

Abstract
We report single-molecule folding studies of a small, single-domain protein, chymotrypsin inhibitor 2 (CI2). CI2 is an excellent model system for protein folding studies and has been extensively studied, both experimentally (at the ensemble level) and theoretically. Conformationally assisted ligation methodology was used to synthesize the proteins and site-specifically label them with donor and acceptor dyes. Folded and denatured subpopulations were observed by fluorescence resonance energy transfer (FRET) measurements on freely diffusing single protein molecules. Properties of these subpopulations were directly monitored as a function of guanidinium chloride concentration. It is shown that new information about different aspects of the protein folding reaction can be extracted from such subpopulation properties. Shifts in the mean transfer efficiencies are discussed, FRET efficiency distributions are translated into potentials, and denaturation curves are directly plotted from the areas of the FRET peaks. Changes in stability caused by mutation also are measured by comparing pseudo wild-type CI2 with a destabilized mutant (K17G). Current limitations and future possibilities and prospects for single-pair FRET protein folding investigations are discussed.
The folding mechanisms of proteins have been intensely studied by using both experimental and theoretical methods. However, because protein folding is extremely complex, no single observable or technique is sufficient to fully describe it. Hence, new technologies have constantly been applied to the folding problem, providing new experimental observables and deeper insights into the problem. Technical developments recently have facilitated crossing the fundamental threshold from ensemble to single-molecule studies, providing access to information unavailable from bulk measurements. Several important aspects of protein folding and dynamics are difficult or impossible to study by using ensemble methods, because of the complexity of protein structures and the stochastic nature of these processes. Key examples of this complexity include (i) the ensemble of unfolded protein molecules, consisting of a large number of nearly degenerate and rapidly interconverting protein conformations (1, 2), (ii) multiple local minima on a rugged energy landscape that can give rise to fluctuations and several conformational substates even for the native protein, and (iii) ensembles of folding pathways and “transition states” for the folding reaction. Hence, the dynamics of folding reactions are expected to be highly stochastic; such stochastic aspects are clearly best studied at single-molecule resolution. Single-molecule methods are also useful to directly monitor static or dynamic distributions in the properties of individual subpopulations, at equilibrium in a mixture of species, and under nonequilibrium conditions. Finally, from a theoretical standpoint, most calculations are carried out at the single-molecule level. Hence, single-molecule experiments provide the most direct experimental benchmarks and tests for theory. In particular, the dramatic increase during recent years of theoretical methods applied to the folding problem makes such single-molecule studies very attractive (3–8).
Single-molecule investigations, however, are still in their infancy and are prone to artifacts because of the conditions under which they are carried out (9). Currently, most single-molecule measurements are conducted on surface immobilized molecules; it is quite clear that surfaces can modify the properties of the solution microenvironment, the photophysics of fluorophores, and conformational properties and the energy landscape of the molecules under study. Methodology has recently been developed to measure fluorescence resonance energy transfer (FRET) on single molecules freely diffusing in solution, under conditions used in conventional ensemble studies (10, 11). Although this method limits the observation time to the diffusion time of each molecule through the laser focal spot (a few hundred microseconds), it permits the rapid gathering of data at single-molecule resolution, on a statistically significant number of molecules.
Materials and Methods
Protein Molecules.
The protein was synthesized by using conformationally assisted ligation of two peptide fragments of chymotrypsin inhibitor 2 (CI2) (12). Peptides corresponding to CI2(1–39) and CI2(40–64) were synthesized separately by using manual solid-phase peptide synthesis (13, 14); the C-terminal (1–39) fragment was synthesized on trityl-associated mercaptopropionic acid-leucine resin to afford a C-terminal thioester (15). For donor-acceptor labeled protein, the N-terminal amine of the (1–39) piece was coupled with tetramethylrhodamine (TMR)-succinimide ester before cleavage from the resin. The peptides were deprotected and cleaved from the resin by using anhydrous hydrofluoric acid, precipitated with ether, dissolved in aqueous acetonitrile, and lyophilized. They were purified by using reverse-phase (RP) (C-18) HPLC and analyzed by using electrospray MS. The above self-associating peptides of CI2 were ligated under folding conditions at room temperature, for 30 min. The product CI2(1–64)M40C was purified by using RP-C18 HPLC and analyzed by electrospray MS. Both unlabeled and TMR-only labeled proteins were synthesized as described. Cy5 modified with a thiopyridyl-activated disulfide was used to label the protein by disulfide exchange with the cysteine thiol group at position 40. A 5-fold excess of Cy5-activated disulfide was used and the reaction was allowed to proceed for 15–20 h. The labeled protein was purified by using RP-C18 HPLC. It was analyzed by using electrospray MS, showing a single species [8,383.2 ± 0.5 Da, for TMR-Cy5-labeled pseudo wild type (pwt) CI2], consistent with a site-specific 1:1 dye/protein reaction. Both Cy5-only-labeled and TMR-Cy5-labeled protein were synthesized in this manner.
Single-Molecule Fluorescence Studies.
The experimental setup, conditions, and data analysis are described in detail elsewhere (10, 11). Briefly, the 514-nm laser light from an argon-ion laser was focused 10 μm inside a solution of the protein, nominally 100 pM in concentration. The emitted fluorescence was separated from the excitation light by using a dichroic mirror (DRLP 530, Omega Optical, Brattleboro, VT), and the resulting light was split into two detection channels by using a second dichroic mirror (DRLP 630). Light passed through a band-pass filter for the TMR channel and a long-pass filter for the Cy5 channel before being detected by two separate Avalanche photodiodes (EG&G, Vandreuil). The two signals were separately and simultaneously recorded as a function of time. Occasionally, fluorescence bursts corresponding to fluorescent molecules diffusing through the excitation volume were observed. These bursts were separated from noise by using a threshold of 30 counts per point, analyzed for FRET efficiency, E, and the results were plotted in the form of a histogram. Here, the factor γ, which accounts for the ratio of donor and acceptor quantum yields and detection efficiencies, is assumed to be 1, as previously discussed (10, 11). The histograms were fit with Gaussians to identify contributions from different subpopulations (11). Interdye distances (R) were calculated according to:
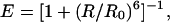 |
1 |
where R0 is the Förster radius, estimated to be 53 Å for the TMR-Cy5 dye pair, using an average value of 2/3 for κ2, the orientation factor (16).
Protein Denaturation Studies.
All denaturation experiments were carried out under solution conditions similar to those used by Fersht and coworkers (19, 23), except that a phosphate buffer (50 mM, pH 6.3) was used instead of MES buffer, because of fluorescence background considerations. Pierce guanidinium chloride (Sequanal grade) was found to be sufficiently background free for these experiments. Tween-20 was used to passivate the glass surface of the sample cell to minimize protein adsorption. Single-molecule experiments were carried out at a series of denaturant concentrations. Data were usually acquired for 5 min, and histograms were calculated and analyzed. Generally, two or three data sets were acquired and analyzed at each denaturant concentration. Ensemble fluorescence denaturation experiments were carried out by using a SPEX Fluoromax-2 fluorescence spectrometer (JY/Horiba, Edison, NJ). Here, FRET or tryptophan fluorescence data were acquired as a function of denaturant concentration, and a two-state model was used to calculate the fraction of the folded state as a function of denaturant; plots of these data are shown in Fig. 4. Sigmoidal fits to the denaturation curves (single molecule and ensemble) resulted in the reported midpoints; errors include estimates from multiple experiments.
Figure 4.

Ensemble and single molecule denaturation curves for pwt and mutant CI2. Symbols show data, lines show sigmoidal fits. Pwt CI2: ensemble tryptophan (○, solid line), ensemble FRET (□, dashed line), sp-FRET (×, dotted line); mutant CI2 sp-FRET (▿, dotted/dashed line).
Results and Discussion
Protein System.
FRET has been used as a tool to study biomolecule conformations and dynamics at the ensemble level for the last three decades. The strong distance dependence (1/R6) of the FRET rate constant allows the measurement of distances on the 20- to 80-Å scale, between donor and acceptor labels on a biomolecule. In the case of protein folding, FRET can provide distance information for pairs of points on the amino acid chain as a function of folding, i.e., it can provide a reaction coordinate that affords a global view of the conformational distributions and dynamics of the protein as it folds. Recently, single-molecule FRET has been used to examine the folding and fluctuations of the surface immobilized peptide, GCN4 (17). It is clear that surface interactions could have modified and broadened the distributions observed under these immobilized conditions, making the interpretation of the results and comparison to existing ensemble measurements more problematic. RNA folding and protein fluctuations under surface immobilized conditions, and DNA hairpin folding in diffusion also have been recently reported (10, 16, 18). In this work, we use diffusion single-pair FRET (sp-FRET) to examine the guanidinium chloride denaturation of freely diffusing CI2 (shown in Fig. 1), a well-established model for protein folding studies.
Figure 1.

CI2 structure, showing points of dye attachment and destabilizing mutation.
The protein CI2 was chosen for several reasons. It has been extensively examined by Fersht and coworkers (19, 20), who provide a large amount of ensemble information with which to compare single-molecule results. This protein also has been the subject of numerous theoretical studies (5–7, 21, 22). The tertiary structure of CI2 contains the three basic units of protein structure: α-helix, β-sheet, and loop elements (Fig. 1). Wild-type (wt) CI2 is very stable, showing a denaturing transition at about 4 M guanidinium chloride, based on changes in tryptophan fluorescence. Kinetic and equilibrium arguments have been used to support a two-state folding mechanism for the protein (23). The structure of the transition state and the mechanism of folding have been probed by using the protein engineering method (19). Furthermore, the truncated form of CI2, used by the Fersht group and in this work, is small (64 aa), which makes it amenable to total or semisynthetic methodologies.
Recently developed conformationally assisted “peptide ligation” methodology using C-α-thioester peptides (12, 24) is a very promising approach for making relatively small proteins, with multiple site-specific mutations, labels, or reactive handles, and was used here. Two fragments, CI2(1–39) and CI2(40–64) were synthesized separately by standard solid-phase peptide synthesis; TMR was site-specifically coupled to the N-terminal amine of CI2(1–39), followed by cleavage, deprotection, and purification. The two fragments then were ligated under folding conditions, allowing a clean and rapid ligation reaction, resulting in wt CI2(1–64) with M40C and N-terminal TMR substitutions. We refer to this amino acid sequence as pwt CI2. The second dye, Cy5, was coupled to the unique cysteine at position 40, affording site-specifically donor-acceptor labeled protein, with a minimum of purification/separation steps. The product was characterized by using HPLC and mass spectral analysis.
Subpopulations of Pwt and Mutant CI2.
A denaturation curve is a plot of the fraction of a given state (e.g., the folded state) as a function of condition (denaturant concentration, temperature, pressure, etc.). For a two-state system, the curve has a sigmoidal shape, and its midpoint corresponds to conditions under which the two states are equally populated; for pwt CI2 (versus guanidinium chloride concentration), this midpoint was found to be at 4 M by ensemble methods (19, 23). Using diffusion sp-FRET, we generated histograms of FRET efficiencies (E) for CI2 as a function of guanidinium chloride concentration. Because they report on the interdye distance, such histograms represent the distribution of conformational states during folding, which can serve as a reaction coordinate for the folding reaction. Fig. 2a shows representative histograms for 3, 4, and 6 M denaturant; these values were chosen to be close to the ensemble denaturation midpoint. Based on the ensemble data, most CI2 molecules should be folded, an equal fraction folded and unfolded, and most molecules unfolded, respectively under these conditions. Large changes are indeed observed between these three histograms. All histograms showed a peak at around zero FRET efficiency, previously shown to arise in part from molecules containing fluorescent donor and photobleached acceptor (10). Because these molecules do not provide relevant FRET (distance) information, only the region above 0.4 E is displayed, and the following discussion refers to peaks in this region.
Figure 2.

(a) sp-FRET histograms of pwt CI2 at 3, 4, and 6 M denaturant. (b) sp-FRET histograms showing comparison of pwt and mutant CI2 (K17G) at 3.5 M denaturant. Solid lines show Gaussian fits to the peaks.
CI2 is believed to fold via a two-state mechanism based on ensemble measurements (23). The sp-FRET histograms directly support such a mechanism. At or below 3 M guanidinium chloride, a peak at around 0.95 E is observed. At high denaturant concentration (6 M), only a peak at around 0.65 E is detected. As intermediate denaturant concentrations are scanned, varying ratios of the two peaks are observed. The properties of the lower efficiency peak change with denaturant concentration (see below). The 0.95 E and the 0.65 E peaks are assigned to the folded and denatured states of the protein, respectively. Note that single-molecule studies confer the ability to examine particular subpopulations of interest. This advantage can be appreciated by comparing mean FRET efficiencies obtained from sp-FRET with the ensemble FRET measurements; the ensemble values are found to be significantly lower (by up to 0.2 E). This observation derives, at least in part, from molecules with nonfluorescent or photobleached acceptor. Because the steady-state ensemble numbers include a contribution from the “zero peak,” they are shifted toward lower FRET efficiency, skewing the true values.
The histograms directly confirm the “two-state” nature of the CI2 folding transition. The upper limit for the observation time per molecule in this experiment is about 1 ms because of the diffusion process and the data are acquired with a 0.2-ms integration time. The observation of two subpopulations indicates that the interconversion of the two forms of the protein must occur on a time scale that is significantly slower than the integration time. This observation is consistent with the folding/unfolding kinetics slower than 100 ms observed in the transition region by ensemble measurements (23).
A mutant (pwt sequence with K17G substitution) with a significantly lower stability also was synthesized, and its single-molecule folding properties were compared with those of the pwt. This helix destabilizing mutation (Fig. 1) has been shown to shift the guanidinium chloride denaturation midpoint to 2.8 M denaturant (19). Hence, it is expected that at 3.5 M denaturant, the observed fraction of the 0.95-E subpopulation should be significantly reduced for the mutant relative to pwt CI2. The sp-FRET histograms (Fig. 2b) show that the fraction of the folded form drops from about 0.5 for the pwt to almost zero for the mutant. Because the solution conditions are identical, this dramatic change must come from protein stability/structure-related changes. This observation strongly supports the assignment of the observed subpopulations to the native and denatured states of CI2. The properties of these subpopulations (mean, width, and area of the FRET peaks) contain information about the protein folding reaction, as discussed in the following sections.
Interdye Distances for the Native and Denatured States.
Average interdye distances in the folded and denatured forms of the protein can be extracted from the mean FRET efficiencies, assuming freely rotating dyes (κ2 = 2/3) and a Förster radius of R0 = 53 Å. Because the single-molecule E values exclude non-FRET contributions (see below), they are used in the discussion below. A value for the mean distance between the two dyes of 31 Å is calculated for the folded form, consistent with a number of about 32 Å estimated from the known crystal structure of the protein (25), and allowing a reasonable additional 7 Å because of the size of the dye/tethers, assuming a right-angled geometry on average. For the unfolded form, the mean calculated distance (from mean E, using the same assumptions for κ2 and R0) is 48 Å. However, changes in the orientation factor κ2 could result in significant changes in R0 and hence skew the calculated value of R (see Eq. 1). A discussion is presented below describing estimation of such changes.
To compute the effects of κ2, it is assumed that the orientations of the donor and acceptor fluorophores are random on the time scale of the measurement (100 μs to 1 ms). This is a reasonable assumption for the denatured subpopulation, because it is rapidly interconverting between conformations and the donor and acceptor are separated by a relatively long distance of 40 aa (26). A second assumption is that the large fluctuations of the peptide chain occur on a longer time scale than the fluorescence lifetime (few ns), allowing the interdye distance to be held constant while analyzing the effects of κ2. Ensemble polarization anisotropy (r) measurements were performed on separate proteins labeled with TMR only (N terminus) and Cy5 only (cysteine 40). It was found that r decreased for TMR (donor) from about 0.09 to 0.07 over a limited range of 3 M to 4.5 M denaturant, with a much smaller increase by 0.005 to 0.0075 from 4.5 M to 6 M denaturant. For Cy5 (acceptor), r stayed constant at about 0.2 over this range. The relatively high anisotropy values indicate that either one or both of the following are true: (i) the fluorophores did not have isotropic freedom of motion, or (ii) the fluorophores' rotational diffusion was on the same time scale as the fluorescence lifetime. The first possibility was investigated by extending the work of Dale and Eisinger (27). An analytical model was developed to describe the fluorophores as rapidly rotating dipoles within an axi-symmetric distribution (a cone), and the cones slowly (compared with donor fluorescence lifetime) and randomly reorienting themselves. For the second possibility, it was necessary to simulate the rotational diffusion directly. It was assumed that the fluorophores had isotropic freedom, but that the rotational diffusion was slow enough to obtain the observed anisotropy. The simulations were performed by a random walk on a sphere and were calibrated by using the Perrin equation (28). In both cases, a decrease in the mean E of 0.03 to 0.05 was found on going from a situation with freely rotating dyes to one with the experimental polarization anisotropy values. Recalculation of the interdye distance for the denatured subpopulation, which included this correction, gave R = 45 Å.
The calculated distances for the denatured state compare reasonably with theoretical predictions of Miller and Goebel (29), who determined an rms distance of about 50 Å for the end-to-end distance of a 40-aa random coil, based on rotational isomeric state model calculations. However, fast distance fluctuations between the donor and acceptor (on the time scale of the donor fluorescence lifetime) will result in a higher average observed transfer efficiency (for the same instantaneous distance distribution) and a corresponding lower average distance (30). Furthermore, other factors such as the particular sequence, dye-protein interactions, the detection efficiency, photobleaching, excluded volume, and solvent interactions also could alter the observed FRET efficiencies; some of these factors are discussed in more detail in the following section.
Shifts in Average FRET Efficiency of the Denatured State.
It is found that the mean FRET efficiency of the denatured subpopulation for the pwt protein shifts as a function of denaturant concentration, whereas the corresponding values for the folded subpopulation are fairly constant. Fig. 3a shows that the denatured E peak shifts from 0.68 ± 0.02 in 4 M denaturant to 0.63 ± 0.01 in 6 M denaturant. Error bars are SDs from multiple measurements. Although the precision of the data are poor and an attempt to fit it to a model will be at best questionable, we might be observing a sigmoidal transition in mean E. This transition is centered at about 4.4 M denaturant and could indicate an increase in protein dimensions with increase in denaturant concentration. Such a “swelling” of the denatured protein can occur because of a reduction in persistent specific structure or an increase in the “solubility” of the protein random coil (1, 31–33). It is noted that this shift occurs over a limited range of denaturant concentration (4 M to 5 M), close to the major unfolding transition region at 4 M denaturant (see the denaturation curves in Fig. 4) and would be at least partly hidden (or convoluted) in any ensemble experiment.
Figure 3.

(a) Shift in the mean E of the denatured subpopulation for pwt CI2. The error bars are single SDs. The solid line is a guide to the eye. (b) Potentials V(R) (free energy functions) for pwt CI2 at 3, 4, and 6 M denaturant. The solid lines represent a smoothing of the data and are meant as a guide to the eye.
However, it is emphasized that several other factors can contribute to this shift and must be taken into account. Although most of the observed E shift seems to occur within a narrow range of 0.5 M in denaturant concentration, an upper bound on changes caused by other factors is calculated by comparing results at 4 M and 6 M denaturant, given the poor precision of the data in Fig. 3. Changes in observed E, (going from 4 M to 6 M denaturant) can be related to nondistance related changes (i.e., R remains constant) as follows:
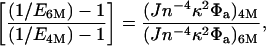 |
2 |
where J is the overlap integral, n is the refractive index, κ2 is the orientation factor, and Φa is the acceptor fluorescence quantum yield. The photophysical properties of the dyes (donor and acceptor absorption and emission) were measured in ensemble at 4 and 6 M denaturant to quantify changes in κ2, J, and φa, and the refractive index change was obtained from the literature (34). The small observed increase in r was translated into a change in κ2 by using the approaches discussed in the previous section, resulting in a decrease of 0.003 E units. Combining these numbers, we estimate that the average E should be reduced because of nondistance effects from 0.68 to 0.65 ± 0.04. It therefore is concluded that together with the relatively large error bars, the nondistance effects could explain the observed shifts and cannot be ruled out. In addition to photophysical effects, an increase in solvent viscosity with increasing denaturant concentration will lead to an increase in time scales of the dipoles' rotational diffusion and the structural fluctuations of the denatured, random coil, amino acid chain. The slowing down of these fluctuations will result in a decrease in the average E. However, we estimate that the contribution of increased viscosity to the observed changes in E is small. Viscosity changes with denaturant concentration will continue above 5 M, whereas the observed E change occurs over a very narrow (4 M to 5 M) concentration range (the E change going from 4 M to 5 M is 0.06 whereas it is <0.01 going from 5 M to 6 M).
Distance Distributions, Widths, and Potential Functions.
sp-FRET histograms give direct access to the E distribution width, thereby providing information on distributions of interdye distances. Distance-related broadening of the FRET peaks could occur because of relatively slow (compared with the binning time scale) interconversion between conformers in the denatured state, especially at lower concentrations of denaturant, and proline isomerization also might play a significant role. However, the distributions are contaminated by several other factors, most significantly shot noise, and one must exercise great caution in interpreting the width of the FRET distribution. For the sake of the following discussion, we ignore these complications and assume contributions from distance only (admittedly, a gross assumption). Under such an assumption, the E histograms can be directly translated into potential functions versus interdye distance. This is done by first converting E distributions into distance distributions assuming κ2 = 2/3 (R0 = 53 Å). The distance distribution then is converted into a potential energy (free energy) function (Fig. 3b) by using the Boltzmann weight Pi = exp(-ΔG/RT) (17), where Pi is the probability of populating bin i at distance Ri, obtained from the distance histogram. The main features to be noted are changes in the position and number of minima as a function of guanidinium chloride; the appearance of the double well at 4 M guanidinium chloride reflects the two-state folding of this protein. Because nondistance factors cause broadening in the histograms, the depicted potential functions show upper limits for the widths and lower limits for the depths of the wells. This finding is consistent with the measured activation barrier of about 19.5 kcal/mol for folding/unfolding of wt CI2 at 4 M denaturant (35). Note that the minimum for the denatured state represents a significant entropic contribution. Furthermore, the limited time resolution (200 μs) prohibits the observation of transiently populated states such as rapidly interconverting conformations of the denatured state under these conditions.
Shot noise dominates the distribution width and therefore the obtainable subpopulation resolving power (10, 11). A limited number of photon counts (up to about 100) are obtained from each molecule as it passes through the detection volume. These counts usually are distributed among 3–5 time bins (in this paper, all photon counting data were binned to 200 μs, with a rejection threshold of 30 counts per bin). For a sample with static heterogeneity (disorder), a reduction in shot-noise broadening could be achieved by respective binning of all donor and acceptor photons collected from each molecule (burst integrated analysis) (36). However, using a detailed comparison between pixel-wise (200-μs bins) time analysis and burst integrated analysis for various threshold values, we find that 200-μs time bins provided the best peak separation. Burst integrated analysis was able to match this performance, but did not exceed it, because of photobleaching considerations. Furthermore, shot-noise broadening places a limit of 10–50 μs on the time resolution achievable for identifying interconverting conformations by sp-FRET histograms, using the current dye pair.
It is relevant to consider here the implications of the ergodic hypothesis, which equates the long-time average behavior of a single molecule with that of the instantaneous average over an ensemble. This is true only if the population of molecules is homogeneous on the single-molecule averaging time scale, which has important consequences for measurements with limited sampling, as is typical for single-molecule measurements. If a molecule does not adequately sample all of its conformational space within the measurement time (for example, because of photobleaching), it will be more representative to rapidly acquire small amounts of data on many single molecules (diffusion or flow methods) rather than to collect a larger amount of data on a small number of molecules. In this respect, the diffusion sp-FRET method offers important information complementary to that obtained from FRET time trajectories acquired on single immobilized molecules.
Denaturation Curves Extracted from FRET Distributions.
Fig. 4 shows a comparison of denaturation curves from ensemble and single-molecule experiments for pwt CI2, showing the cooperative, two-state folding/unfolding transitions. The single-molecule denaturation curves were directly extracted from the sp-FRET histograms. Here, in marked contrast to the procedure used in ensemble measurements, the fraction of the protein in the folded form was directly calculated as the ratio of the area of the 0.95 E peak to the sum of the areas of the 0.95 and the 0.65 E peaks; a transition midpoint of 3.7 ± 0.1 M denaturant is observed for the pwt protein. This result is in good agreement with our ensemble FRET measurements on the donor-acceptor labeled protein (midpoint = 3.8 ± 0.2 M denaturant) and ensemble denaturation using tryptophan fluorescence for the unlabeled protein (midpoint = 3.9 ± 0.2 M denaturant). The ensemble midpoint from the tryptophan fluorescence experiments of Fersht and coworkers for wt CI2 is 4.0 M denaturant (19). Although, the midpoints for the dye-labeled and unlabeled protein are within experimental error of each other, there might be a small decrease in protein stability because of attachment of the dyes. Several other factors could affect the position of the single-molecule midpoint, such as different photophysics, photochemistry, or diffusion time between the two forms of the protein. However, the present analysis results in numbers that are in good agreement with ensemble data, showing that the single-molecule method is reliable, and that dye attachment has not seriously compromised protein stability. Fig. 4 shows a comparison of the single-molecule denaturation curve for the pwt and K17G mutant protein, clearly showing the destabilizing effect of the mutation. The mutant yields a midpoint of 2.5 M denaturant, again comparing reasonably well with the ensemble FRET value of 2.65 M and ensemble tryptophan value (2.95 M, unlabeled) measured here, and the literature tryptophan value of 2.8 M for CI2 K17G (19).
In equilibrium ensemble experiments, direct information on the fraction of the protein in a particular state is not available because only average properties are measured. Instead, a model and extrapolation-dependent analysis must be used to calculate such fractions. This analysis sometimes can be problematic. For example, changes in the value of the observable for a given state as a function of condition (e.g., the shifts in E peak position v/s denaturant observed in this work) often are corrected for by extrapolating from conditions where only one state exists. Nontrivial changes in the observable, especially in the transition region, can produce significant question marks in interpretation, as pointed out by Dill and Shortle (1). Because single-molecule methods can directly follow changes in subpopulations (individual states or ensembles), a more accurate representation of the process is obtained. Evidently, the effects discussed here will be much more pronounced in situations where a large nonlinear change in a property is observed, or when the folding mechanism is complex, involving additional steps.
Conclusions
We have described the use of diffusion ratiometric sp-FRET methodology to the study of protein folding at single-molecule resolution. Analysis of the folded and unfolded subpopulations of CI2 during guanidinium chloride denaturation provides the first single-molecule denaturation curves for a single-domain protein, and changes in protein stability caused by mutation are observed by using this methodology. Comparison of the single-molecule denaturation curves with those from bulk measurements shows that the sp-FRET method is reliable for studying protein folding. In short, this work validates the diffusion sp-FRET methodology as a valuable tool in such studies. Such a validation opens up possibilities for future studies on novel and more complex systems, paving the way for exploration and discovery of new folding phenomena by single-molecule spectroscopy.
Improvements in dye pairs, single-molecule data acquisition, data analysis, and surface/gel immobilization methods will be crucial to fully exploit this technique. Issues such as the influence of dyes on the folding reaction and the contribution of dye photophysics to the observed FRET changes also must be addressed in detail. Immobilization of molecules can afford long time trajectories and more detailed information, especially under nonequilibrium conditions. However, such immobilization generally will change the static and dynamic distributions of molecular conformations caused by variations in the protein energy landscape induced by the local environment. The diffusion methodology presented here can be used to generate benchmark single-molecule data and characterize the unperturbed folding potentials, which will be crucial for comparison during the optimization of surface chemistries and immobilization methods.
Because multiple factors (protein structure, solvent viscosity, dye solvation, etc.) contribute to changes in dye and FRET properties, it is important to deconvolute the variations in these properties for a particular subpopulation (denatured state in this work) of protein molecules. Control experiments ideally should be carried out at single-molecule resolution on individual subpopulations of molecules. Further, simultaneous intensity, lifetime, and polarization anisotropy measurements will help to identify, quantify, and separate out contributions from κ2 and dynamic distance fluctuations. Here, the development of a detection system with four time-correlated single-photon counting Avalanche photodiodes, auto and cross-correlation methods, and the observation of long time trajectories of single immobilized molecules will be essential in future work to further clarify these complicated issues.
Acknowledgments
We are grateful to David King for high-resolution protein mass spectral analysis and David Wemmer for useful discussion. Financial support for this work was provided by the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory under U.S. Department of Energy Contract DE-AC03–76SF00098, Office of Naval Research Contract N0001498F0402, National Science Foundation Grant CHE-971–4390, National Institutes of Health Grants RO1 GM49220–09 and GM59380, and the Skaggs Institute for Chemical Biology. T.A.L. is supported by a National Science Foundation graduate fellowship.
Abbreviations
- CI2
chymotrypsin inhibitor 2
- FRET
fluorescence resonance energy transfer
- sp-FRET
single-pair FRET
- TMR
tetramethylrhodamine
- wt
wild type
- pwt
pseudo wt
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.090104997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.090104997
References
- 1.Dill K A, Shortle D. Annu Rev Biochem. 1991;60:795–825. doi: 10.1146/annurev.bi.60.070191.004051. [DOI] [PubMed] [Google Scholar]
- 2.Smith L J, Fiebig K M, Schwalbe H, Dobson C M. Fold Design. 1996;1:R95–R106. doi: 10.1016/S1359-0278(96)00046-6. [DOI] [PubMed] [Google Scholar]
- 3.Brooks C L., 3rd Curr Opin Struct Biol. 1998;8:222–226. doi: 10.1016/s0959-440x(98)80043-2. [DOI] [PubMed] [Google Scholar]
- 4.Alm E, Baker D. Curr Opin Struct Biol. 1999;9:189–196. doi: 10.1016/S0959-440X(99)80027-X. [DOI] [PubMed] [Google Scholar]
- 5.Muñoz V, Eaton W A. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galzitskaya O V, Finkelstein A V. Proc Natl Acad Sci USA. 1999;96:11299–11304. doi: 10.1073/pnas.96.20.11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alm E, Baker D. Proc Natl Acad Sci USA. 1999;96:11305–11310. doi: 10.1073/pnas.96.20.11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Onuchic J N, Luthey-Schulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 9.Weiss S. Science. 1999;283:1676–1683. doi: 10.1126/science.283.5408.1676. [DOI] [PubMed] [Google Scholar]
- 10.Deniz A A, Dahan M, Grunwell J R, Ha T, Faulhaber A E, Chemla D S, Weiss S, Schultz P G. Proc Natl Acad Sci USA. 1999;96:3670–3675. doi: 10.1073/pnas.96.7.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dahan M, Deniz A A, Ha T J, Chemla D S, Schultz P G, Weiss S. Chem Phys. 1999;247:85–106. [Google Scholar]
- 12.Beligere G S, Dawson P E. J Am Chem Soc. 1999;121:6332–6333. [Google Scholar]
- 13.Merrifield R B. J Am Chem Soc. 1963;85:2149–2154. [Google Scholar]
- 14.Schnölzer M, Alewood P, Jones A, Alewood D, Kent S B. Int J Pept Protein Res. 1992;40:180–193. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 15.Hackeng T M, Griffin J H, Dawson P E. Proc Natl Acad Sci USA. 1999;96:10068–10073. doi: 10.1073/pnas.96.18.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ha T, Ting A Y, Liang J, Caldwell W B, Deniz A A, Chemla D S, Schultz P G, Weiss S. Proc Natl Acad Sci USA. 1999;96:893–898. doi: 10.1073/pnas.96.3.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia Y, Talaga D S, Wai Leung L, Lu H S M, DeGrado W F, Hochstrasser R M. Chem Phys. 1999;247:69–83. [Google Scholar]
- 18.Ha T, Zhuang X, Kim H D, Orr J W, Williamson J R, Chu S. Proc Natl Acad Sci USA. 1999;96:9077–9082. doi: 10.1073/pnas.96.16.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 20.Oliveberg M, Tan Y J, Silow M, Fersht A R. J Mol Biol. 1998;277:933–943. doi: 10.1006/jmbi.1997.1612. [DOI] [PubMed] [Google Scholar]
- 21.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 22.Ladurner A G, Itzhaki L S, Daggett V, Fersht A R. Proc Natl Acad Sci USA. 1998;95:8473–8478. doi: 10.1073/pnas.95.15.8473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson S E, Fersht A R. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 24.Dawson P E, Muir T W, Clark-Lewis I, Kent S B. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 25.Mcphalen C A, James M N G. Biochemistry. 1987;26:261–269. doi: 10.1021/bi00375a036. [DOI] [PubMed] [Google Scholar]
- 26.Englert A, LeClerc M. Proc Natl Acad Sci USA. 1978;75:1050–1051. doi: 10.1073/pnas.75.3.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dale R E, Eisinger J, Blumberg W E. Biophys J. 1979;26:161–193. doi: 10.1016/S0006-3495(79)85243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lakowicz J R. Principles of Fluorescence Spectroscopy. New York: Plenum; 1999. [Google Scholar]
- 29.Miller W G, Goebel C V. Biochemistry. 1968;7:3925–3935. doi: 10.1021/bi00851a021. [DOI] [PubMed] [Google Scholar]
- 30.Haas E, Steinberg I Z. Biophys J. 1984;46:429–437. doi: 10.1016/S0006-3495(84)84040-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alonso D O, Dill K A. Biochemistry. 1991;30:5974–5985. doi: 10.1021/bi00238a023. [DOI] [PubMed] [Google Scholar]
- 32.Pollack L, Tate M W, Darnton N C, Knight J B, Gruner S M, Eaton W A, Austin R H. Proc Natl Acad Sci USA. 1999;96:10115–10117. doi: 10.1073/pnas.96.18.10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plaxco K W, Millett I S, Segel D J, Doniach S, Baker D. Nat Struct Biol. 1999;6:554–556. doi: 10.1038/9329. [DOI] [PubMed] [Google Scholar]
- 34.Nozaki Y. Methods Enzymol. 1972;26:43–50. doi: 10.1016/s0076-6879(72)26005-0. [DOI] [PubMed] [Google Scholar]
- 35.Jackson S E, Fersht A R. Biochemistry. 1991;30:10436–10443. doi: 10.1021/bi00107a011. [DOI] [PubMed] [Google Scholar]
- 36.Schaffer J, Volkmer A, Eggeling C, Subramaniam V, Striker G, Seidel C A M. J Phys Chem A. 1999;103:331–336. [Google Scholar]