

Abstract
The capacity of the corneal epithelium to adapt to hypertonic challenge is dependent on the ability of the cells to upregulate the expression and activity of cell membrane-associated Na-K-2Cl cotransporter1 (NKCC1). Yet, the signaling pathways that control this response during hypertonic stress are still unclear. We studied stress-induced changes in proliferation and survival capacity of SV40-immortalized human (HCEC) and rabbit (RCEC) corneal epithelial cells as a function of (i) the magnitude of the hypertonic challenge, (ii) differential changes in activation of mitogen-activated protein kinase (MAPK), and (iii) the extent of p38MAPK interaction with NKCC1. Cells were incubated in hypertonic (up to 600 mOsm) media for varying time periods up to 24 h. Phosphorylated forms of p44/42, p38, and stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) MAPK were immunoprecipitated from cell lysates, and the amount of each activated NKCC1-associated MAPK was evaluated by Western blot/ECL assay. DNA integrity was assessed by electrophoresis in a 2% agarose gel. Cell survival and proliferation were evaluated based on three criteria: protein content, cell count, and the MTT assay. Exposure to media of 325–350 mOsm increased proliferation of HCEC up to 75%, whereas this response was limited to <16% in RCEC. At higher osmolarities, cell proliferation decreased in both species. SAPK/JNK activity increased 150-fold in HCEC and <10-fold in RCEC, while DNA fragmentation occurred only in HCEC. Compared to HCEC, the better RCEC survival rate was associated with higher p38MAPK activity and near complete restoration of p44/42MAPK activity after the first 30 min. In both cell lines, the amount of phospho-NKCC1 that coimmunoprecipitated with phospho-p38MAPK was proportional to the magnitudes of their respective activation levels. However, no such associations occurred between amounts of phosphorylated p44/42MAPK or SAPK/JNK and phospho-NKCC1. Under isotonic conditions, with bumetanide-induced inhibition of RCEC and HCEC NKCC1 activities, p44/42MAPK activity declined by 40 and 60%, respectively. Such declines led to proportional decreases in cell proliferation. Survival of hypertonicity-stressed corneal epithelial cells depends both on p38MAPK activation capacity and the ability of p38MAPK to stimulate NKCC1 activity through protein-protein interaction. The level of NKCC1 activation affects the extent of cell volume recovery and, in turn, epithelial survival capacity.
Keywords: corneal epithelial cells, p38MAPK, p44/42MAPK, SAPK/JNK, NKCC1, regulatory volume increase, hypertonic challenge, cell proliferation
1. Introduction
The barrier function provided by the corneal epithelium is essential for preservation of normal vision. In this capacity, the corneal epithelium protects the corneal stroma from permeation of noxious and infectious agents. Changes in proliferation and death (i.e., apoptosis) of the corneal epithelial cells potentially compromises the efficacy of the barrier by disrupting tight junctional integrity between the cells, thus resulting in the loss of optical transparency (Lu et al., 2001).
During an osmotic challenge, cells are able to function normally, provided that they are able to sustain their volume within narrow (3%) boundaries; this is achieved through activation of various regulatory volume mechanisms (Bortner and Cidlowski, 1998). The specific mechanism that mediates maintenance of isotonic cell volume during hypertonic stress is referred to as regulatory volume increase (RVI) (Hoffmann and Dunham, 1995; Hoffmann and Simonsen, 1989; Strange, 2004). Such control is requisite for cell cycle progression and proliferation (Anbari and Schultz, 1993; Burg, 2002). Should such regulation be inadequate, cell proliferation declines and apoptosis may ensue (Bortner and Cidlowski, 1998; Maeno et al., 2000; Okada et al., 2001). Exposure to a hypertonic stress that doubles normal osmolarity is one of the well-known triggers of apoptosis (Rosette and Karin, 1996). Osmotic shrinkage directly activates the apoptotic machinery in a variety of cell lines (Burg, 2002; Lang et al., 2002; Moran et al., 2000; Terada et al., 2001), the effect being most prominent in cells that lack or have limited capacity for RVI (Bortner and Cidlowski, 1996; Bortner and Cidlowski, 1998). Apoptosis, which occurs predominantly in proliferating cells, includes the following successive stages: (i) reduction of cell volume and detachment from neighboring cells, (ii) mitochondrial dysfunction, (iii) condensation of chromatin and (iv) fragmentation of DNA (Kerr et al., 1972; King and Cidlowski, 1995; Michea et al., 2002).
The Na-K-2Cl cotransporter (NKCC), an essential component of the membrane ionic transport mechanism underlying RVI activation (Geck and Pfeiffer, 1985; Jensen et al., 1993), has two isoforms—NKCC1 and NKCC2 (Russell, 2000). The isoform mediating RVI in secretory epithelia, such as the corneal epithelium, is NKCC1. In SV40-immortalized rabbit corneal epithelial cells (RCEC), maintenance of regulatory volume mechanisms during chronic hypertonic conditions is dependent on upregulation and protein expression of the NKCC1 gene (Bildin et al., 2000). However, SV40-immortalized human corneal epithelial cells (HCEC) are unable to provide such a safeguard under similar conditions. This deficit is due to a much more rapid decline in proliferative activity in HCEC than in RCEC during exposure to chronic hypertonic stress. Such a limitation in HCEC is in concordance with their inability to maintain their RVI capacity during exposure to a hypertonic challenge.
Upregulation of NKCC1 function is dependent on an increase in its phosphorylation status, which may occur through stimulation of a limb of the mitogen-activated protein kinase (MAPK) superfamily or via a volume-sensitive kinase (Klein et al., 1999; Kurihara et al., 2002; Liedtke and Cole, 2002). Therefore, control of NKCC1 expression and activity is essential for chronic/acute hypertonic stress adaptation, which is required for timely restoration of the corneal epithelia’s barrier function in response to such challenges.
During hypertonic stress in corneal epithelia, the identity of and interplay between the signaling pathways that induce apoptosis are unclear. Differential activation by hypertonic stress of three major subfamilies of MAPK cascades determines cell survival and proliferative capacity. In most cell lines, two of these subfamilies—stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) and p38MAPK—induce cell cycle delay, cellular repair, or apoptosis in response to a hypertonic challenge (Kyriakis and Avruch, 1996). Another subfamily—p44/42 MAPK—is primarily responsible for transduction of mitogenic signals induced by growth factor; inhibition of p44/42MAPK activity accelerates cell death (Lee et al., 2005). However, in human limbal epithelial cells, hyperosmolarity-induced activation of the p44/42MAPK pathway induces inflammation (Li et al., 2006). These considerations suggest that the dynamic balance between the growth factor-activated p44/42MAPK pathway and the stress-activated p38MAPK and SAPK/JNK pathways is cell-type specific and may be critical for determining whether a cell survives or undergoes apoptosis (Xia et al., 1995). During exposure to an acute hypertonic challenge (i.e., 600 mOsm for 30 min), sufficient activation of the p38MAPK pathway is essential for maintenance of epithelial cell volume as well as for recovery of epithelial layer barrier function. On the other hand, restoration of p44/42MAPK activity seems to be dependent on the extent of cell volume recovery (Bildin et al., 2003). Neither the identity of nor the interactions between the hypertonic stress-activated signaling pathways that determine cell fate have been delineated in either HCEC or RCEC. Furthermore, the nature of the hypertonicity-induced MAPK pathway interaction with NKCC1 has not been described.
The present study demonstrates that 24-hour exposure of HCEC to moderate (up to 350mOsm) increases in tonicity stimulates cell proliferation but does not induce cell death; however, greater hypertonic stress does activate apoptosis. Survival of hypertonicity-stressed HCEC and RCEC depends on both the magnitude of p38MAPK activation and on stimulation of NKCC1 activity. These effects are associated with p38MAPK/NKCC1 protein-protein interaction, which are needed to elicit an adequate RVI response for survival.
2. Materials and methods
2.1. Cell culture and treatment
SV40-adenovirus-immortalized HCEC and RCEC (generous gifts from Dr. Araki-Sasaki, Kumamoto University Kumamoto, Japan) were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 5 ng/ml epidermal growth factor (EGF), 5 μg/ml insulin, and an antibiotic mixture of penicillin and streptomycin. Cells were grown in an atmosphere of 5% CO2 and 95% ambient air at 37°C. Tonicity of the media was increased by adding NaCl or sucrose.
2.2. Cell proliferation analysis
Cell proliferation was assessed by three separate methods: protein content measurement, cell count, and the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. To monitor proliferation by protein content, cells exposed to hypertonic (325–600 mOsm) media for 24 h were lysed and protein amount measured using the Micro BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA), as previously described, and normalized to the protein amount under isotonic conditions. For cell count assays, cells were plated in 60-mm plates, as described above, and cultured in a 300 mOsm medium for 24 h. Cells were then exposed to hypertonic (325–600 mOsm) media. Trypsin-EDTA (0.05%) was used to detach cells from the plates. An aliquot of this cell suspension was taken, and cells were counted in a hemocytometer and exposed to trypan blue to monitor viability. Measurement of mitochondrial succinate dehydrogenase (SDH) activity was achieved using the MTT assay kit according to manufacturer’s instructions (American Type Culture Collection, Manassas, VA, USA). In brief, corneal epithelial cells were seeded in 96-well plates (Falcon) at a density of 104 cells/well in isotonic (300 mOsm) medium for 24 h. Isotonic medium was replaced by hypertonic medium, as indicated above, for an additional 24 h. Cells were incubated with 10 μl MTT reagent for 2–4 h at 37°C. Following purple precipitate development, cells were solubilized by adding DTT detergent reagent (100 μl) to each well and kept in the dark overnight. Absorbance at 570 nm was measured using a Fusion™ Universal Microplate Analyzer (Perkin-Elmer, Boston, MA, USA).
2.3. DNA fragmentation ladder assay
Genomic DNA was isolated using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA), as described by the manufacturer. DNA integrity was analyzed by electrophoresis in high-resolution 2% agarose gel (Bortner et al., 1995). Apoptosis was evaluated based on DNA fragmentation (“laddering”), which was quantified using Sigmagel software (Jandel Scientific, San Rafael, CA, USA).
2.4. Cell lysate preparation
Lysates were prepared using a previously described method, with slight modifications (Bildin et al., 2000). In brief, cells were plated at a density of 105 cells in 60-mm culture plates (Fisher Scientific, Pittsburgh, PA, USA) and grown to 60–70% confluence. Cells were deprived of serum and growth factors for 24 h before treatment. After exposure to hypertonic media, cells were washed twice with phosphate-buffered saline (PBS) and homogenized on ice with homogenization buffer—20 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1.5 mM EGTA, 2.5 mM sodium pyrophosphate, 10 mM β-glycerolphosphate, and 1 mM Na3VO4)—supplemented with a protease inhibitor mixture—1 mM benzamidine, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were sonicated (three times at 40 mV for 10 s each) and centrifuged at 12 000 rpm for 15 min at 4°C to pellet cellular debris. To obtain plasma membrane-enriched fractions, the supernatant was centrifuged at 32 000 rpm for 30 min at 4°C. The resulting pellets were resuspended in 100 μl of 2% sodium dodecylsulphate (SDS). The total protein concentrations of the resulting whole cell and membrane-enriched fraction lysates were measured with the Micro BCA protein assay kit. Lysates were dissolved in equal volumes of Laemmli sample buffer—12.5 mM Tris/HCl (pH 6.8), 4% SDS, 20% glycerol, 2.5% β-mercaptoethanol, and 0.1% bromophenol blue.
2.5. Kinase activity assay and immunoprecipitation
Activity of p38MAPK, p44/42MAPK, and SAPK/JNK kinases was evaluated using kinase assay kits in accordance with manufacturer’s instructions (Cell Signaling Technology, Inc., Beverly, MA). Immunoprecipitation experiments were performed as previously described (Du et al., 2006). In brief, corneal epithelial cells grown in 60-mm culture plates were exposed to isotonic media (300 mOsm) in the presence or absence of 50 μM bumetanide, or to hypertonic media for the indicated time period. After harvesting the cells, a sample was centrifuged at 13 000 rpm at 4°C for 15 min. Cell lysates were subjected to immunoprecipitation. Supernatant was pre-cleared with 50 μl of 50% vol/vol of Proteins A and G (Santa Cruz Biotechnology) with the appropriate control IgG for 30 min at 4°C, followed by centrifugation at 2500 rpm for 30 s. The Micro BCA protein assay kit was used to determine protein concentration. Equal amounts of protein were subjected to immunoprecipitation with R5 anti-phospho-NKCC1 antibody (generously provided to us by B. Forbush, Yale University). In vitro p38MAP kinase activity was measured in equal amounts of protein, immunoprecipitated with immobilized p38MAPK (Thr180/Tyr182) antibody. The kinase reaction was performed in the presence of ATF2 fusion protein and cold ATP. Phosphorylation of ATF2 was measured by Western blot using phospho-ATF2Thr71. For extracellular signal-regulated kinase (Erk)1/2 activity, Elk-1 (Ser383) was used as the substrate following immunoprecipitation with immobilized anti-Erk1/2/ antibody. With the JNK assay, equal amounts of protein were immunoprecipitated with c-Jun (Ser63) fusion protein beads overnight. After washing the beads, JNK kinase assays were performed according to manufacturer’s instructions (New England Biolabs, Ipswich, MA). Samples were loaded on an SDS-polyacrylamide gel, and immunoblotting was performed with an antibody against phospho-specific c-Jun. Control immunoprecipitation was performed with an equivalent amount of cell extract in the absence of primary antibody. Immunoprecipitated proteins were subjected to Western blot analysis.
2.6. Western blot analysis
Western blot analysis was performed as previously described (Bildin et al., 2000). Briefly, resolved proteins were electrotransferred overnight to PVDF membranes. After blocking with nonfat dry milk, blots were exposed for 2 h at room temperature to T4 anti-NKCC1 (Developmental Studies Hybridoma Bank, University of Iowa, IA, USA) (1:10 000), R5 anti-phospho-NKCC1 (1:5000), and anti-p38 (Cell Signaling, Beverly Technology, Inc., MA, USA), or to either phospho-specific monoclonal antibody ERK1/2 (Tyr-204), p38MAPK (Thr-180/Tyr-182), Elk-1, ATF-2, or c-Jun (Cell Signaling, Beverly Technology, Inc., MA, USA). Immunoreactive bands were detected with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000) and the ECL Plus kit (Amersham Biosciences, Piscataway, NJ, USA). MAPK activities were quantified by scanning the X-ray film and quantifying band density using Sigmagel software (Jandel Scientific, San Rafael, CA, USA). Exposed membranes were stained with colloidal gold (Bio-Rad Laboratories, Hercules, CA, USA) to verify that each lane contained a similar amount of protein.
3. Results
3.1. Effects of hypertonicity on HCEC and RCEC proliferation and death
To assess whether increases in medium tonicity affect proliferation and viability of HCEC and RCEC, we measured the effects of a 24-h increase in medium tonicity on cell protein content, cell number, and mitochondrial activity during the cells’ logarithmic growth phase. The results shown in Figures 1A and B indicate that biphasic changes occurred in HCEC, as determined by protein content and cell count, respectively. At tonicities of 300–350 mOsm, HCEC cell proliferation was stimulated by up to 175%; at higher tonicities, cell proliferation was progressively suppressed and cell death observed. On the other hand, RCEC were nearly insensitive to changes in medium tonicity up to 375 mOsm; however, at higher tonicities, cell death occurred. Nevertheless, it should be noted that at 600 mOsm, the decline in RCEC viability, as assessed by protein measurement, was significantly less than that of HCEC—5 and 56%, respectively (Fig. 1A). Cell counts of RCEC and HCEC revealed comparable viability (2 and 54%, respectively) (Fig. 1B). These results correspond to the individual decline reported for Erk1/2 activity in hypertonicity-stressed HCEC (Bildin et al., 2003). Further, they are in agreement with our previous finding that the RVI capacity of RCEC declines less than does that of HCEC during exposure to such challenges (Bildin et al., 2000). Another commonly used approach for evaluating cell proliferation and viability is the MTT test, which monitors mitochondrial SDH activity. Figure 1C shows that mitochondrial activity—normalized to cell number—increased at values >350 mOsm in both cell lines. It is noteworthy that, at values up to 350 mOsm, there was ~15% decline of mitochondrial activity normalized to protein content in HCEC (Fig. 1C), even though the cell numbers rose (Figs. 1A and B). However, at values >375 mOsm, increases in HCEC mitochondrial activity normalized to protein content were much greater than those for RCEC.
Fig. 1.
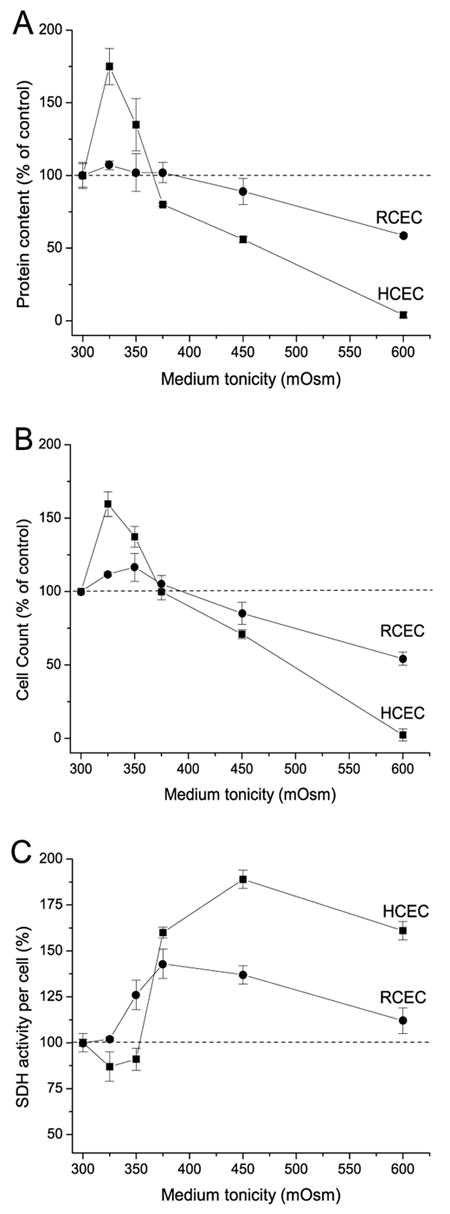
Hypertonicity-induced species-specific changes in corneal epithelial cell proliferation. HCEC and RCEC, initially cultured in 300 mOsm medium, were subsequently exposed to hypertonic (325–600 mOsm) media for an additional 24 h. (A) Protein content normalized to the protein amount under isotonic conditions (%) was used as an index of cell number. (B) Cell proliferation status, as determined by cell count with the aid of a hemocytometer and trypan blue to monitor viability. (C) Hypertonicity-dependent changes in SDH activity, normalized to protein content. SDH activity was evaluated based on results of the MTT assay. Cell viability was defined as a ratio (%) of mitochondrial SDH activity of treated cells to untreated cells. Data are presented as the mean ±SEM of three independent experiments.
To evaluate whether there are differences between HCEC and RCEC resistance to different kinds of osmotic stress, we compared protein contents of cells and mitochondrial activity after 4 h of incubation in a 600 mOsm medium supplemented with either NaCl or sucrose. Figure 2A shows that the protein content of RCEC was more invariant than that of HCEC, suggesting that RCEC are better able to withstand hyperosmolar stress than are HCEC. Only in HCEC, 4 h exposure to hyperosmolar conditions induced by NaCl supplementation resulted in a greater suppression of cell survival than that induced by sucrose—70 and 50%, respectively. However, these results are not reflective of those obtained with the MTT test. In HCEC, there was an initial increase (up to 60%) in MTT activity; in RCEC, MTT activity decreased by 50% after the first 30 min and remained unchanged thereafter. The disparity in results shown in Figures 1 and 2 suggests that the MTT test reflects biphasic changes in mitochondrial activity in dying cells. This test is, therefore, of limited value for evaluating the effect of hypertonic stress on cell proliferation and viability. In other words, the MTT test is reflective only of mitochondrial activity, which is not always an indicator of cell number.
Fig. 2.
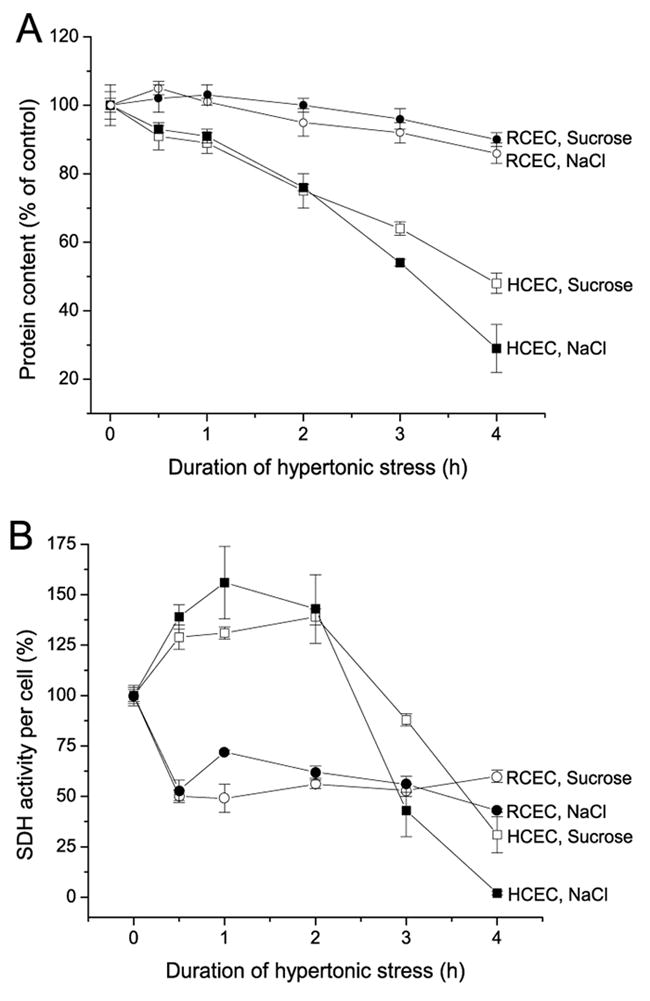
Time-dependent effects of hyperosmolarity on RCEC and HCEC proliferation. Corneal epithelial cells were initially cultured in a 300 mOsm medium, which was replaced by 600 mOsm medium, in which cells were cultured for up to 4 h. Osmolarity was increased by supplementation with NaCl or sucrose. (A) Percentage of total protein from treated cells normalized to the protein content under isotonic conditions, as an indicator of cell proliferation. (B) Time-dependent changes in SDH activity normalized to protein content. Cell viability was defined as the ratio (expressed as a %) of mitochondrial SDH activity of treated cells to untreated cells. Experiments were performed in triplicate for each osmolarity. Values are reported as means ±SEM.
3.2 Relationship between hypertonicity-induced HCEC and RCEC death and differential activation of SAPK/JNK
We investigated (i) whether there is a correlation between development of apoptosis in HCEC and RCEC and (ii) the adaptive capabilities of HCEC and RCEC during a 6 h exposure to hypertonic stress. This was accomplished by analyzing DNA integrity after exposure of cells to a hypertonic (NaCl, 600 mOsm) medium. Figure 3 (inset) shows the time-dependent losses of HCEC DNA integrity. However, DNA laddering was not observed in RCEC. Quantification of these data (Fig. 3) demonstrates that after a 6 h exposure to hypertonicity, nearly 50% of HCEC DNA was fragmented. These results indicate that hypertonicity-induced apoptosis develops more rapidly in HCEC than in RCEC, a finding consistent with the lesser capability of HCEC to sustain their RVI capacity and proliferate in a hypertonic environment.
Fig. 3.
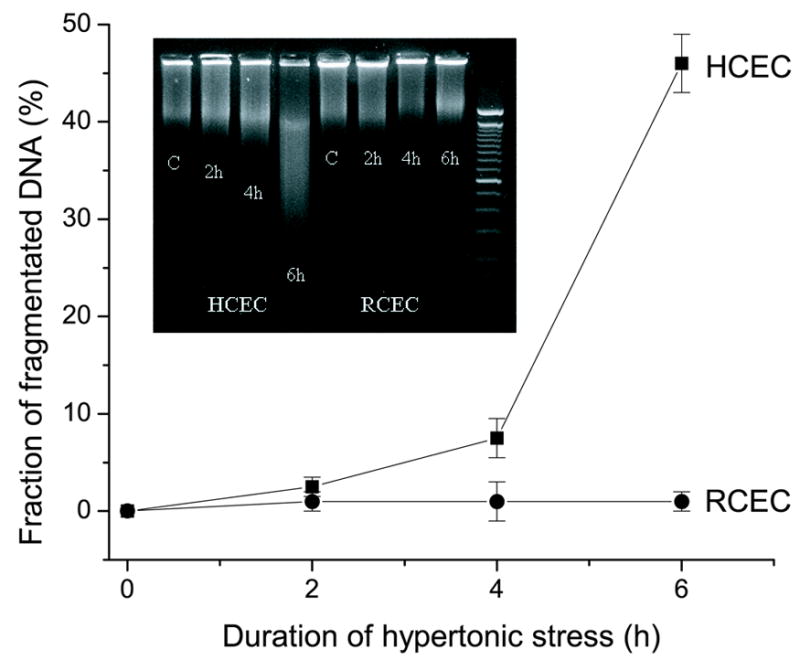
Comparison of hypertonicity-induced DNA fragmentation in HCEC and RCEC. DNA integrity was analyzed by high-resolution 2% agarose gel electrophoresis following exposure of RCEC and HCEC to 600 mOsm medium for up to 6 h. Experiments were performed in triplicate for each osmolarity. Values are reported as means ±SEM. Inset: Representative DNA fragmentation blot.
Figure 4 (insets A and B) shows a representative Western blot analysis of the time dependence of SAPK/JNK activation in HCEC and RCEC, respectively. After the indicated periods of exposure to hypertonic media, cells were harvested and lysed. Then, the N-terminal portion of the c-Jun transcription factor was used to selectively “pull down” SAPK/JNK from cell lysates. This was followed by separation on SDS-PAGE. Activity of SAPK/JNK was measured on the basis of its ability to phosphorylate c-Jun. Figure 4 (inset A) demonstrates that HCEC SAPK/JNK activity increased ~150-fold during the first 4 h of hypertonic stress and continued to increase for another 2 h. At the same time, the maximum increase of RCEC SAPK/JNK activity remained <10-fold (Fig. 4, inset B). The correspondence between the time courses for initiation of apoptosis and activation of SAPK/JNK suggests this signaling pathway to be a mediator of this response.
Fig. 4.
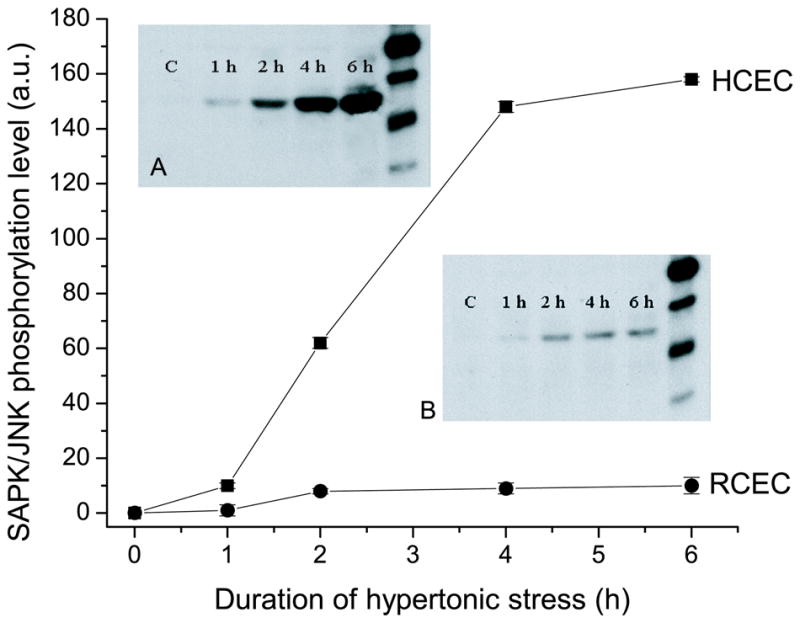
Time-dependent effects of hypertonicity on SAPK/JNK activity. RCEC and HCEC were exposed to 600 mOsm medium for up to 6 h. SAPK/JNK was pulled down by the N-terminal portion of the c-Jun transcription factor, and its activity evaluated based on its ability to phosphorylate c-Jun. Inset: Representative Western blot of typical experiments performed on “pull-downs” obtained from HCEC (inset A) and RCEC (inset B) lysates. Data are presented as the mean ±SEM of independent experiments carried out in triplicate for each osmolarity.
3.3 Dependence of p44/42MAPK, p38MAPK and SAPK/JNK activities on NKCC inhibition
Increases in NKCC1 activity and expression are important factors in maintaining isotonic cell volume during the cell’s exposure to a hypertonic challenge. Proper isotonic maintenance depends on the ability of cells to upregulate NKCC1 gene expression under osmotic stress (Schliess et al., 2002). Conversely, the failure of cells to adequately adapt to osmotic stresses is associated with declines in NKCC1 phosphorylation status or activity (O'Donnell et al., 1995). The resulting cell shrinkage can lead to the development of apoptosis (Friis et al., 2005). Accordingly, we evaluated whether bumetanide-induced inhibition of NKCC1 is responsible for the cell signaling changes that produce alterations in cell proliferation, ultimately leading to programmed cell death. Figure 5 (A–C) demonstrates typical changes in HCEC and RCEC Erk1/2 (i.e., p44/42MAPK), p38MAPK, and SAPK/JNK activities after 5 min of exposure to isotonic medium in the absence and presence of 50 μM bumetanide. In each figure, the upper panel shows the effect of NKCC1 inhibition on the ability of “pulled down” kinases to selectively phosphorylate Elk-1, ATF-2, and c-Jun. Graphic representation of these data shows that there were (i) >40 and 60% declines in p44/42MAPK activity and (ii) greater than four- and three-fold increases in p38MAPK activity in RCEC and HCEC, respectively. On the other hand, SAPK/JNK was only activated by ~25% in both cell lines. These results are in agreement with previous data that indicate that cell shrinkage induced by NKCC1 inhibition leads to (i) declines in Erk1/2 activity and cell proliferation and (ii) activation of p38MAPK and RVI, resulting in recovery of cell volume (Bildin et al., 2003; Bildin et al., 2000). Such restoration is sufficient to stimulate Erk1/2 activity and increase cell proliferation.
Fig. 5.
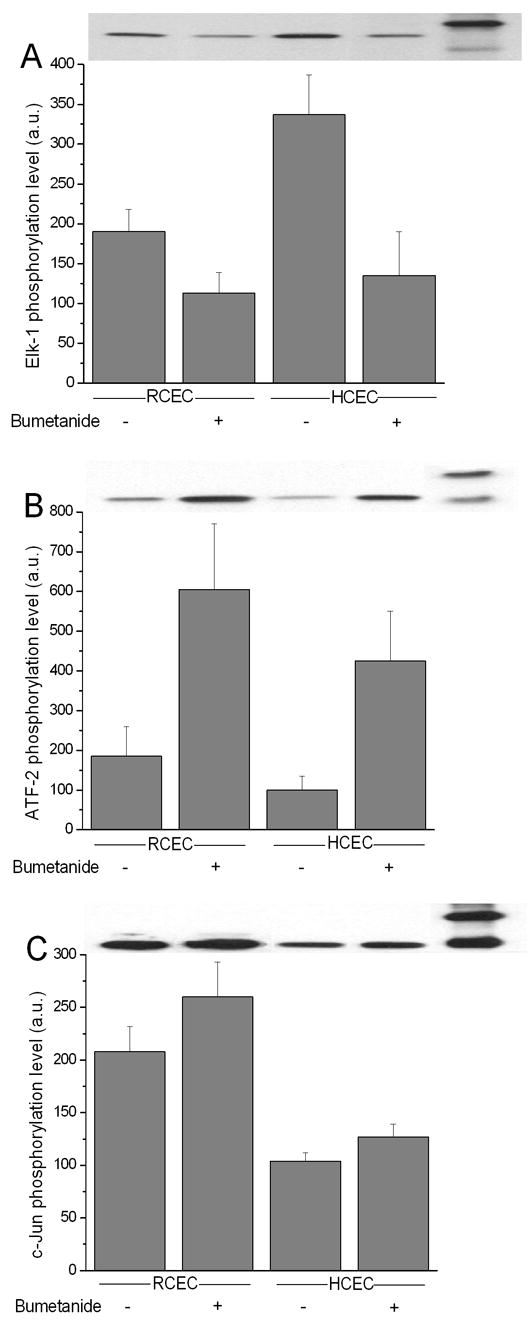
Changes in MAPK limb activities in RCEC and HCEC induced by inhibition of NKCC1. Under isotonic conditions, RCEC and HCEC were exposed to 50μM bumetanide for 5 min. Activities of p44/42, p38, and SAPK/JNK MAPK were measured based on results of Western blot analysis of the phosphorylation status of Elk-1 (A), ATF-2 (B), and c-Jun (C), respectively. Summaries of experiments performed in triplicate and values are reported as means ±SEM. Representative Western blot of typical experimental results are shown above each summary figure.
3.4 Interaction between NKCC1 and p38MAPK in hypertonicity-stressed cells
It is possible that SAPK/JNK activation leads to apoptosis, whereas p38MAPK activation may—through its interaction with NKCC1—induce expression of an adaptive response to hypertonic stress. Our finding that there is an association between hypertonic stress-induced NKCC1 and p38MAPK activation led us to explore the nature of this interaction. Conceivably, this kinase merely activates NKCC1 via phosphorylation. Another possibility is that p38MAPK is involved in inducing NKCC1 trafficking to the cell membrane. Accordingly, we determined if acute hypertonic stress leads to changes in (i) association of p44/42MAPK, p38MAPK, and JNK/SAPK with a cell membrane-enriched fraction containing NKCC1 and (ii) amount of coimmunoprecipitation of NKCC1 with these phosphorylated kinases. Figure 6A (upper panel) shows a representative Western blot comparing time-dependent accumulation of NKCC1 in a cell membrane-enriched fraction, isolated from HCEC and RCEC, following exposure to 600 mOsm hypertonic stress. Figure 6B summarizes the results obtained from such experiments. Under isotonic conditions, RCEC contains ~two-fold greater total NKCC1 protein content than does HCEC. Hypertonic conditions induced a 2.4-fold increase in the amount of total NKCC1 in HCEC after 30 min exposure. In RCEC, there was only a 30% increase in cotransporter accumulation. Figure 6A (lower panel) shows the time-dependent changes in amounts of total p38MAPK associated with membrane samples of the same membrane-enriched fraction. Quantification of both cell lines shows that there were similar increases (~5.5-fold) in total p38MAPK/cell membrane association after the first 5 min (Fig. 6C). However, in RCEC, such associations declined four-fold after 30 min of hypertonic challenge, whereas there were no significant changes in HCEC. There were no traces of SAPK/JNK in cell membrane-enriched fractions, and the insignificant p44/42MAPK contamination of the samples remained unchanged during hypertonic exposure (data not shown). Taken together, these data document that there are hypertonic stress-induced increases in cell membrane-associated total p38MAPK and NKCC1. However, kinetics and patterns of changes in association differ between the two cell lines. Despite the higher expression level of RCEC NKCC1 under non-stimulated conditions, hypertonic stress triggers more translocation of NKCC1 to HCEC plasma membrane than to RCEC plasma membrane. These results suggest that the adaptive response of RCEC to hypertonic challenge does not result from increased membrane accumulation of total NKCC1 or p38MAPK during such stress.
Fig. 6.
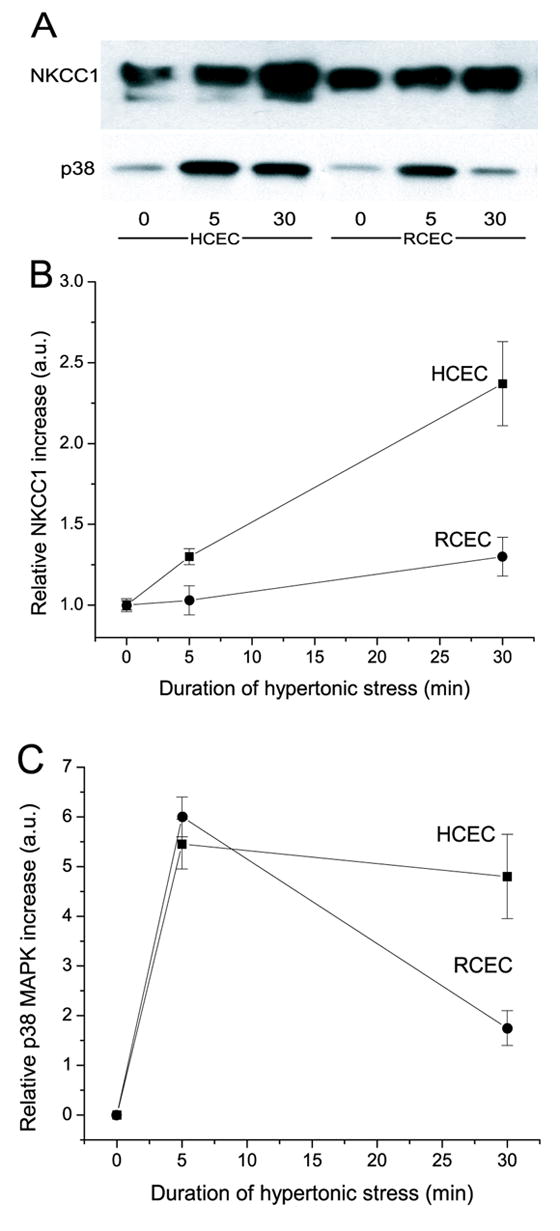
Time-dependent changes in RCEC and HCEC cell membrane-associated NKCC1 and p38MAPK content. Cells were exposed to 600 mOsm stress for up to 30 min. Plasma membrane-enriched fraction was assayed for total NKCC1 and p38MAPK content using Western blot analysis. (A) Blots from a typical experiment. Upper panel indicates NKCC1 content detected with T4 antibody. Bottom panel shows p38MAPK content using anti-p38MAPK antibody. Panels (B) and (C) provide graphic summaries of the representative results shown in panel (A). Experiments were performed in triplicate at each osmolarity. Results are reported as means ±SEM.
To verify if, in hypertonicity-stressed cells, interaction of p38MAPK with NKCC1 is dependent on kinase activity, we immunoprecipitated phospho-p38MAPK and analyzed its ability to phosphorylate substrate (ATF-2) and varying amounts of coimmunoprecipitated NKCC1. Figure 7A and B show the changes in p38MAPK activity and phospho-p38MAPK/NKCC1 interaction in HCEC and RCEC, respectively, during exposure for 28 min to hypertonic stress. There was a 20-fold increase in p38MAPK activity and a nearly eight-fold rise in the amount of phospho-NKCC1 that associated with phospho-p38MAPK in HCEC after 8 min (Fig. 7A). This trend was maintained for up to 28 min, with insignificant changes in p38MAPK activity and interaction with NKCC1. There was a remarkable (>90-fold) increase in p38MAPK activity and an increase of nearly 40-fold in its association with NKCC in RCEC after 2 min. Thereafter, in RCEC, there was a ~4.5-fold decrease in both p38MAPK activity and in amounts of associated phospho-NKCC1. No significant amounts of phosphorylated p44/42MAPK and SAPK/JNK coimmunoprecipitated with p38MAPK/NKCC1. Additionally, phospho-NKCC1 was never “pulled-down” by anti-phospho-p44/42MAPK or SAPK/JNK monoclonal antibodies. These results suggest that the p38MAPK/NKCC1 interaction is strongly related to p38MAPK phosphorylation status, and that hypertonicity-induced NKCC1 activation is dependent on that interaction. Therefore, in RCEC, the initial hypertonicity-induced increases in p38MAPK activity and its association with NKCC1 were ~four- and six-fold greater, respectively, than those observed in HCEC. These large differences between HCEC and RCEC in p38MAP kinase activation and NKCC1 association dwindled after 28 min, but were still evident.
Fig. 7.
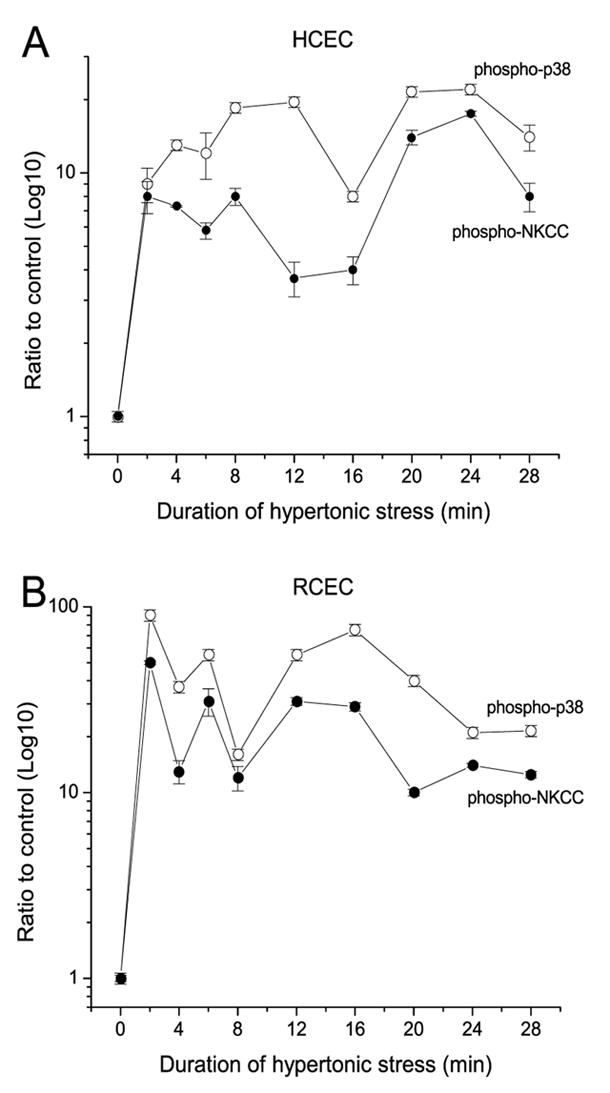
Hypertonicity-induced changes of p38MAPK activity and NKCC1 content in RCEC and HCEC cell membrane-enriched fractions. Cells were exposed to 600 mOsm stress for up to 28 min, and assayed at each of the indicated times for phosphorylated p38MAPK and NKCC1. Results for HCEC (A) and RCEC (B) are plotted on a semi-logarithmic scale and normalized to the control as a function of time. Experiments were performed in triplicate for each osmolarity. Data are reported as means ±SEM.
4. Discussion
In this study, SV40-immortalized RCEC and HCEC were used, rather than primary cultures of their counterparts. The results obtained with these lines are highly relevant, since previous studies indicate that transformation does not alter growth factor control of proliferation, cell cycle progression, or phenotypic cytokeratin profile expression (Kang 2001; Araki-Sasaki et al, 1993; Araki-Sasaki, 1995). Another consideration for the validity of their use is that physiological responses to challenges are more consistent in these cell lines, since the cells are in a more uniform functional state prior to reaching confluence than are those in primary culture.
Accordingly, the effects of a 24-h exposure to hypertonic stress (325–600 mOsm) on proliferation and viability of HCEC and RCEC were evaluated based on measurements of protein content, cell count, and results of the MTT test. In HCEC, there was ~75% stimulation of cell proliferation during incubation in media of 350 mOsm (Fig. 1). Such stimulation could be related to the increases in NKCC1 activity induced by this hypertonic challenge, since NKCC1 activation promotes proliferation. The dependence of an increase in proliferation on NKCC1 activation is indicated by the finding that this response is suppressed by bumetanide (Bildin et al., 2000). This association was described in human skin fibroblasts, in which stimulation of bumetanide-sensitive NKCC by different mitogens was shown to be essential for cell proliferation (Panet and Atlan, 1991), and the NKCC inhibitors—bumetanide and furosemide—were shown to inhibit cell proliferation (Panet et al., 1994). Additional evidence for the dependence of proliferation on amount of NKCC1 comes from the finding that gene overexpression augments cell proliferation in human skin fibroblasts (Panet et al., 2000). In the current study, however, the association between tonicity-induced increases in NKCC1 activity and proliferation is species-specific. Only in HCEC did a relatively small increase in tonicity (up to 350 mOsm) stimulate proliferation, whereas RCEC were insensitive to changes of up to 400 mOsm. A possible explanation for this disparity is that the RCEC NKCC1 plasma membrane content is much higher than in HCEC under isotonic conditions (Fig. 6A). This difference is reflected in the observation that RCEC recover their relative isotonic cell volume more completely and more rapidly (1.6-fold) than do HCEC during acute hypertonic challenge (Bildin et al., 2000). Such a difference in extent and rapidity of recovery suggests that RCEC restore cell contacts more rapidly than do HCEC. Restoration of cell contacts, in turn, suppresses proliferation and maintains the cells in a quiescent state. It is conceivable that a delay in restoration of cell contacts in HCEC accounts for why the ERK pathway activation was only observed in HCEC (data not shown).
At tonicities >375 mOsm, progressive cell death occurred first in HCEC and then in RCEC. These data are in agreement with those of other mammalian cell reports, which indicate that osmotic shrinkage slows cell proliferation as result of (i) blocks at the G1 and G2/M checkpoints, and (ii) S-phase prolongation (Burg, 2002; Dmitrieva et al., 2001; Michea et al., 2000). Increases in osmolality to levels >550 mOsm/kg induced apoptosis. However, at 600 mOsm, the decline in viability of RCEC was significantly less than that of HCEC—5 and 56%, respectively—as determined by protein content measurement. This ability of RCEC to better withstand such a challenge is consistent with the capacity of RCEC to upregulate NKCC1 mRNA and protein expression; HCEC did not induce such a response (Bildin et al., 2000).
Three of the many indices used to evaluate cell proliferation rates are measurements of protein content, cell count, and characterization of mitochondrial activity (the MTT test). However, our results indicate that MTT test is not an accurate indicator of cell proliferation under the studied conditions. The discrepancy lies in the fact that increases in mitochondrial activity were not correspondingly associated with changes in protein amounts and cell number (Figs. 1 and 2). At concentrations <375 mOsm, protein content and cell number (Figs. 1A and B) declined. However, the ratio of mitochondrial SDH activities to protein content increased (Fig. 1C). Another disparity is that in the 300–350 mOsm range, proliferation—based on protein content and cell number—increased, while the normalized MTT assay did not reflect such an increase. In other words, for HCEC in <350 mOsm medium, the MTT level fell ~15%, even though Figures 1A and B show that the cell number rose (up to 75%). Furthermore, >375 mOsm, HCEC increases in SDH activity per cell were much greater than those of RCEC, even though protein content and cell number (i.e., cell viability) declined more markedly in HCEC. Similar types of discrepancies are evident from the results shown in Figures 2A and 2B. A tenable explanation for why changes in mitochondrial activity do not necessarily parallel those of proliferation is based on a study performed in cultivated yeast. In that study, increases in medium osmolarity over the same range employed in this study led to a five-fold activation of 6-phosphofructo-2-kinase and, ultimately, increases in SDH activity, with no corresponding effect on proliferation (Dihazi et al., 2004). Another example of a disparity between the results obtained with the two assays is that decreased MTT-reducing activity was observed in human endothelial cells 1 h immediately following inhibition of NADPH oxidase activity, whereas cell viability was invariant, as estimated by other methods (Balcerczyk et al., 2005). These considerations, coupled with those in this study, indicate that the MTT assay is not an accurate measure of proliferation, since mitochondrial activity can increase despite no change (or even a decline) in proliferation. Another example in which the MTT assay results do not mirror changes in proliferation is that mitochondrial activity increases prior to apoptosis. Therefore, the MTT assay should not be used to evaluate changes in cell viability resulting from conditions that could induce apoptosis.
Increasing medium osmolarity to >500 mOsm can trigger apoptosis, as indicated by nucleosomal DNA laddering (King and Cidlowski, 1995; Michea et al., 2000; Rosette and Karin, 1996; Terada et al., 2001). The DNA fragmentation ladder assay shown in Figure 3 reveals a progressive time-dependent increase in HCEC DNA fragmentation. After 6 h incubation in 600 mOsm, ~50% of the HCEC DNA was fragmented. However, such an effect was not observed in RCEC. The earlier onset of apoptosis in HCEC than in RCEC is consistent with the lower capacity of HCEC to undergo RVI and to proliferate in a hypertonic medium (Bildin et al., 2000). The greater resilience of RCEC versus RCEC to withstand hypertonic challenges is in agreement with findings from other studies that indicate that maintenance of cell volume regulatory capacity correlates with resistance to apoptotic cell death during hypertonic stress. Only cells that exhibit a loss in cell volume degrade their DNA (Bortner and Cidlowski, 1996). Although nonlymphoid cells lines (e.g., L cells, COS, HeLa, and GH3) showed a similar initial reduction in cell volume in response to hypertonic conditions, they subsequently maintained volume or regulated back to a near normal cell volume. In other words, an essential adaptation to hypertonic stress is a sustained cell volume regulatory capacity, since offsetting shrinkage protects against induction of apoptosis.
The characterization of stress-activated signaling pathways that induce regulatory volume responses or apoptosis is complex for a variety of reasons. For one thing, there may be crosstalk between the various pathways, coupled with the possibility that the same pathway may mediate different responses in different species. These pathways include MAPK cascades, which are triggered by osmotic shrinkage to induce apoptosis (Lang et al., 1998). Both ERKs and the stress-activated kinase p38 have been implicated in controlling cell survival (Junger et al., 2003). On the other hand, a number of other reports suggest that SAPK/JNK pathway activation is an important determinant for inducing cell survival or apoptosis (Malek et al., 1998; Reinehr et al., 2002; Xia et al., 1995). Accordingly, we analyzed the time dependence of SAPK/JNK activation in HCEC and RCEC. As can be seen in Figure 4, SAPK/JNK activity increased maximally >150-fold after the first 4 h of exposure to hypertonic stress in HCEC. However, in RCEC, this effect was limited to an increase of <10-fold even after 6 h. The correspondence between the time courses of SAPK/JNK activation and apoptosis development (DNA fragmentation shown in Fig. 3) suggests that this signaling pathway mediates this response. The inability of HCEC to restore their relative isotonic cell volume through RVI activation during acute hypertonic exposure leads to programmed cell death. This finding is consistent with those showing that osmotic shrinkage directly impinges on the apoptotic machinery in a variety of cell types (Burg, 2002; Lang et al., 2002; Moran et al., 2000; Terada et al., 2001). The relationship between osmotic shrinkage and apoptosis induction seems to be the most pronounced in cells that are incapable of offsetting osmotic-induced shrinkage through RVI (Bortner and Cidlowski, 1996).
During exposure to a hypertonic challenge, RCEC adapt better than do HCEC. This is because RCEC are able to effectively offset shrinkage through NKCC1 upregulation of gene protein expression and ion transport activity (Bildin et al., 2000). Here, we determined if, under isotonic conditions, bumetanide-induced inhibition of NKCC1 activity, followed by cell shrinkage, induces changes in cell signaling that either compensate for losses in NKCC1 activity by maintaining proliferative capacity or induce programmed cell death. Figure 5 (A–C) contrasts changes in HCEC and RCEC Erk1/2 (i.e., p44/42MAPK), p38MAPK, and SAPK/JNK activities after 5 min of cell exposure to isotonic medium in the absence and presence of 50 μM bumetanide. Even such a short-term blockage of NKCC1 activity caused 34 and 53% decreases in 86Rb uptake (Bildin et al., 2000), and >40 and 60% declines in p44/42MAPK activity in RCEC and HCEC, respectively (Fig. 5A). These declines also led to proportional decreases in proliferation of the two different cell types. At the same time, >four- and three-fold stimulation of p38MAPK activity in RCEC and HCEC, respectively, occurred (Fig. 5B). As we suggested, the extent and rate of recovery of p44/42MAPK activity and cell proliferation are dependent on the magnitude of p38MAPK activation and on the extent of restoration of cell volume (Bildin et al., 2003). On the other hand, the increases of SAPK/JNK in both cell lines were limited to only ~25%. This small increase suggests that this stress-activated kinase is not directly involved in mediating NKCC1-induced cell volume regulation. Taken together, these results and previous data are in agreement with one another, and show that the cell shrinkage induced by NKCC1 inhibition leads to (i) declines in Erk1/2 activity and cell proliferation and (ii) p38MAPK and RVI activation, resulting in recovery of cell volume. Such restoration is sufficient to induce Erk1/2 activity and cell proliferation.
Our findings that there is an association between hypertonic stress-induced p38MAPK and NKCC1 activation led us to investigate the nature of this interaction. It is conceivable that this kinase activates NKCC1 by direct phosphorylation or that p38MAPK is involved in inducing NKCC1 trafficking to the cell membrane. Figure 6A documents the time-dependent accumulation of total NKCC1 in cell membrane-enriched fractions isolated from HCEC and RCEC. Surprisingly, during exposure to 600 mOsm hypertonic medium, the rate of NKCC1 accumulation into this fraction was >1.6-fold higher in HCEC than in RCEC. However, in isotonic medium, the NKCC1 content was ~two-fold greater in RCEC than in HCEC; this difference closely parallels the difference in NKCC1 mRNA amounts between these cell lines (Bildin et al., 2000). In contrast, the results shown in Figure 6A indicate that cell membrane fraction-associated total p38MAPK increased ~5.5-fold after the first 5 min in both cell lines. Regarding NKCC1 mobilization into this fraction, the increase was ~two-fold higher in HCEC than that in RCEC. In addition, after 30 min of exposure, the relative amounts of total p38MAPK associated with the cell membrane-containing fraction isolated from HCEC membrane remained nearly unchanged, whereas it fell nearly four-fold in RCEC. However, NKCC1 trafficking into this cell membrane-enriched fraction remained unchanged in both cell lines (Fig. 6A). Taken together, these results make it unlikely that p38MAPK is involved in the NKCC1 trafficking necessary to activate RVI in response to hypertonic stress.
To evaluate if, in hypertonicity-stressed cells, interaction of p38MAPK with NKCC1 is dependent on its kinase activity, phospho-p38 from cell lysates were immunoprecipitated. Thereafter, analyses were performed to determine (i) p38MAPK’s ability to phosphorylate a substrate (ATF-2) and (ii) if phospho-NKCC1 coimmunoprecipitates in the same membrane-containing fraction. After 8 min in HCEC, there was a 20-fold increase in p38MAPK activity and a nearly eight-fold rise in the amount of phospho-NKCC1 associated with this kinase (Fig.7A). These values—with small deviations—remained unchanged for the next 20 min. In the first 2 min, p38MAPK activation reached a maximum of 90-fold above the pre-stressed level for RCEC. This increase was associated with ~50-fold increase in its association with phospho-NKCC1 (Fig. 7B). After 28 min of exposure, there were ~4.5-fold decreases in both p38MAPK activity and amounts of associated phospho-NKCC1. Such changes in p38MAPK phosphorylation status after 30 min of hyperosmotic challenge are in agreement with results presented in Figure 6B, which shows that the total p38MAPK membrane-associated amounts were relatively unchanged in HCEC. In contrast, these amounts decreased in a manner similar to the declines in phospho-p38MAPK level in RCEC. Furthermore, there is a very good correspondence between the kinetics of hypertonicity-induced changes in p38MAPK activity, stimulation of 86Rb uptake, RVI responses, p44/42MAPK activity recovery (Bildin et al., 2003; Bildin et al., 2000), and survival of HCEC and RCEC.
Taken together, our data suggest that p38MAPK/NKCC1 interaction is fundamentally related to p38MAPK phosphorylation status and that hypertonicity-induced NKCC1 activation is dependent on that interaction. The rapid and very substantial p38MAPK activation leads to increases in NKCC1 activity, which determines the kinetics and extent of RVI-mediated recovery to the isotonic condition. These events determine if corneal epithelial cells will be able, under conditions of hyperosmolar adversity, to recover their p44/42MAPK activity, proliferate, adapt to stress, and avoid SAPK/JNK activation, i.e., avoid apoptosis. In other words, survival of hypertonicity-stressed corneal epithelial cells depends both on p38MAPK activation capacity and on the ability of p38MAPK to stimulate NKCC1 activity through protein-protein interaction. It is still unclear what the immediate substrate is for p38MAPK in RCEC and HCEC. There may be intervening steps between p38MAPK activation and NKCC1 phosphorylation. The level of NKCC1 activation affects the extent of cell volume recovery and, in turn, epithelial survival capacity. Figure 8 summarizes our current understanding of the mechanisms controlling cell fate in HCEC and RCEC during exposure to hypertonic stress.
Fig. 8.
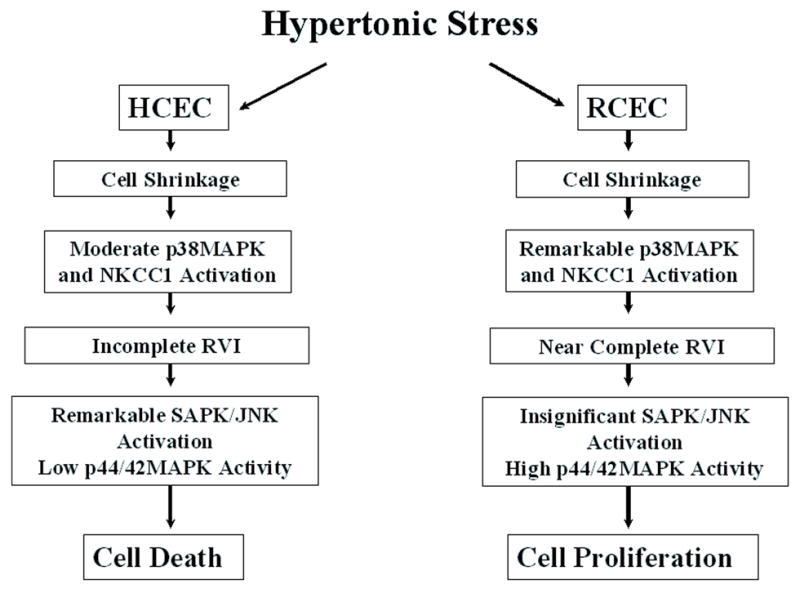
Differential responses of HCEC and RCEC to hypertonic stress. Summary of the mechanisms accounting for apoptosis or proliferation in HCEC and RCEC during hypertonic stress.
Acknowledgments
This work was supported in part by grants EY04795 (PR) and by an Unrestricted Grant from Research to Prevent Blindness, Inc., NY (KP). The discussions with Victor N. Bildin are appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anbari K, Schultz RM. Effect of sodium and betaine in culture media on development and relative rates of protein synthesis in preimplantation mouse embryos in vitro. Mol Reprod Dev. 1993;35:24–28. doi: 10.1002/mrd.1080350105. [DOI] [PubMed] [Google Scholar]
- Araki K, Ohashi Y, Sasabe T, Kinoshita S, Hayashi K, Yang XZ, Hosaka Y, Aizawa S, Handa H. Immortalization of rabbit corneal epithelial cells by a recombinant SV40-adenovirus vector. Invest Ophthalmol Vis Sci. 1993;34:2665–2671. [PubMed] [Google Scholar]
- Araki-Sasaki K, Ohashi Y, Sasabe T, Hayashi K, Watanabe H, Tano Y, Handa H. An SV40-immortalized human corneal epithelial cell line and its characterization. Invest Ophthalmol Vis Sci. 1995;36:614–621. [PubMed] [Google Scholar]
- Balcerczyk A, Soszynski M, Rybaczek D, Przygodzki T, Karowicz-Bilinska A, Maszewski J, Bartosz G. Induction of apoptosis and modulation of production of reactive oxygen species in human endothelial cells by diphenyleneiodonium. Biochem Pharmacol. 2005;69:1263–1273. doi: 10.1016/j.bcp.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Bildin VN, Wang Z, Iserovich P, Reinach PS. Hypertonicity-induced p38MAPK activation elicits recovery of corneal epithelial cell volume and layer integrity. J Membr Biol. 2003;193:1–13. doi: 10.1007/s00232-002-2002-8. [DOI] [PubMed] [Google Scholar]
- Bildin VN, Yang H, Crook RB, Fischbarg J, Reinach PS. Adaptation by corneal epithelial cells to chronic hypertonic stress depends on upregulation of Na:K:2Cl cotransporter gene and protein expression and ion transport activity. J Membr Biol. 2000;177:41–50. doi: 10.1007/s002320001098. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Cidlowski JA. Absence of volume regulatory mechanisms contributes to the rapid activation of apoptosis in thymocytes. Am J Physiol. 1996;271:C950–961. doi: 10.1152/ajpcell.1996.271.3.C950. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Cidlowski JA. A necessary role for cell shrinkage in apoptosis. Biochem Pharmacol. 1998;56:1549–1559. doi: 10.1016/s0006-2952(98)00225-1. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Oldenburg NB, Cidlowski JA. The role of DNA fragmentation in apoptosis. Trends Cell Biol. 1995;5:21–26. doi: 10.1016/s0962-8924(00)88932-1. [DOI] [PubMed] [Google Scholar]
- Burg MB. Response of renal inner medullary epithelial cells to osmotic stress. Comp Biochem Physiol A Mol Integr Physiol. 2002;133:661–666. doi: 10.1016/s1095-6433(02)00203-9. [DOI] [PubMed] [Google Scholar]
- Dihazi H, Kessler R, Eschrich K. High osmolarity glycerol (HOG) pathway-induced phosphorylation and activation of 6-phosphofructo-2-kinase are essential for glycerol accumulation and yeast cell proliferation under hyperosmotic stress. J Biol Chem. 2004;279:23961–23968. doi: 10.1074/jbc.M312974200. [DOI] [PubMed] [Google Scholar]
- Dmitrieva NI, Michea LF, Rocha GM, Burg MB. Cell cycle delay and apoptosis in response to osmotic stress. Comp Biochem Physiol A Mol Integr Physiol. 2001;130:411–420. doi: 10.1016/s1095-6433(01)00439-1. [DOI] [PubMed] [Google Scholar]
- Du JW, Zhang F, Capó-Aponte Tachado SD, Zhang J, Yu FX, Sack RA, Koziel H, Reinach PS. AsialoGM1-mediated IL-8 release by human corneal epithelial cells requires co-expression of TLR5. Invest Ophthalmol Vis Sci. 2006 doi: 10.1167/iovs.06-0250. In press. [DOI] [PubMed] [Google Scholar]
- Friis MB, Friborg CR, Schneider L, Nielsen MB, Lambert IH, Christensen ST, Hoffmann EK. Cell shrinkage as a signal to apoptosis in NIH 3T3 fibroblasts. J Physiol. 2005;567:427–443. doi: 10.1113/jphysiol.2005.087130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geck P, Pfeiffer B. Na+ + K+ + 2Cl− cotransport in animal cells--its role in volume regulation. Ann N Y Acad Sci. 1985;456:166–182. doi: 10.1111/j.1749-6632.1985.tb14862.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Dunham PB. Membrane mechanisms and intracellular signalling in cell volume regulation. Int Rev Cytol. 1995;161:173–262. doi: 10.1016/s0074-7696(08)62498-5. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Simonsen LO. Membrane mechanisms in volume and pH regulation in vertebrate cells. Physiol Rev. 1989;69:315–382. doi: 10.1152/physrev.1989.69.2.315. [DOI] [PubMed] [Google Scholar]
- Jensen BS, Jessen F, Hoffmann EK. Na+, K+, Cl− cotransport and its regulation in Ehrlich ascites tumor cells. Ca2+/calmodulin and protein kinase C dependent pathways. J Membr Biol. 1993;131:161–178. doi: 10.1007/BF02260106. [DOI] [PubMed] [Google Scholar]
- Junger H, Edelman DB, Junger WG. Hypertonicity promotes survival of corticospinal motoneurons via mitogen-activated protein kinase p38 signaling. J Mol Neurosci. 2003;21:111–120. doi: 10.1385/JMN:21:2:111. [DOI] [PubMed] [Google Scholar]
- Kang SS, Wang L, Kao WW, Reinach PS, Lu L. Control of SV-40 transformed RCE cell proliferation by growth-factor-induced cell cycle progression. Curr Eye Res. 2001;23:397–405. doi: 10.1076/ceyr.23.6.397.6965. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KL, Cidlowski JA. Cell cycle and apoptosis: common pathways to life and death. J Cell Biochem. 1995;58:175–180. doi: 10.1002/jcb.240580206. [DOI] [PubMed] [Google Scholar]
- Klein JD, Lamitina ST, O'Neill WC. JNK is a volume-sensitive kinase that phosphorylates the Na-K-2Cl cotransporter in vitro. Am J Physiol. 1999;277:C425–431. doi: 10.1152/ajpcell.1999.277.3.C425. [DOI] [PubMed] [Google Scholar]
- Kurihara K, Nakanishi N, Moore-Hoon ML, Turner RJ. Phosphorylation of the salivary Na(+)-K(+)-2Cl(−) cotransporter. Am J Physiol Cell Physiol. 2002;282:C817–823. doi: 10.1152/ajpcell.00352.2001. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays. 1996;18:567–577. doi: 10.1002/bies.950180708. [DOI] [PubMed] [Google Scholar]
- Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, Haussinger D. Functional significance of cell volume regulatory mechanisms. Physiol Rev. 1998;78:247–306. doi: 10.1152/physrev.1998.78.1.247. [DOI] [PubMed] [Google Scholar]
- Lang KS, Fillon S, Schneider D, Rammensee HG, Lang F. Stimulation of TNF alpha expression by hyperosmotic stress. Pflugers Arch. 2002;443:798–803. doi: 10.1007/s00424-001-0768-7. [DOI] [PubMed] [Google Scholar]
- Lee JS, Lee JJ, Seo JS. HSP70 deficiency results in activation of c-Jun N-terminal Kinase, extracellular signal-regulated kinase, and caspase-3 in hyperosmolarity-induced apoptosis. J Biol Chem. 2005;280:6634–6641. doi: 10.1074/jbc.M412393200. [DOI] [PubMed] [Google Scholar]
- Li DQ, Luo L, Chen Z, Kim HS, Song XJ, Pflugfelder SC. JNK and ERK MAP kinases mediate induction of IL-1beta, TNF-alpha and IL-8 following hyperosmolar stress in human limbal epithelial cells. Exp Eye Res. 2006;82:588–596. doi: 10.1016/j.exer.2005.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke CM, Cole TS. Activation of NKCC1 by hyperosmotic stress in human tracheal epithelial cells involves PKC-delta and ERK. Biochim Biophys Acta. 2002;1589:77–88. doi: 10.1016/s0167-4889(01)00189-6. [DOI] [PubMed] [Google Scholar]
- Lu L, Reinach PS, Kao WW. Corneal epithelial wound healing. Exp Biol Med (Maywood) 2001;226:653–664. doi: 10.1177/153537020222600711. [DOI] [PubMed] [Google Scholar]
- Maeno E, Ishizaki Y, Kanaseki T, Hazama A, Okada Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc Natl Acad Sci U S A. 2000;97:9487–9492. doi: 10.1073/pnas.140216197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek AM, Goss GG, Jiang L, Izumo S, Alper SL. Mannitol at clinical concentrations activates multiple signaling pathways and induces apoptosis in endothelial cells. Stroke. 1998;29:2631–2640. doi: 10.1161/01.str.29.12.2631. [DOI] [PubMed] [Google Scholar]
- Michea L, Combs C, Andrews P, Dmitrieva N, Burg MB. Mitochondrial dysfunction is an early event in high-NaCl-induced apoptosis of mIMCD3 cells. Am J Physiol Renal Physiol. 2002;282:F981–990. doi: 10.1152/ajprenal.00301.2001. [DOI] [PubMed] [Google Scholar]
- Michea L, Ferguson DR, Peters EM, Andrews PM, Kirby MR, Burg MB. Cell cycle delay and apoptosis are induced by high salt and urea in renal medullary cells. Am J Physiol Renal Physiol. 2000;278:F209–218. doi: 10.1152/ajprenal.2000.278.2.F209. [DOI] [PubMed] [Google Scholar]
- Moran TJ, Gray S, Mikosz CA, Conzen SD. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res. 2000;60:867–872. [PubMed] [Google Scholar]
- O'Donnell ME, Martinez A, Sun D. Endothelial Na-K-Cl cotransport regulation by tonicity and hormones: phosphorylation of cotransport protein. Am J Physiol. 1995;269:C1513–1523. doi: 10.1152/ajpcell.1995.269.6.C1513. [DOI] [PubMed] [Google Scholar]
- Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD) J Physiol. 2001;532:3–16. doi: 10.1111/j.1469-7793.2001.0003g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panet R, Atlan H. Stimulation of bumetanide-sensitive Na+/K+/Cl− cotransport by different mitogens in synchronized human skin fibroblasts is essential for cell proliferation. J Cell Biol. 1991;114:337–342. doi: 10.1083/jcb.114.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panet R, Marcus M, Atlan H. Overexpression of the Na(+)/K(+)/Cl(−) cotransporter gene induces cell proliferation and phenotypic transformation in mouse fibroblasts. J Cell Physiol. 2000;182:109–118. doi: 10.1002/(SICI)1097-4652(200001)182:1<109::AID-JCP12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Panet R, Markus M, Atlan H. Bumetanide and furosemide inhibited vascular endothelial cell proliferation. J Cell Physiol. 1994;158:121–127. doi: 10.1002/jcp.1041580115. [DOI] [PubMed] [Google Scholar]
- Reinehr R, Graf D, Fischer R, Schliess F, Haussinger D. Hyperosmolarity triggers CD95 membrane trafficking and sensitizes rat hepatocytes toward CD95L-induced apoptosis. Hepatology. 2002;36:602–614. doi: 10.1053/jhep.2002.35447. [DOI] [PubMed] [Google Scholar]
- Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- Schliess F, Schafer C, vom Dahl S, Fischer R, Lordnejad MR, Haussinger D. Expression and regulation of the Na(+)/K(+)/2Cl(−) cotransporter NKCC1 in rat liver and human HuH-7 hepatoma cells. Arch Biochem Biophys. 2002;401:187–197. doi: 10.1016/S0003-9861(02)00047-4. [DOI] [PubMed] [Google Scholar]
- Strange K. Cellular volume homeostasis. Adv Physiol Educ. 2004;28:155–159. doi: 10.1152/advan.00034.2004. [DOI] [PubMed] [Google Scholar]
- Terada Y, Inoshita S, Hanada S, Shimamura H, Kuwahara M, Ogawa W, Kasuga M, Sasaki S, Marumo F. Hyperosmolality activates Akt and regulates apoptosis in renal tubular cells. Kidney Int. 2001;60:553–567. doi: 10.1046/j.1523-1755.2001.060002553.x. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]