (Fig. 1d). The additional peak at 0 E (zero-peak) observed in these experiments is due to nonfluorescent acceptor (due to photobleaching or other causes) in some of the observed molecules. From the mean FRET efficiency, the average distance between the donor and the acceptor in this monomeric state was estimated to be ≈43 Å (For distance estimation from these experiments, see SI Text). Similar experiments done in the presence of 6 M GdmCl showed a significantly unfolded population of polypeptides. We estimate that this shift in the peak position corresponds to an ≈20 Å increase in the distance between the two dyes upon going from native to denatured conditions, and the mean distance in the denatured state was estimated to be ≈ 63 Å. Thus, upon dilution into buffer, NM adopted a compact monomeric form.
The SM-FRET data could be indicative of one of the two possibilities, that native monomeric NM (i) adopts a single native conformation with well defined structural elements (and hence very few or no conformational fluctuations), or (ii) fluctuates either slowly or quickly between multiple FRET-distinguishable conformations. To distinguish among these alternatives, we first investigated the conformational behavior of NM in the presence of a denaturant.
Progressive Noncooperative Expansion Observed Upon Denaturation.
In previous equilibrium SM-FRET unfolding experiments with globular proteins (19, 23), two FRET peaks were observed. As denaturant concentrations increased, a lower FRET peak corresponding to an unfolded species appeared and grew. This was mirrored by a decrease in the high FRET peak corresponding to the folded protein. A plot of the fractional population of the folded state showed a rather sharp sigmoidal transition as a function of denaturant concentration indicative of cooperative unfolding. A very different transition was observed with SM-FRET measurements of NM. As GdmCl concentrations increased from 0 to 6 M, we observed only a single nonzero FRET peak (Fig. 2). This peak smoothly and gradually transitioned from a high FRET value (at low concentrations of denaturant) to a low FRET value (at high concentrations of denaturant). Conversion to the low-FRET form appeared complete only at ≈3 M denaturant.
Fig. 2.

FRET-efficiency histograms of NM at various concentrations of GdmCl. Dotted lines are drawn to show the progressive shift in the FRET-peak from 0.8 to 0.3.
For NM, the progressive shift in the FRET-peak coupled with the broad non-sigmoidal shape of the transition (Fig. 3a) provides evidence for a continuous expansion of the protein in the presence of denaturant, rather than a distinct two-state transition (23, 31). Thus, the observed compact monomeric NM species represents an ensemble of states that are characterized by surprisingly strong, but non-cooperative interactions, rather than a single or a small group of distinct structural entities.
Fig. 3.

Progressive E-shifts and Gaussian simulations support fast dynamics. (a) Plot of FRET efficiency vs. denaturant concentration. The solid line is the exponential fit to guide the eye. (b) Expected FRET-efficiency distribution calculated by taking shot-noise into account for the two limiting cases of fast (solid line) and slow (dashed line) conformational averaging with respect to the observation time (0.5 ms) of a Gaussian chain (for mean E of 0.8). Observed histogram is shown in gray.
Simple Simulations Support Fast Polypeptide Chain Dynamics in NM.
To understand whether fluctuating multiple FRET-distinguishable states could be accessible to natively unfolded NM, we modeled its energy-transfer distribution using simulations within a simple Gaussian-chain approximation (23, 31–33) (see Methods and SI Text). Fig. 3b shows (dashed line) the broad FRET-histogram expected if the resulting conformational distribution is frozen on the timescale of the FRET experiments (23). Regardless of the precise details of the structural model used, it is expected that a similarly broad distribution of conformations might be generally accessible to and observed for an “unstructured” and flexible protein chain. For example, an excluded volume-limit model would also show a broad distribution, with some deviations due to correlated chain repulsions (34). However, because a relatively narrow peak was observed for native NM, any multiple FRET-distinguishable conformations must not be stable on the experimental timescale (≈50–100 μs). The solid line in Fig. 3b shows the relatively narrow simulated FRET-histogram expected if averaging over multiple conformations is complete within the experimental time scale, showing very reasonable agreement with the experimental data. Thus, our SM-FRET data, taken together with the simple simulations, are consistent with native NM occupying an ensemble of rapidly fluctuating or interconverting conformations.
FCS: Observation of Fast Fluctuations in NM.
To directly test for the presence of such fast fluctuations in native NM, we took advantage of the ability of aromatic amino acids to quench Alexa Fluor 488 fluorescence (35, 36) and the fact that the N domain is unusually abundant in tyrosines. In FCS measurements, the fluctuations of the fluorescence signals from freely diffusing molecules are recorded for a small-ensemble of molecules with low nanomolar concentrations in a confocal geometry as for the SM-FRET experiments (27, 28). The autocorrelation function of this fluctuating fluorescence signal can provide information about (i) conformational fluctuations on nanosecond to microsecond time scales (37–39) that arise due to side chain movements and (ii) translational diffusion (tens of microseconds to several milliseconds, depending on molecular size). We first established that the fast fluorescence fluctuations (sub-microsecond) in Alexa Fluor 488-labeled NM were not due to dye photo-physics, rotational motion, an imperfect confocal geometry, or from oligomers, but rather must arise due to monomeric polypeptide chain dynamics (see SI Text).
Next, we carried out FCS experiments on several preparations of NM molecules, each individually labeled with Alexa Fluor 488 at a different position (amino acids 21, 38, 51, 77, 96, 121, 137, and 184). Significant fast decay amplitudes were observed on the 20–300 ns timescale for residue positions 21, 38, 51, 77, 96, and 121, which are located in the region that becomes the core of the amyloid fiber when NM acquires its prion state (Fig. 4a). The amplitude was largest for position 51 (0.6, compared with 0.3–0.5 for most other positions). In the case of position 137, which lies at the interface of the core and unstructured regions, only a small amplitude (≈0.1) was observed. This amplitude variation correlates well with the number of nearby tyrosine residues (see SI Table 1), with position 51 having six tyrosines within a 10-residue sequence separation, and position 137 having none within this range. For position 184, with no tyrosines within 71 residues, no sub-microsecond decay amplitude was observed (Fig. 4b).
Fig. 4.

FCS autocorrelation of NM labeled with Alexa Fluor 488 at 21 (a) and 184 (b) under native condition showing the fluctuation and diffusion components. (Insets) The complete autocorrelation curves.
The FCS data on monomeric NM also revealed the presence of at least two well separated components in the fast decays (SI Table 1), a faster component in the range of 20–40 ns, and a slower component in the range of 150–250 ns. The presence or absence of these two decay components for different labeling positions provides insights into their origins. Alexa Fluor 488 at all positions from 21 to 121 (all of which have one or more tyrosines within a 10-residue separation) exhibited both faster and slower dynamics, whereas the fluorophore at position 137 (no tyrosines closer than a 24-residue separation) exhibits only fast dynamics. As noted above, the fluorophore at 184 (no tyrosines closer than a 71 residues separation) does not sense any sub-microsecond fluctuations. These observations suggest that the faster decay component originates from quenching due to relatively proximal (short-range) tyrosines, whereas the slower component originates from distant (long-range) tyrosines. We note that, although we only resolved two components in the decays, additional unresolved components due to quenching from multiple tyrosines and specific transient structures may also be present. Finally, in the presence of GdmCl concentrations of 3 M or higher, these fluctuation components essentially disappear, either because the average quenching efficiency drops as a result of protein expansion or because of an absence of fluctuations within the timescale range that we are examining. These results establish that the monomeric form of NM adopts an ensemble of relatively unordered states with rapid conformational fluctuations.
Discussion
We have used single-molecule and small-ensemble techniques to directly study the conformational properties of NM in physiological buffers in the non-prion monomeric state. Our results demonstrate that natively unfolded NM is not a completely denatured random coil under these conditions. Instead, it occupies an ensemble of rapidly interconverting compact conformations. The mean interresidue distance between amino acid residues 21 and 121 is ≈43 Å based on our SM-FRET data. This is significantly lower than a value of ≈72 Å calculated from a reported correlation between mean radii of gyration and number of amino acids for denatured polypeptides (40), and may also be somewhat lower than the value expected for a polymer chain in Flory's theta solvent (47 Å), in which intrachain and chain-solvent interactions counterbalance (http://www.pnas.org/cgi/content/full/0611503104/DC1SI Text). From the FCS diffusion time, we estimate a value of 45 ± 5 Å for the hydrodynamic radius of native NM (SI Text), which is in agreement with previous dynamic light scattering results (15). Interestingly, this size is very similar to ≈50 Å estimated for a denatured 250-residue random coil polypeptide (41). Therefore, our results support the view that two distinct structural units (N, compact; M, expanded) are juxtaposed in NM. Upon addition of denaturant, the compact native N undergoes a progressive noncooperative transition to a more expanded form with a ≈63 Å interdye separation, presumably moving the conformational distribution of the entire NM region closer to that which would be occupied by a denatured globular protein. This transition is not complete until 3 M GdmCl, indicating that relatively strong forces may govern the collapsed state.
NM dynamics comprising short- and long-range conformational fluctuations of the polypeptide chain were revealed in our FCS measurements. The time scale of the short-range conformational dynamics is similar to previous estimates of short-range contact formation in unfolded polypetides (42, 43). However, it should be noted that a more detailed molecular interpretation of NM chain dynamics is complicated by the existence of a number of tyrosines at varying length scales, varying relative stiffness around different amino acid residues and excluded-volume effects (44). Our results and analysis also establish evidence that native NM does not exist as a single conformer, but rather populates an ensemble of multiple rapidly interconverting conformers.
Natively unfolded or intrinsically unstructured proteins (45) have been classified as either less structured (random-coil like) or more-structured (premolten globule like), based on a “double-wavelength plot” of [θ]222 vs. [θ]200 obtained from circular dichroism (CD) experiments (46–48). Although our previous CD data obtained during the lag-phase indicated that NM is largely unfolded (15), there are experimental limitations in interpreting such CD data. First, small oligomers would always be present at the micromolar concentrations required for CD experiments. Secondly, the large random coil signals from the highly charged unstructured M region would mask signals from any small ordered region in the N region. Natively unfolded proteins have also been classified based on their dimensions. Random-coil like natively unfolded proteins have dimensions very similar to those of denatured globular proteins, whereas those of premolten globule, like natively unfolded proteins, are significantly smaller. Our finding that the N region of native NM is significantly more compact than a corresponding denatured protein (40) then identifies it with the premolten globule class of natively unfolded proteins. It has been noted that a combination of low mean hydrophobicity and high charge is a prerequisite for maintaining a flexible, unstructured state under physiological conditions (49). The amyloid core of Sup35 (N region) contains only 8% hydrophobic and 4% charged amino acids. Our results indicate that the core N region remains compact and flexible despite lacking both hydrophobic and charged residues. When N is separated from M and diluted into buffer, it immediately assembles into amyloid fibers with no lag phase. Thus, the highly charged extended M region appears essential for allowing N to maintain a monomeric state.
The compact, unstructured form of NM is reminiscent of early (burst-phase) collapsed intermediates observed during protein folding from denaturant (50). Given the low occurrence of hydrophobic residues in NM, it is likely that its collapse occurs when polar residues in the N-segment (Q and N) form hydrogen bonds through backbone and side chain interactions, and possibly by π-stacking interactions among the many tyrosine residues. In support of such a mechanism, the hydrogen bond disrupting solvent DMSO significantly reduced the FRET efficiency (and hence increased the dimensions) of NM (data not shown). Our hypothesis of hydrogen bonding-mediated chain collapse is consistent with recent FRET results from unstructured Gly–Ser repeat peptides (51), as well as a recent FCS study of scaling behavior (52) and simulations of poly(Q) peptides (53). The conformational flexibility in the polypeptide is probably governed by the interplay between hydration of polar side chains of Gln, Asn, and Tyr (54, 55) and intramolecular hydrogen bonds. Additionally, in light of a recent viewpoint on the nature of protein unfolded states, transient interconverting polyproline type II and β-sheet structures might also be present in native NM (56).
Our finding that the N domain undergoes conformational fluctuations on a time scale well below a millisecond explains why an oligomeric species is required for nucleation of NM prion assembly (14, 16). The likelihood of independent monomers finding the correct prion conformation at the same time and nucleating assembly via such preformed structures (as suggested for another prion) (57) would seem infinitesimally small. In the context of an oligomeric assembly, rapidly fluctuating conformations would facilitate chance encounters between the critical sequence elements whose interaction has been shown to be sufficient to drive nucleation (13). These oligomers would then reorganize themselves to form the nucleating species that is capable of rapid assembly into amyloid fibers. Thus, relative stability of the collapsed state in the monomeric species and the ease with which the monomers transit to on-pathway oligomeric intermediates may determine how amyloidogenic a protein is.
In conclusion, we have investigated the structural ensemble of a natively unfolded yeast prion, which forms amyloid under physiological conditions. Our single-molecule experiments provide a picture of the structure and dynamics of the monomer, from which the amyloid state ensues. Future work with additional mutants and detailed FCS experiments might also resolve slower microsecond conformational dynamics (arising due to specific local structure) that may be currently obscured by triplet-state dynamics and molecular diffusion. The combined methodology described here can be used to probe the structural properties of monomeric forms of other amyloidogenic and natively unfolded proteins. Finally, we anticipate that these methods will now also prove useful in understanding the energy landscape of the aggregation process, including characterizing the effects of mutations on the stability and structures of monomers and the formation of oligomeric intermediates during amyloid assembly.
Materials and Methods
Protein Expression, Purification, and Fluorescence Labeling.
Mutagenesis, protein expression, and purification were performed according to reported procedures (13). Fluorescence labeling of Cys mutants was achieved by reaction with the thiol-reactive (maleimide) fluorescent dyes. For details of labeling, purification, and ensemble fluorescence experiments see SI Text.
Single-Molecule Fluorescence, FCS Measurements, and Modeling.
Single-molecule measurements were performed as described (19, 20). Briefly, measurements were carried out by using a home-built laser confocal microscope. Excitation was achieved by focusing the 488-nm line of a tunable argon-ion laser inside the sample solution, 20 μm from the glass surface, using a water immersion objective. For fluorescence coincidence measurements, excitation was achieved by two overlapped laser beams, the 488-nm line of an argon-ion laser and 633-nm line of a He-Ne laser. Two-color fluorescence emissions were isolated and separately detected using two avalanche photodiodes. FCS experiments were performed on the same setup with a 35-μm-diameter pinhole and using a pseudo-cross-correlation mode to allow sub-microsecond correlation measurements. Modeling was performed using IGOR (Wavemetrics, Lake Oswego, OR), and provided shot-noise limited distributions for a random coil Gaussian chain model under fast or slow fluctuation regimes. For details of the experimental setup, data collection, analysis, and modeling, see SI Text.
Supplementary Material
Acknowledgments
We thank Prof. Peter Schultz for providing access to a spectrofluorometer; J. C. Vanderschans for assistance with labeled-protein purification; and Prof. Marcia Levitus, Dr. Yann Gambin, and members of the S.L. laboratory for critical comments on the manuscript. This research was supported by National Institutes of Health (NIH) Grant GM066833 (to A.A.D), the DuPont–MIT Alliance and NIH Grant GM25874 (to S.L.), and a postdoctoral fellowship from the Alexander von Humboldt Foundation (to E.A.L).
Abbreviations
- SM-FRET
single-molecule FRET
- FCS
fluorescence correlation spectroscopy.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0611503104/DC1.
References
- 1.Prusiner SB. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caughey B. Nat Med. 2000;6:751–754. doi: 10.1038/77476. [DOI] [PubMed] [Google Scholar]
- 3.Chien P, Weissman JS. Nature. 2001;410:223–227. doi: 10.1038/35065632. [DOI] [PubMed] [Google Scholar]
- 4.Shorter J, Lindquist S. Nat Rev Genet. 2005;6:435–450. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 5.Tuite MF, Cox BS. Nat Rev Mol Cell Biol. 2003;4:878–889. doi: 10.1038/nrm1247. [DOI] [PubMed] [Google Scholar]
- 6.Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu HX, Kandel ER. Cell. 2003;115:893–904. doi: 10.1016/s0092-8674(03)01021-3. [DOI] [PubMed] [Google Scholar]
- 7.Si K, Lindquist S, Kandel ER. Cell. 2003;115:879–891. doi: 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- 8.Teravanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Ingevechtomov SG, Smirnov VN. Mol Microbiol. 1993;7:683–692. doi: 10.1111/j.1365-2958.1993.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 9.Chernoff YO, Derkach IL, Ingevechtomov SG. Curr Genet. 1993;24:268–270. doi: 10.1007/BF00351802. [DOI] [PubMed] [Google Scholar]
- 10.Kishimoto A, Hasegawa K, Suzuki H, Taguchi H, Namba K, Yoshida M. Biochem Biophys Res Commun. 2004;315:739–745. doi: 10.1016/j.bbrc.2004.01.117. [DOI] [PubMed] [Google Scholar]
- 11.Collins SR, Douglass A, Vale RD, Weissman JS. PLoS Biol. 2004;2:1582–1590. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 13.Krishnan R, Lindquist SL. Nature. 2005;435:765–772. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 15.Scheibel T, Lindquist SL. Nat Struct Biol. 2001;8:958–962. doi: 10.1038/nsb1101-958. [DOI] [PubMed] [Google Scholar]
- 16.Shorter J, Lindquist S. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 17.Jahn TR, Parker MJ, Homans SW, Radford SE. Nat Struct Mol Biol. 2006;13:195–201. doi: 10.1038/nsmb1058. [DOI] [PubMed] [Google Scholar]
- 18.Platt GW, McParland VJ, Kalverda AP, Homans SW, Radford SE. J Mol Biol. 2005;346:279–294. doi: 10.1016/j.jmb.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 19.Deniz AA, Laurence TA, Beligere GS, Dahan M, Martin AB, Chemla DS, Dawson PE, Schultz PG, Weiss S. Proc Natl Acad Sci USA. 2000;97:5179–5184. doi: 10.1073/pnas.090104997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deniz AA, Laurence TA, Dahan M, Chemla DS, Schultz PG, Weiss S. Ann Rev Phys Chem. 2001;52:233–253. doi: 10.1146/annurev.physchem.52.1.233. [DOI] [PubMed] [Google Scholar]
- 21.Michalet X, Weiss S, Jager M. Chem Rev. 2006;106:1785–1813. doi: 10.1021/cr0404343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuler B. Chem Phys Chem. 2005;6:1206–1220. doi: 10.1002/cphc.200400609. [DOI] [PubMed] [Google Scholar]
- 23.Schuler B, Lipman EA, Eaton WA. Nature. 2002;419:743–747. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 24.Tinnefeld P, Sauer M. Angew Chem Int Ed. 2005;44:2642–2671. doi: 10.1002/anie.200300647. [DOI] [PubMed] [Google Scholar]
- 25.Bilsel O, Matthews CR. Curr Opin Struct Biol. 2006;16:86–93. doi: 10.1016/j.sbi.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Magde D, Elson E, Webb WW. Phys Rev Lett. 1972;29:705–708. [Google Scholar]
- 27.Hess ST, Huang SH, Heikal AA, Webb WW. Biochemistry. 2002;41:697–705. doi: 10.1021/bi0118512. [DOI] [PubMed] [Google Scholar]
- 28.Medina MA, Schwille P. BioEssays. 2002;24:758–764. doi: 10.1002/bies.10118. [DOI] [PubMed] [Google Scholar]
- 29.Heinze KG, Rarbach M, Jahnz M, Schwille P. Biophys J. 2002;83:1671–1681. doi: 10.1016/S0006-3495(02)73935-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li HT, Ying LM, Green JJ, Balasubramanian S, Klenerman D. Anal Chem. 2003;75:1664–1670. doi: 10.1021/ac026367z. [DOI] [PubMed] [Google Scholar]
- 31.Sherman E, Haran G. Proc Natl Acad Sci USA. 2006;103:11539–11543. doi: 10.1073/pnas.0601395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gopich I, Szabo A. J Chem Phys. 2005;122:014707. doi: 10.1063/1.1812746. [DOI] [PubMed] [Google Scholar]
- 33.Gopich IV, Szabo A. J Phys Chem B. 2003;107:5058–5063. [Google Scholar]
- 34.Tran HT, Pappu RV. Biophys J. 2006;91:1868–1886. doi: 10.1529/biophysj.106.086264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marme N, Knemeyer JP, Sauer M, Wolfrum J. Bioconj Chem. 2003;14:1133–1139. doi: 10.1021/bc0341324. [DOI] [PubMed] [Google Scholar]
- 36.Vaiana AC, Neuweiler H, Schulz A, Wolfrum J, Sauer M, Smith JC. J Am Chem Soc. 2003;125:14564–14572. doi: 10.1021/ja036082j. [DOI] [PubMed] [Google Scholar]
- 37.Chattopadhyay K, Elson EL, Frieden C. Proc Natl Acad Sci USA. 2005;102:2385–2389. doi: 10.1073/pnas.0500127102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chattopadhyay K, Saffarian S, Elson EL, Frieden C. Proc Natl Acad Sci USA. 2002;99:14171–14176. doi: 10.1073/pnas.172524899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neuweiler H, Doose S, Sauer M. Proc Natl Acad Sci USA. 2005;102:16650–16655. doi: 10.1073/pnas.0507351102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kohn JE, Millet IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Siefert S, Thiyagarajan P, Sosnick TR, et al. Proc Natl Acad Sci USA. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 42.Krieger F, Fierz B, Bieri O, Drewello M, Kiefhaber T. J Mol Biol. 2003;332:265–274. doi: 10.1016/s0022-2836(03)00892-1. [DOI] [PubMed] [Google Scholar]
- 43.Kubelka J, Hofrichter J, Eaton WA. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 44.Flory PJ. Statistical Mechanics of Chain Molecules. New York: Hanser; 1969. [Google Scholar]
- 45.Dyson HJ, Wright PE. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 46.Uversky VN. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uversky VN. Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 48.Uversky VN, Fink AL. Biochim Biophys Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 49.Uversky VN, Gillespie JR, Fink AL. Proteins Struct Funct Genet. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 50.Sadqi M, Lapidus LJ, Munoz V. Proc Natl Acad Sci USA. 2003;100:12117–12122. doi: 10.1073/pnas.2033863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moglich A, Joder K, Kiefhaber T. Proc Natl Acad Sci USA. 2006;103:12394–12399. doi: 10.1073/pnas.0604748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Proc Natl Acad Sci USA. 2006;103:16764–16769. doi: 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang XL, Vitalis A, Wyczalkowski MA, Pappu RV. Proteins Struct Funct Bioinform. 2006;63:297–311. doi: 10.1002/prot.20761. [DOI] [PubMed] [Google Scholar]
- 54.Balbirnie M, Grothe R, Eisenberg DS. Proc Natl Acad Sci USA. 2001;98:2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dunitz JD. Science. 1994;264:670–670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- 56.Shi ZS, Chen K, Liu ZG, Ng A, Bracken WC, Kallenbach NR. Proc Natl Acad Sci USA. 2005;102:17964–17968. doi: 10.1073/pnas.0507124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lansbury PT. Chem Biol. 1995;2:1–5. doi: 10.1016/1074-5521(95)90074-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
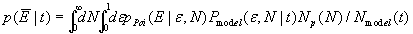{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
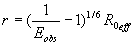{kind=link}