

Abstract
Although the molecular enzymology of DNA replication is well characterised, how and why it occurs in discrete nuclear foci is unclear. Using fission yeast, we show that replication takes place in a limited number of replication foci, whose distribution changes with progression through S phase. These sites define replication factories which contain on average 14 replication forks. We show for the first time that entire foci are mobile, able both to fuse and re-segregate. These foci form distinguishable patterns during S phase, whose succession is reproducible, defining early-, mid- and late-S phase. In wild-type cells, this same temporal sequence can be detected in the presence of hydroxyurea (HU), despite the reduced rate of replication. In cells lacking the intra-S checkpoint kinase Cds1, replication factories dismantle on HU. Intriguingly, even in the absence of DNA damage, the replication foci in cds1 cells assume a novel distribution that is not present in wild-type cells, arguing that Cds1 kinase activity contributes to the spatio-temporal organisation of replication during normal cell growth.
Keywords: nuclear organisation, replication foci, replication timing, S-phase checkpoint
Introduction
In animal cells, DNA replication takes place in discrete subnuclear domains, called replication foci. During progression through S phase, these foci undergo reproducible changes in size, number and position. At the beginning of mammalian S phase, replication occurs in numerous small foci throughout the nucleus, whereas in cells approaching G2 phase, a limited number of large foci are observed clustered at the nuclear envelope or around the nucleolus (for a review, see Gilbert, 2001). These observations led to the conclusion that replication is both temporally and spatially organised in metazoan cells.
Several attempts have been made to establish the molecular basis of replication foci (Cook, 2001). Replication foci have been shown to contain DNA polymerases, replication accessory factors and nascent DNA from several replication forks; moreover they resist solubilisation by high salt or other chaotropes (Tubo and Berezney, 1987). The average number of replication forks per focus in cultured metazoan cells ranges from 12 to 40, possibly reflecting variations in cell type and culture conditions (Berezney et al, 2000; Cook, 2001). Such observations led to the notion of a replication factory in which clustered forks are coordinately processed.
Although the functional significance of fork juxtaposition has never been tested directly, it is thought to provide a means to duplicate chromatin states: specific types of chromatin would replicate in specialised replication factories found at temporally and spatially distinct sites (Gilbert, 2001; Taddei et al, 2004). Quality control during DNA replication is also critically important for cell division, and the spatial juxtaposition of replisomes may help coordinate the sensing of DNA damage, the activation of checkpoints or even repair factor recruitment.
Improved imaging and tagging techniques have recently shown that replication foci are conserved from metazoans to budding yeast (Pasero et al, 1997; Lengronne et al, 2001; Meister et al, 2005; Kitamura et al, 2006). Nonetheless, to date, there have been no reports of temporally regulated patterns of replication foci for a single-celled eukaryote, and the factors that establish and modulate their patterning remain unknown (Gilbert, 2001; Taddei et al, 2004).
The spatial appearance of replication foci is likely to be determined by two general features of the genome: chromatin organisation and replication timing. These in turn are thought to either influence or result from transcription. In support of this, a good, but not absolute, correlation of transcriptionally active chromatin with early replication has been reported for complex organisms such as flies (Schubeler et al, 2002), particularly when transcriptional activity is integrated over ∼180 kb (MacAlpine et al, 2004). Similarly, genome-wide analyses in human cells showed that early replicating zones overlap with transcriptionally active GC-rich isochors, which span megabases (Jeon et al, 2005; reviewed in Schwaiger and Schubeler, 2006). Not surprisingly, these large-scale correlations fail when one analyses the smaller eukaryotic genomes of budding and fission yeast. A thorough genome-wide study of DNA replication in budding yeast revealed no global correlation between transcriptional activity and replication timing (Raghuraman et al, 2001). Nonetheless, the binding of general chromatin repressors, such as the Silent Information Regulatory complex, did suppress or delay origin firing locally, for example near telomeres (Ferguson and Fangman, 1992; Stevenson and Gottschling, 1999; Zappulla et al, 2002). In apparent contrast, centromeric heterochromatin in fission yeast was found to replicate early, although telomeres replicated late (Kim et al, 2003). Given this lack of coherence, it appears that something other than transcriptional activity is the major determinant of sequential initiation events.
Intra-S checkpoints are an additional mechanism implicated in the regulation of origin firing. Checkpoint controls were shown to prevent the initiation of late-firing origins and to be help maintain the integrity of stalled replication complexes under conditions of replicational stress (Santocanale and Diffley, 1998; Dimitrova and Gilbert, 2000; Shechter and Gautier, 2005; Feng et al, 2006). This mechanism was extensively explored in budding yeast, where both the checkpoint kinase Mec1, Rad53 and the Dbf4/Cdc7 kinase participate in late origin control (reviewed in Duncker and Brown, 2003; Raveendranathan et al, 2006). Moreover, in a rad53 mutant, late origins were reported to fire earlier than in wild-type cells, even during an unperturbed S phase (Shirahige et al, 1998), although this was a contested result.
In this study, we show that DNA replication factories are present in the unicellular eukaryote Schizosaccharomyces pombe. These replication factories display reproducible spatial and temporal patterns throughout S phase, showing specific localisation relative to the nucleolus and the nuclear periphery. Detailed analysis of the dynamics of replication factories indicated that the succession of patterns arises mainly from the de novo appearance and disappearance of foci, although foci are highly mobile and able to both fuse and split. We find that replication focal patterns progress even in the presence of hydroxyurea (HU), reflecting a systematic, yet very slow succession of origin firing events. However, this was not observed in the intra-S checkpoint mutant cds1, in which replication factories rapidly dismantle in the presence of HU. Surprisingly, we found that even in an unperturbed S phase the pattern of replication foci formation was altered in cells lacking the intra-S checkpoint kinase Cds1. Our data, thus, argue that intra-S checkpoint kinases participate in the regulation of origin firing, contributing to defined temporal patterns of replication.
Results
A system to study the organisation of replication in living fission yeast
To analyse the spatial organisation of DNA replication in living fission yeast, we generated a strain expressing the well-characterised replication marker PCNA fused to GFP. The tagged PCNA protein was expressed from endogenous PCNA promoter and integrated at the ura4 locus on chromosome III. The fusion protein was present at about 70% (±6%) of the untagged endogenous PCNA level (Figure 1A, lane 4), and had no effect on cell growth or resistance to genotoxic treatment (UV, gamma irradiation, HU or MMS; data not shown). Such strains appeared indistinguishable from wild-type strains. On the other hand, when we disrupted the wild-type pcn1 gene in the EGFP-PCNA expressing haploids, we observed an increased sensitivity to UV irradiation, even though the growth rates and DNA damage checkpoints were normal, and cells were not hypersensitive to HU, MMS or γ irradiation (data not shown). We conclude that EGFP-PCNA efficiently complements PCNA for replicative functions, yet it may hinder repair of UV-induced lesions when expressed alone. For this reason, only haploids expressing both the endogenous and the fused proteins were used in the experiments described below.
Figure 1.
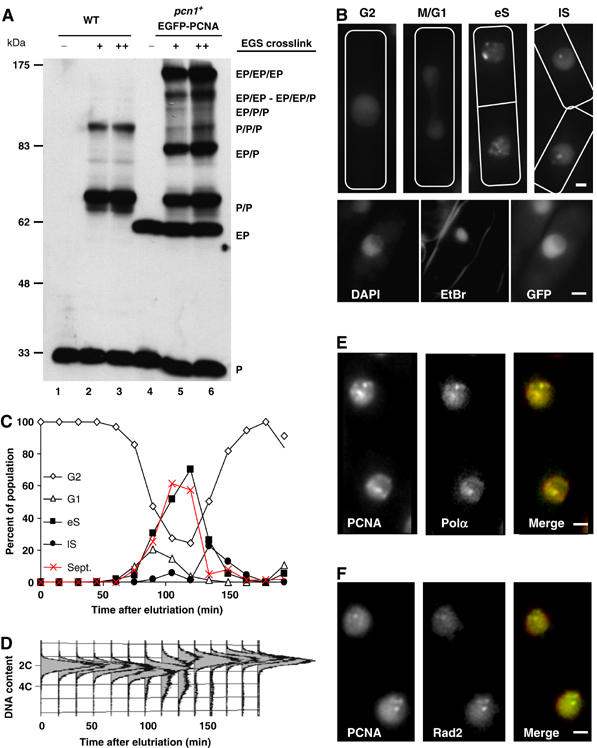
Characterisation of the EGFP-PCNA expression in fission yeast. (A) EGFP-PCNA interacts with itself and with endogenous PCNA to form multimers. After protein extraction from wild-type (lanes 1–3) and EGFP-PCNA-expressing (lanes 4–6) strains, extracts were crosslinked (lanes 2, 3, 5 and 6) with EGS. Extracts were subjected to electrophoresis and PCNA was detected by Western blot. The different multimers are indicated to the right: P, wild-type PCNA; E, EGFP-PCNA. (B) EGFP-PCNA patterns in asynchronously growing cells are shown as deconvolved images. Four different fluorescence patterns can be observed at distinct points in the cell cycle defined by the shape of the cell and presence of a septum (indicated by the drawings). Late S (lS) and early S (eS) nuclei show a brighter punctate pattern. The crescent with lower fluorescence corresponds to the nucleolus, as shown by selective EtBr staining (bottom panels). (C, D) Punctate EGFP-PCNA patterns coincide with DNA replication. The four patterns described above (B) were counted in elutriated cells synchronously traversing the cell cycle. Concomitantly, septation was scored and DNA content evaluated by FACS analysis (D). (E) ECFP-PCNA spots colocalise with DNA polα subunit B in S phase. Images are taken from asynchronously growing cells expressing ECFP-PCNA (red) and a YFP fusion to polα subunit B (green); colocalisation is shown in yellow. (F) As panel E, except that ECFP-PCNA is coexpressed with a YFP fusion to the Flap Endonuclease homolog Rad2. All bars=2 μm.
As wild-type PCNA is found as a homotrimeric complex that can slide along DNA duplexes, we next confirmed that the fusion protein was able to trimerise. In vitro cross-linking experiments were previously used to demonstrate that recombinant PCNA purified from Escherichia coli is able to form trimers (Piard et al, 1998). We used the same procedure to test PCNA oligomerisation into dimeric and trimeric complexes in the soluble fraction of wild-type S. pombe protein extracts (Figure 1A, lanes 2 and 3). In cells expressing both EGFP-PCNA and endogenous PCNA, EGS-crosslinking produced seven bands with molecular weights corresponding to all possible dimeric and trimeric complexes (Figure 1A, lanes 5 and 6). Finally, we used an in situ chromatin binding assay (Kearsey et al, 2000), in which GFP-tagged unfixed spheroplasts are washed with a non-ionic detergent and visualised by fluorescence microscopy: tightly bound chromatin proteins are retained, whereas soluble proteins are washed away. In this experiment, the bulk of EGFP-PCNA, like PCNA, was chromatin-bound exclusively during S phase (Supplementary Figure 1). Together, these results argue that in the presence of endogenous PCNA, EGFP-PCNA behaved like the wild-type protein, rendering it a useful marker for replication in living cells.
Replication-specific PCNA foci in fission yeast
The fusion protein EGFP-PCNA is exclusively nuclear, occupying primarily the non-nucleolar part of the fission yeast nucleus (Figure 1B; and data not shown). PCNA fluorescence was diffuse during the M, G1 and G2 phases of the cell cycle, but became punctate in early S phase, which was identified by the cell's binucleate septated morphology (Figure 1B, eS). Similarly, foci were observed in very small mononucleated cells (late S phase or lS, Figure 1B), whereas cells expressing an unrelated YFP-lacI fusion protein, or another GFP-fusion protein, did not give a punctate fluorescence pattern at any stage of the cell cycle (Meister et al, 2003).
The sequential appearance of focal patterns was confirmed on a cell population synchronised by elutriation. Starting from a G2-phase population, cells were imaged every 15 min and DNA content was monitored by FACS (Figure 1C and D). G1 and eS cells appeared simultaneously and rapidly, and a strong correlation could be drawn between entry into S phase, septation and the appearance of PCNA foci. The lS pattern, on the other hand, correlated with the termination of DNA replication as determined by FACS profiling (Figure 1D).
To confirm that the S-phase PCNA pattern reflects replication foci, we tagged the second subunit of DNA polymerase α (polα) with YFP. This fusion had no effect on cell growth or sensitivity to classical DNA damaging agents (data not shown). Using high-resolution microscopy, we could show that YFP-polα colocalised with ECFP-PCNA (Figure 1E). Similar results were obtained with Rad2, the flap endonuclease FEN1 homologue (Figure 1F). We conclude that the foci formed by fluorescently-tagged PCNA are indeed sites of genomic DNA replication.
PCNA foci correspond to replication factories in S. pombe
Using asynchronously growing cells, we analysed spot number in single nuclei using novel software designed for this purpose (see Materials and methods). Cells were imaged on an agarose pad and image stacks were deconvolved using Metamorph® software. First, single nuclei were delineated by a combination of threshold and in-focus plane detection, and thereafter subnuclear spots were identified by image filtering (Figure 2A). The use of automated spot detection software avoids user-dependent bias and ensures impartial quality control on stacks taken at different times. Using this method, we analysed replication in asynchronously growing wild-type cultures. Under these conditions, about 50% of all nuclei showed PCNA foci, and in all nuclei, only a limited number of PCNA foci were observed (Figure 2B). Indeed, the average number of spots per S-phase nucleus was 5.5, with a maximum of 14 and a minimum of 1 (n=310; Figure 2B). As at least 76 replication forks must be active throughout S phase to replicate the entire genome of S. pombe, we conclude that each PCNA focus contains at least several replication forks, stemming from at most seven origins (see below). Thus, an S-phase-specific formation of replication factories also occurs in fission yeast.
Figure 2.
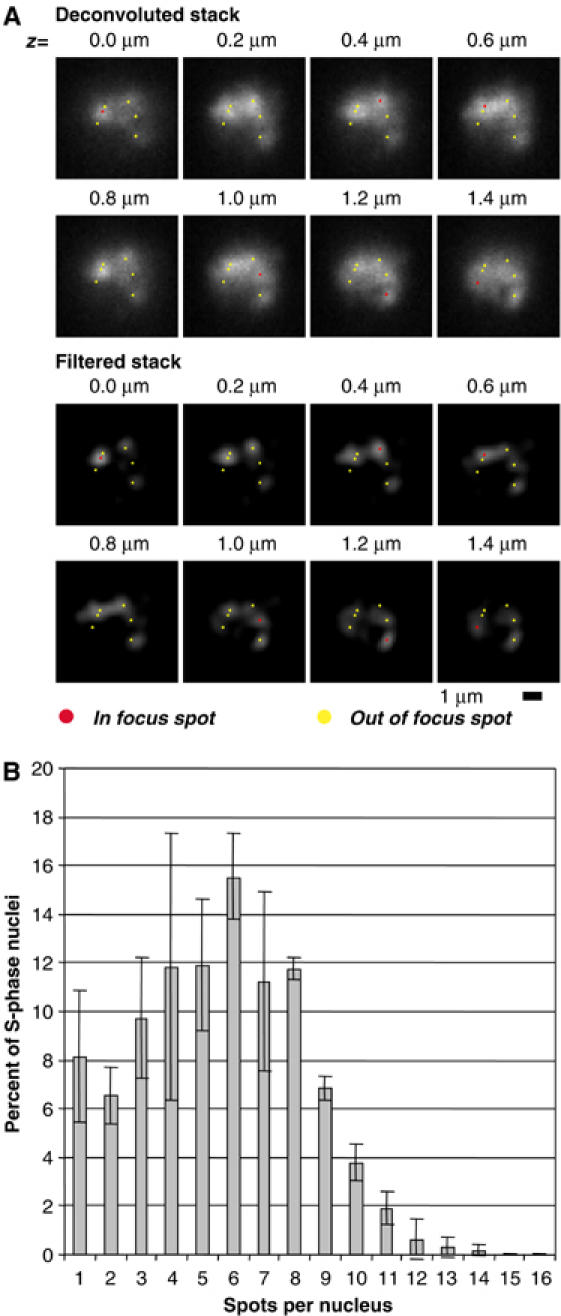
Replication occurs in a low number of replication factories. (A) Example of EGFP-PCNA spot detection in a single S-phase nucleus showing six spots. The original deconvolved stack is first filtered before spot detection by local maxima detection (in 3D). Spots are checked after detection by an overlay of spot position on the original stacks. (B) Replication foci number in an asynchronously growing population of EGFP-PCNA expressing S. pombe cells using automated spot detection.
Spatio-temporal organisation of DNA replication in S. pombe
As discussed above, higher eukaryotic replication foci are found in distinct patterns that occur in a reproducible sequence during S phase (reviewed in Cook, 2001; Gilbert 2001). To characterise possible replication patterns in fission yeast, we took 3D images of exponentially growing cell cultures and attempted to classify replicating nuclei by their patterns of PCNA foci. We classified 350 nuclei from two independent experiments, taking deconvolved images with maximal resolution at a single time point.
We could identify four distinct patterns, labelled a to d (Figure 3A). In pattern a, replication foci appeared in the extranucleolar region of the nucleus, but did not pervade the entire extranucleolar region (Figure 3A, pattern a). This pattern was present in about 20% of replicating nuclei. In pattern b, representing 40% of the replicating cells, the entire extranucleolar space was filled with PCNA foci. Pattern c, found in 25% of the replicating cells (Figure 3A, pattern c), showed large replication foci at the edge of the nucleolus and a small number of bright spots in the rest of the nucleus. Finally, in pattern d, only a few bright spots located at the edges of the nucleolus were observed.
Figure 3.
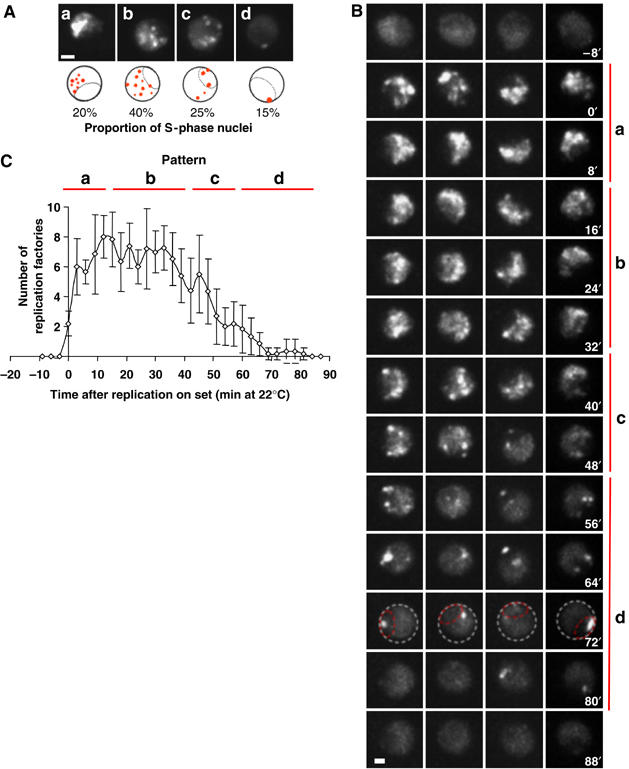
Spatio-temporal organisation of replication foci. (A) Four successive replication factories pattern based on EGFP-PCNA fluorescence can be observed during S phase (a–d) as described in the text. The nucleolus forms a subnuclear domain as indicated. Bar=2 μm. (B) Selected images from time-lapse microscopy (see Supplementary Movie 1) of EGFP-PCNA expressing fission yeast cells grown at 22°C. Pattern succession in four live cells is shown during an entire S phase, synchronised relative to initial PCNA foci appearance. The patterns a–d are seen to arise in sequential order. Bar=2 μm. (C) The number of replication factories was scored at 4-min intervals extracted from time-lapse imaging throughout an entire S phase (n=6).
To ascertain whether these patterns were temporally ordered, we performed time-lapse analysis by recording asynchronously growing cells during an entire S phase at 22°C. Cells were imaged every 4 min to avoid bleaching and artefacts owing to fluorescence excitation. Indeed, we confirmed that repetitive exposure to 475 nm light at 4 min intervals and growth in a closed observation chamber had almost no effect on subsequent cell division; cell-cycle duration was similar with or without fluorescence excitation (data not shown).
Time-lapse series were temporally aligned with respect to the appearance of PCNA spots. After alignment, a strikingly reproducible succession of patterns a to d can be observed for different cells (Figure 3B and Supplementary Movie 1). The relative proportions of the four patterns scored in a sequential analysis of S phase were quite similar to the proportions scored in the asynchronous culture (data not shown). The last pattern (d) is particularly striking as all cells ended S phase with bright PCNA spots in or around the nucleolus (encircled, Figure 3B).
The number of PCNA foci were scored for the different patterns observed during time-lapse recordings through S phase. As the z resolution of these images is lower than in single time-point capture and automated spot counting, these were counted manually. Results were nonetheless very similar to those obtained by either automated or manual counting on asynchronous cultures. Soon after the onset of S phase and the appearance of PCNA spots, that is, in patterns a and b, the numbers of foci are relatively high (averaging 7–9; Figure 3C). As patterns c and d appear, the number of replication factories decreases progressively reaching on average only two perinucleolar spots in pattern d. Similar distributions were obtained from asynchronously growing cells that were independently categorised by their spatial pattern prior to counting foci (Supplementary Figure 2). We conclude that the number and localisation of replication factories varies in a reproducible manner during S phase in fission yeast, reminiscent of the spatial and temporal regulation observed in higher eukaryotes (Taddei et al, 2004).
Dynamics of individual replication factories
To study replication foci dynamics, cells were imaged at high frequency in 3D as they progressed through S phase (every 6 s). Although the small size and large number of replication factories in patterns a and b precluded frame-by-frame spot tracking, the movies of early replicating nuclei show that the replication factories are highly mobile (see Supplementary Movie 2). The mid-to-late-S-phase replication factories could be tracked because of their larger size and smaller number. To visualise replication factories in time, each time-point stack was first projected along the z-axis. These projected frames were then grouped to create a ‘time' stack in which each plane is one timepoint. This stack is then projected horizontally along the time axis, forming a kymograph (Figure 4A). Kymographs showed that single replication foci were able to traverse the nuclear radius in less than 1.5 min (Figure 4B, white arrowheads). Because different foci moved in independent directions at the same time, such that their paths occasionally cross each other (Figure 4), focus movement cannot be accounted for by nuclear rotation. As further confirmation of this, we monitored EGFP-PCNA movement in S-phase cells treated with thiabendazole (TBZ), which depolymerises cytoplasmic microtubules and eliminates nuclear rotation. Replication factory movement in not inhibited by the presence of TBZ (Supplementary Movie 4) and quantification of focus mobility shows that the radii of constraint are reduced by only 20%, indicating that the movement of replication foci is primarily due to their own displacement rather than general nuclear rotation (Supplementary Figure 4).
Figure 4.
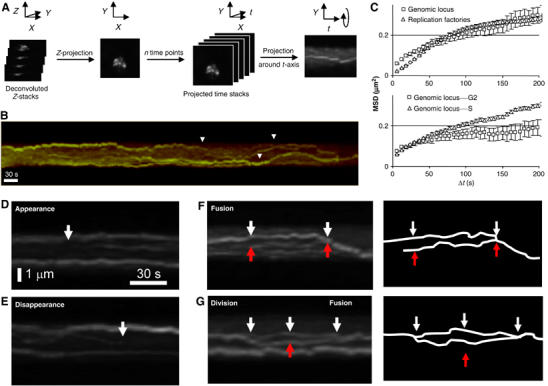
In vivo dynamics of EGFP-PCNA-tagged replication factories. (A) A scheme for the construction of individual kymograph is shown: each 3D image stack taken at 6-s intervals, fluorescence is deconvolved, then z-projected. Time stacks are built from z-projections and projected around t axis. (B) Kymograph over 10 min of an individual fission yeast nucleus showing the EGFP-PCNA transition from pattern c–d at 30°C. Large movements can be observed (arrows) for individual replication factories (see Supplementary movies 2 and 3). (C) MSD analysis on aligned nuclei compares the movement of individual EGFP-PCNA spots (diamonds, n=20) with that of a lacO-tagged late replicating genomic locus (ars2.1; (Kim and Huberman, 2001), squares, n=6). In the upper graph, we see that the MSD plateau of a genomic locus and of replicating foci are similar, and we calculate a radius of constraint for each as 0.65 μm (Gartenberg et al, 2004). The diffusion coefficient (Dmax) reflects the initial slope of the curve, indicating that replication factories diffuse less rapidly. The lower panel compares the same genomic locus in G2- and S-phase cells, indicating a significant restriction in the radius of constraint in G2 phase. (D, E) Enlargements of kymographs of fission yeast nuclei in mid-S phase show the appearance (D) or disappearance (E) of an individual replication factory (arrows). (F) As panel D, but showing the merging of replication factories (arrows). (G) As panel E, but showing division and merging of one replication factory (arrows).
By comparing the average mobility of 20 mid-to-late-S-phase replication factories with the dynamics of a lacop-tagged gene (Straight et al, 1996), we found that single loci have a slightly higher Dmax, but have the same spatial constraints as EGFP-PCNA-tagged replication foci (Figure 4C). The MSD curves further indicated that the mobility of a tagged DNA locus decreases in G2 phase as compared with S phase (Figure 4C; see Discussion).
Mammalian DNA replication patterns are thought to progress through sequential patterns of replication foci by de novo assembly, rather than by shifting their position (Sporbert et al, 2002). A similar de novo assembly and disappearance of replication foci can be observed in fission yeast. In Figure 4B, we show an example of the transition from pattern c to d, coincident with the disappearance of a replication factory. Moreover, time-lapse imaging allowed us to show that fission yeast replication factories also fuse (Figure 4F) and split into two distinct and smaller replication foci, before later merging again, phenomena that were not previously reported (Figure 4G; see Supplementary Movie 3). Such dynamics are likely to be facilitated by the large degree of mobility scored for replication foci (see MSD curves).
Replication factories patterns during HU treatment
We next asked to what degree replication factory patterns and their transitions require normal replication rates. To impair the progression of DNA synthesis, we used concentrations of HU ranging from 12 to 50 mM. Asynchronously growing cells treated with different concentrations of HU were observed immediately after its addition. No replication factory breakdown was observed at any concentration (data not shown), consistent with ChIP studies in budding yeast that show DNA polymerase persistence at HU-arrested forks for up to 1 h (Cobb et al, 2003). The number of cells with PCNA spots increased with time, as cells entered S phase (compare 0, 2 and 4 h; Figure 5A), indicating that replication factories are not disassembled, despite a significant reduction in DNA replication rates. By 4 h most wild-type cells accumulate with an early S-phase pattern, presumably because cells have progressed through G2 and M in the presence of HU, and then stall upon entry into S phase. However, some cells could still be observed with a late S pattern as long as 4 h after adding HU.
Figure 5.
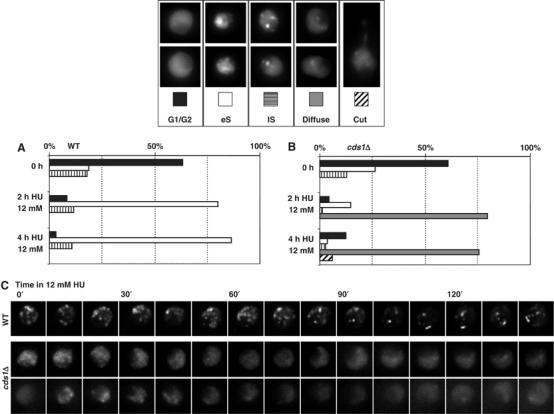
Effect of HU treatment on replication factories in wild-type and cds1Δ cells. (A) Examples of the EGFP-PCNA distribution are shown above the graphs. In wild-type cells replication foci are maintained on HU and early S patterns accumulate after 2–4 h on HU. For each strain, >150 nuclei were scored. (B) As in panel A, but for the cds1Δ strain, in which we score the disassembly of replication factories in cells incubated on HU (cf. patterns at 0, 2 and 4 h of HU treatment). (C) Time-lapse imaging was performed on wild-type and cds1Δ fission yeast cells bearing EGFP-PCNA at 30°C. Cells were treated with incubated in 12 mM HU and 3D stacks were taken at 10 min intervals. Projections of individual frames are shown. Replication factories are lost by 1 h on HU in the cds1Δ mutant, while pattern progression proceeds in wild-type cells that had already entered S phase. A large fraction of the cells in an asynchronous population of fission yeast are in G2 phase, and these accumulate in early S phase on 12 mM HU.
We examined the kinetics of replication pattern progression on HU by performing time-lapse microscopy on living cells over 2.5 h in the presence of 12 mM HU (Figure 5C). Wild-type cells that have already entered S phase were seen to progress slowly through the four previously described replication patterns during the first 2 h on HU (Figure 5C). In some cells replication factories disappeared slowly but completely, suggesting that they were able to terminate DNA replication (Figure 5C). Indeed, even in 50 mM HU a progression from mid-to-late-S-phase patterns could be scored, including disappearance of the characteristic perinucleolar foci (data not shown). Therefore, we conclude that S-phase cells slowly complete replication on HU in the presence of reduced nucleotide pools and activated S-phase checkpoint kinases. The conserved progression of replication factory patterns argues for the maintenance of a normal temporal organisation of replication despite the induction of the intra-S checkpoint by HU treatment.
Replication factories disassemble in cds1Δ cells on HU
In both budding and fission yeast, replication fork integrity is thought to be maintained by the intra-S checkpoint when replication itself is compromised. In the absence of S-phase checkpoint factors, replication forks undergo catastrophic collapse, which precludes the restart of DNA replication even after HU removal (Kim and Huberman, 2001; Lopes et al, 2001; Sogo et al, 2002; Meister et al, 2005). We therefore examined the organisation of replication factories in intra-S checkpoint mutants during an HU block. Fission yeast has well-defined checkpoint transducers, namely Cds1, which mediated the S-phase checkpoint and Chk1, which is activated during the G2/M checkpoint. SpCds1 is the functional homolog of mammalian Chk1 (S-phase checkpoint, activated by ATR), whereas SpChk1 function is carried out by Chk2 (G2/M checkpoint, activated by ATM). Unlike wild-type cells, replication factories were generally diffuse in a cds1Δ mutant strain after 4 h on HU, and distinct patterns were rarely observed (compare 0, 2, and 4 h; Figure 5B). In many cells, replication factories disappeared completely, without passing first through the late replication pattern (Figure 5B, 4 h). Further analysis using time-lapse recordings showed that pattern progression did not occur and that PCNA foci became dismantled within an hour after HU addition (Figure 5C, cds1Δ). Cells were then blocked in a G2-like phase with no visible replication factories, and ultimately underwent mitotic catastrophe (cut phenotype, Figure 5B). These observations are consistent with studies in budding yeast, in which the loss of functional replication forks and MCM association are observed when rad53 mutants are treated with HU (Cobb et al, 2005). Similar fork collapse was observed in cds1 fission yeast cells treated with HU (Meister et al, 2005).
A novel replication pattern in S-phase checkpoint mutants
We next analysed the effect of checkpoint gene deletion on pattern organisation and replication factory number during an unperturbed S phase. Asynchronous cells bearing full deletions of checkpoint kinase genes and expressing EGFP-PCNA were imaged, and patterns were identified within individual nuclei of S-phase cells.
In both wild-type and chk1Δ fission yeast cells the four standard patterns of replication foci were found to be present in similar proportions (Figure 6A and B). In contrast, in cells lacking cds1, we observe a novel pattern of replication foci (n, Figure 6A and B). Notably, in 35% of the replicating cds1 cells PCNA foci appeared at the same time both in the nucleolus and in non-nucleolar nucleoplasm, a phenomenon that arose in <2% of a wild-type population. Similarly, in strains lacking Rad3, the S. pombe ATR kinase, this new pattern was scored in ∼10% of replicating cells (Figure 6B).
Figure 6.
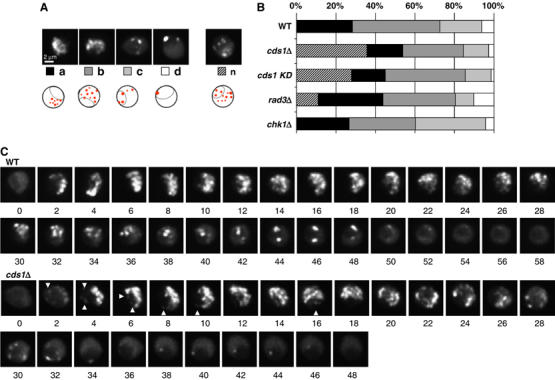
Loss of intra-S phase checkpoint kinase activity influences replication factory patterns in asynchronously growing cells. (A) The four standard replication foci patterns a–d are shown with a sketch of their relationship to the nucleolus. A new pattern (n) can be observed in cds1Δ asynchronously growing cells in the absence of HU, as shown here. (B) Proportions of the different patterns from panel A, relative to the total number of nuclei harbouring PCNA foci in asynchronously growing cultures in the absence of HU. The striped bar indicates the appearance of a novel pattern n in checkpoint mutants. (C) Time-lapse imaging of EGFP-PCNA expressing wild-type and cds1Δ fission yeast cells was performed throughout S phase at 1-min intervals at 30°C. Arrows represent the appearance of nucleolar replication foci in cells bearing numerous non-nucleolar foci.
As rad3Δ cells have been shown to accumulate DNA damage, as monitored by the colocalisation of Rad22-YFP with PCNA in foci, PCNA foci cannot be unambiguously attributed to replication in this strain (Noguchi et al, 2004; Meister et al, 2005). On the other hand, cds1Δ cells showed very few spontaneous Rad22-YFP foci, making it unlikely that the appearance of the new PCNA pattern resulted from the processing of DNA damage. This suggests that the cds1 mutation specifically interferes in the temporal organisation of origin firing. To see if the kinase activity of Cds1 was required for wild-type pattern repartition, we performed a similar analysis of replication foci patterns in a strain bearing a point mutation in the Cds1 active site (Boddy et al, 2000). The kinase-dead mutant also produced pattern ‘n' in 25% of replicating nuclei, as opposed to <2% in a wild type strain (Figure 6B). This argues that Cds1 kinase activity is essential for the patterning of replication foci during an unchallenged S phase.
The clear qualitative differences between replication foci in wild-type or G2/M checkpoint mutant (chk1) and the intra-S checkpoint mutants, led us to perform a quantitative analysis of foci in these strains. Using the software described above, we counted EGFP-PCNA foci in replicating nuclei of the cds1Δ and control strains. In the intra-S phase kinase mutant, we saw a 15% drop in the average number of foci throughout S phase (t-test; P=0.002), which was not the case for chk1Δ mutants (P=0.3; data not shown). We compared the distributions of foci number per nucleus pooled for each strain from all phases of S-phase and found that wild-type and cds1Δ distributions were significantly different (Kolmogorov–Smirnov test; P=2 × 10−5; Supplementary Figure 3).
Finally, we used time-lapse microscopy to confirm that the novel pattern of foci appeared during an unperturbed S phase in cds1Δ mutant cells. Cells were imaged at 30°C every minute, and shortly after entry into S phase, the new focal pattern, which combines intranucleolar foci with numerous non-nucleolar factories, could be observed (arrows in Figure 6C, cds1Δ). We could not detect this pattern in time-lapse imaging of wild-type cells at any point in S phase (Figure 6C, WT). Thus, replication focal patterns change in both a qualitative and quantitative manner in the cds1 intra-S checkpoint mutant, arguing that the kinase modulates either directly or indirectly both the spatial and temporal organisation of DNA replication foci.
Discussion
Replication factories in a unicellular eukaryote
Many studies have examined the organisation of replication factories in mammalian cells using DNA labelling, immunofluorescence and GFP technology, although what controls their number and intranuclear distribution remains unknown (reviewed by Berezney et al, 2000; Gilbert, 2001). Indeed, it is well established that PCNA forms foci at sites of active DNA replication based on colocalisation with BrdU pulse-labeled DNA. Using a functional EGFP-PCNA fusion, we present the first kinetic analysis of replication factories tracked in living yeast cells over time, allowing us to make a number of novel observations about the spatial organisation of replication.
First, we find that in fission yeast S-phase-specific DNA replication occurs in discrete subnuclear foci, which are present in a limited number. This argues that multiple forks must assemble at these sites. In a random S-phase population we scored on average 5.5 replication foci per nucleus, with a maximum of 14. Given that the fission yeast genome has 13.8 Mbp, we calculate that 76 active forks would be needed to replicate the entire genome during a 60-min S phase at 30°C, assuming a progression rate of 3 kb min−1 (Raghuraman et al, 2001). Thus, with ∼5.5 foci per nucleus, an average factory would contain 14 replication forks, initiating from seven bidirectional origins. If origins are spaced at 20–30 kb (Feng et al, 2006; Patel et al, 2006), then each focus represents ∼250 kb of coordinately replicated DNA. The estimated number of 14 replication forks per focus is remarkably similar to that calculated for mammalian cells (Ma et al, 1998), and the large size of coordinately replicated domains is also similar to that found in flies. These values may therefore reflect structural limitations on the number of forks tolerated by a replication factory.
Replication is organised in space and time
As in mammalian cells, S. pombe replication foci assume spatially distinct patterns that are temporally ordered, and distinguishable by size and number of foci, subnuclear distribution, as well as their order of appearance (Figure 2). Time-lapse microscopy shows that nucleoplasmic, non-nucleolar foci appear first in early S phase in wild-type cells (pattern a), increasing in number to form pattern b. By late S phase, the number of foci drops, and the remaining foci concentrate within the nucleolus or at the nucleolar periphery (pattern c). Finally, S phase ends reproducibly with 1–2 large spots at the nucleolar border (pattern d). In a recent study, only ∼10% of fission yeast origins were found to be late firing (Heichinger et al, 2006). These late replication origins were mainly subtelomeric, in agreement with a previous study which scored telomeres as very late replicating using 2D gel analysis (Kim and Huberman, 2001). We also detect a significant level of colocalisation of late foci with Taz1 (data not shown).
The sensitivity with which one detects replicating DNA genome-wide depends critically on the method employed. When labelled nucleotides ratio is used, the detection scale can only range from 1 to 2 (Heichinger et al, 2006). If origin firing is stochastic as indicated by single DNA molecule analysis (Patel et al, 2006), the background of non-firing origins will reduce the sensitivity of detection. As here we analyse replication on a single-cell level, stochastic events can be readily scored. This sensitivity is further enhanced by the presence of multiple PCNA molecules at each replication fork. It is remarkable that even if the firing of a given origin is a stochastic event, replication patterns are reproducible, suggesting that chromatin regions maintain a robust replication timing derived from the stochastic firing of multiple potential origins contained therein.
Replication factories move
We show here for the first time that replication factories move in a diffusive manner and that they occasionally fuse or segregate. This contrasts with reports on mammalian cells, which argue that replication factories are nearly immobile (Sporbert et al, 2002). Indeed, changes in foci positions in mammalian cell nuclei are thought to depend entirely on the progression of replication forks; FRAP experiments suggest that replication factors are detached from the DNA before reloading (Sporbert et al, 2002). In yeast, we monitor replication factories that can cross half of the nucleus in less than 30 s, without changing intensity. This makes it very unlikely that their movement reflects a progressive reloading of PCNA. We see that the radius of constraint of replication focus movement is similar to that of a tagged genomic locus, arguing that replication itself is not the source of movement. Instead, this mobility could reflect the absence of nuclear lamins and an internal nuclear substructure (Taddei et al, 2004). However, as the high-resolution dynamics of replication foci has not been studied in mammalian cells, one cannot exclude that such movements also exist in higher eukaryotes.
Replication factory stability upon nucleotide depletion requires an intra-S checkpoint
When wild-type cells were treated with hydroxyurea, the PCNA foci did not change significantly, providing a visual demonstration that replication forks are maintained under these conditions. Even more importantly, despite the activation of a checkpoint response, the temporal succession of replication patterns was maintained on HU, although patterns progress at a much slower rate. This is consistent with recent studies in budding yeast (Feng et al, 2006; Raveendranathan et al, 2006), which argue for a slow progression of replication forks in the presence of HU. This phenomenon was also reported for fission yeast based on FACS analysis (Kim and Huberman, 2001). Here, we extend their conclusion by showing that growth in the presence of low HU concentrations does not alter the spatio-temporal ordering of origin firing, despite a dramatic extension in the time it takes to complete S phase.
We also analysed the effect of HU on replication factories in strains lacking an active intra-S phase checkpoint kinase Cds1. No pattern succession was observed in this mutant: PCNA foci accumulated throughout the nucleoplasm and then progressively disappeared after 1 h. Cells were then blocked in a G2-like phase and ultimately underwent mitotic catastrophe (Figure 5). This is consistent with the major structural changes of replication forks observed in this mutant during an HU block (Meister et al, 2005). The arrest of replication by HU in a budding yeast rad53 mutant has been shown to induce the loss of the replicative helicase MCM, leading subsequently to fork collapse (Cobb et al, 2005; Feng et al, 2006). Thus, the loss of replication foci we observe may well reflect the loss of functional replication forks altogether. The effect of HU on origin firing in checkpoint mutants has been recently analysed in fission yeast using an assay that detects ssDNA. This study argues that 39% of the origins are suppressed by Cds1 in the presence of HU (Feng et al, 2006). Increased origin firing followed by rapid collapse would not be detectable by our assay, although we are able to score a global loss of functional replication forks.
Intra-S phase checkpoint as a ‘rheostat' for initiating forks
In the absence of any external insult such as HU, we observed that checkpoint mutants, and most notably those lacking the Cds1 kinase, have a novel pattern of replication factories: foci were observed simultaneously in the nucleoplasm and in or near the nucleolus. This was essentially never seen in wild-type cells. This new pattern appeared to be an overlap of late- and early-replication patterns, as if the early and the mid-to-late-S-phase patterns were superimposed, even though the number of foci per nucleus averaged over the entire S phase did not vary significantly (Figure 6). This new pattern was present both in a cds1Δ strain and in a kinase-dead cds1 point mutant. We can rule out that the abnormal focal pattern reflects repair events, as the cds1Δ cells suffer only a minor increase of DNA damage during a normal S phase, as judged by the appearance of Rad22 foci (Meister et al, 2005). We do not propose that the cds1Δ pattern reflects a complete loss of regulated origin timing, but rather an altered efficiency of firing combined with impaired fork progression, such that early and late patterns overlap. As the observed change in pattern occurred in unchallenged cells, the intra-S checkpoint kinases must be at least partially active during normal S-phase progression.
One signal for checkpoint activation during normal S phase could be the presence of an excessive amount of single-stranded DNA (ssDNA). Recent work has shown that the intra-S phase checkpoint response requires a threshold level of stalled forks for activation, suggesting that the total amount of nuclear ssDNA may be integrated to promote a checkpoint response, possibly owing to the titration of RFA (Shimada et al, 2002). During an unchallenged S phase, the purpose of the intra-S checkpoint could be to avoid the firing of too many origins, which could itself lead to nucleotide exhaustion and unwanted fork stalling (Shechter and Gautier, 2005). In this model, the most readily unwound or activated origins would fire first at the beginning of S phase until a critical threshold of ssDNA is reached. This might trigger a low-level S-phase checkpoint kinase activity, that in turn would inhibit subsequent origin firing. Supporting this model, S-phase checkpoint activity in the absence of DNA damage has been monitored in Xenopus egg extracts and in human cell lines (Miao et al, 2003; Marheineke and Hyrien, 2004; Shechter et al, 2004; Sorensen et al, 2004). This ssDNA feedback system would be self-regulatory in nature: once early replicons reach completion, ssDNA levels should decrease and relieve checkpoint activation, allowing late origins to fire. In this way, checkpoint kinases would help limit the concomitant firing of an excessive number of replication origins, preventing fork stalling and chromosome breakage. In this model, the key difference between early and late firing origins is their propensity to initiate replication, which may be determined by other factors such as chromatin structure (reviewed in Schwaiger and Schubeler, 2006). This second chromatin-based control may explain why we observe a persistence of the very late focal pattern in the cds1Δ mutant.
In summary, we describe here, the spatio-temporal organisation of replication in a genetically tractable organism, and show that mutations in the intra-S phase checkpoint kinase gene cds1 alter the visible pattern of replication foci. These mutations do not, however, eliminate focus formation. Importantly, the system provides the means to identify and characterise factors responsible for the spatial organisation of genomic replication.
Materials and methods
S. pombe strains and culture conditions
All constructs used in this study are derived from previously described strains (Meister et al, 2003, 2005), except strains expressing Rad2-YFP and Polα subunit B fused to YFP. A list of all strains used in this study is in Supplementary data. Cells were cultured at 30°C in rich medium unless otherwise indicated (YE; Moreno et al, 1991) and HU was used at a 12 or 50 mM final concentration (US Biologicals).
EGS crosslink of total extracts
Cells cultured in rich medium were collected by centrifugation, washed in water and broken with glass beads in 60 μl lysis buffer (50 mM Tris–HCl pH 8, 150 mM NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol, 50 mM NaF, 1 mM Na3VO4, 0.1% NP-40 and 10% glycerol). The EGS (ethylene glycol-bis(succinic acid n-hydroxysuccinimide ester) (Sigma Chemicals, St Louis, MO) crosslinking method was used as previously described (Piard et al, 1998). A 40 μg volume of total extract was incubated for 5 min in 50 mM EGS, 20 mM NaPO4 pH 7.5 and 0.15 M NaCl at rt. Reactions were quenched with 50 mM Tris pH 7.5. After boiling in sample buffer, 10 μg were loaded on a 15–8% gradient acrylamide gel, followed by Western blotting with a PVDF membrane and anti-PCNA (PC10; Santa Cruz Biotechnology, Santa Cruz, CA).
FACS staining
Ethanol-fixed cells were treated as previously described. DNA was stained with propidium iodide at a final concentration of 4 μg/ml. Flow sorting was performed on a BDFacsCalibur.
Imaging of cells
GFP-PCNA expressing cells were imaged either on agarose (still images) or in a lectin-coated Ludin chamber (Griffonia simplifolia for movies). Imaging was performed on an Olympus IX70 microscope with a CoolSnapHQ cooled CCD camera and a TillVision polychrome II monochromator as light source. Usually 21-plane stacks were used for still images, with a step size ranging from 0.2 to 0.45 μm. Imaging was done either at room temperature for single-stack acquisition or whole S phase time-lapse acquisition (4-min intervals, Figures 1, 2 and 3 and 5), whereas a temperature-controlled chamber was used for rapid 6 s interval acquisition (Figures 4 and 6; Life Imaging Services, Reinach, CH). Image processing and deconvolution were performed using Metamorph (Universal Imaging) and ImageJ (NIH). Final statistical analysis was performed using R (Ihaka and Gentleman, 1996). MSD is described in Supplementary data.
Spot counting on individual nuclei
The spot-detection algorithm developed for this paper is based on Kruizinga and Petkov (2000), but is extended to 3D. In brief, the signal is convolved with a finite, discrete 3D Difference of Gaussians (DoG) kernel, whose frequency cut-off is calculated from the expected, sampled spot diameter. The result of convolution is half-wave rectified to set all negative responses to the filter to 0. A 3D local maximum operator is then run on the result of rectification and all returned local maxima are recorded as spot candidates. As a large fraction of all local maxima is due to noise (and partially to the finite support of the DoG kernel), we define a real spot as an outlier of the observed intensity distribution of noise-induced local maxima. To limit the computation overhead, all the steps of the algorithm are limited to a small volume around each of the nuclei containing sufficient background to allow a robust intensity discrimination. Segmentation of the nuclei is the first step of the algorithm and uses a modified isovalue algorithm. Statistically significant spots are assigned to the nucleus containing them for further statistics. The algorithm for spot counting is part of a larger software package written in house: Qu. Qu is written in the MATLAB programming language (The MathWorks, Inc, Natick, MA), and can be obtained upon request.
Supplementary Material
Supplementary data
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Acknowledgments
We thank the Baldacci and Gasser Laboratories and D Schübeler for helpful discussions and critical reading of the manuscript. This work was supported in part by an EMBO short-term Fellowship to PM, by the ARC grant 3471 to GB and by the Institut Curie PIC. PM received support from the French MRT and the Association pour la Recherche sur le Cancer (ARC). The Gasser Laboratory is supported by the Swiss NCCR program ‘Frontiers in Genetics', Oncosuisse and the Novartis Research Foundation.
References
- Berezney R, Dubey DD, Huberman JA (2000) Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 108: 471–484 [DOI] [PubMed] [Google Scholar]
- Boddy MN, Lopez-Girona A, Shanahan P, Interthal H, Heyer WD, Russell P (2000) Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol Cell Biol 20: 8758–8766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM (2003) DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J 22: 4325–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb JA, Schleker T, Rojas V, Bjergbaek L, Tercero JA, Gasser SM (2005) Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev 19: 3055–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PR (2001) Principles of Nuclear Structure and Function. Wilmington, Delaware, USA: Wiley-Liss Inc [Google Scholar]
- Dimitrova DS, Gilbert DM (2000) Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol 2: 686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncker BP, Brown GW (2003) Cdc7 kinases (DDKs) and checkpoint responses: lessons from two yeasts. Mutat Res 532: 21–27 [DOI] [PubMed] [Google Scholar]
- Feng W, Collingwood D, Boeck ME, Fox LA, Alvino GM, Fangman WL, Raghuraman MK, Brewer BJ (2006) Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat Cell Biol 8: 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BM, Fangman WL (1992) A position effect on the time of replication origin activation in yeast. Cell 68: 333–339 [DOI] [PubMed] [Google Scholar]
- Gartenberg MR, Neumann FR, Laroche T, Blaszczyk M, Gasser SM (2004) Sir-mediated repression can occur independently of chromosomal and subnuclear contexts. Cell 119: 955–967 [DOI] [PubMed] [Google Scholar]
- Gilbert DM (2001) Making sense of eukaryotic DNA replication origins. Science 294: 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heichinger C, Penkett CJ, Bahler J, Nurse P (2006) Genome-wide characterization of fission yeast DNA replication origins. EMBO J 25: 5171–5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R (1996) R: A language and environment for statistical computing. J Comput Graph Stat 5: 299–314 [Google Scholar]
- Jeon Y, Bekiranov S, Karnani N, Kapranov P, Ghosh S, MacAlpine D, Lee C, Hwang DS, Gingeras TR, Dutta A (2005) Temporal profile of replication of human chromosomes. Proc Natl Acad Sci USA 102: 6419–6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey SE, Montgomery S, Labib K, Lindner K (2000) Chromatin binding of the fission yeast replication factor mcm4 occurs during anaphase and requires ORC and cdc18. EMBO J 19: 1681–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Dubey DD, Huberman JA (2003) Early-replicating heterochromatin. Genes Dev 17: 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Huberman JA (2001) Regulation of replication timing in fission yeast. EMBO J 20: 6115–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura E, Blow JJ, Tanaka TU (2006) Live-cell imaging reveals replication of individual replicons in eukaryotic replication factories. Cell 125: 1297–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruizinga P, Petkov N (2000) Computational model of dot-pattern selective cells. Biol Cybern 83: 313–325 [DOI] [PubMed] [Google Scholar]
- Lengronne A, Pasero P, Bensimon A, Schwob E (2001) Monitoring S phase progression globally and locally using BrdU incorporation in TK(+) yeast strains. Nucleic Acids Res 29: 1433–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- Ma H, Samarabandu J, Devdhar RS, Acharya R, Cheng PC, Meng C, Berezney R (1998) Spatial and temporal dynamics of DNA replication sites in mammalian cells. J Cell Biol 143: 1415–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine DM, Rodriguez HK, Bell SP (2004) Coordination of replication and transcription along a Drosophila chromosome. Genes Dev 18: 3094–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marheineke K, Hyrien O (2004) Control of replication origin density and firing time in Xenopus egg extracts: role of a caffeine-sensitive, ATR-dependent checkpoint. J Biol Chem 279: 28071–28081 [DOI] [PubMed] [Google Scholar]
- Meister P, Poidevin M, Francesconi S, Tratner I, Zarzov P, Baldacci G (2003) Nuclear factories for signalling and repairing DNA double strand breaks in living fission yeast. Nucleic Acids Res 31: 5064–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister P, Taddei A, Vernis L, Poidevin M, Gasser SM, Baldacci G (2005) Temporal separation of replication and recombination requires the intra-S checkpoint. J Cell Biol 168: 537–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Seiler JA, Burhans WC (2003) Regulation of cellular and SV40 virus origins of replication by Chk1-dependent intrinsic and UVC radiation-induced checkpoints. J Biol Chem 278: 4295–4304 [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol 194: 795–823 [DOI] [PubMed] [Google Scholar]
- Noguchi E, Noguchi C, McDonald WH, Yates JR III, Russell P (2004) Swi1 and swi3 are components of a replication fork protection complex in fission yeast. Mol Cell Biol 24: 8342–8355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P, Braguglia D, Gasser SM (1997) ORC-dependent and origin-specific initiation of DNA replication at defined foci in isolated yeast nuclei. Genes Dev 11: 1504–1518 [DOI] [PubMed] [Google Scholar]
- Patel PK, Arcangioli B, Baker SP, Bensimon A, Rhind N (2006) DNA replication origins fire stochastically in fission yeast. Mol Biol Cell 17: 308–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piard K, Baldacci G, Tratner I (1998) Single point mutations located outside the inter-monomer domains abolish trimerization of Schizosaccharomyces pombe PCNA. Nucleic Acids Res 26: 2598–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL (2001) Replication dynamics of the yeast genome. Science 294: 115–121 [DOI] [PubMed] [Google Scholar]
- Raveendranathan M, Chattopadhyay S, Bolon YT, Haworth J, Clarke DJ, Bielinsky AK (2006) Genome-wide replication profiles of S-phase checkpoint mutants reveal fragile sites in yeast. EMBO J 25: 3627–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santocanale C, Diffley JF (1998) A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395: 615–618 [DOI] [PubMed] [Google Scholar]
- Schubeler D, Scalzo D, Kooperberg C, van Steensel B, Delrow J, Groudine M (2002) Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat Genet 32: 438–442 [DOI] [PubMed] [Google Scholar]
- Schwaiger M, Schubeler D (2006) A question of timing: emerging links between transcription and replication. Curr Opin Genet Dev 16: 177–183 [DOI] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J (2004) ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 6: 648–655 [DOI] [PubMed] [Google Scholar]
- Shechter D, Gautier J (2005) ATM and ATR check in on origins: a dynamic model for origin selection and activation. Cell Cycle 4: 235–238 [PubMed] [Google Scholar]
- Shimada K, Pasero P, Gasser SM (2002) ORC and the intra-S-phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev 16: 3236–3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahige K, Hori Y, Shiraishi K, Yamashita M, Takahashi K, Obuse C, Tsurimoto T, Yoshikawa H (1998) Regulation of DNA-replication origins during cell-cycle progression. Nature 395: 618–621 [DOI] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602 [DOI] [PubMed] [Google Scholar]
- Sorensen CS, Syljuasen RG, Lukas J, Bartek J (2004) ATR, Claspin and the Rad9–Rad1–Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle 3: 941–945 [PubMed] [Google Scholar]
- Sporbert A, Gahl A, Ankerhold R, Leonhardt H, Cardoso MC (2002) DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol Cell 10: 1355–1365 [DOI] [PubMed] [Google Scholar]
- Stevenson JB, Gottschling DE (1999) Telomeric chromatin modulates replication timing near chromosome ends. Genes Dev 13: 146–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Belmont AS, Robinett CC, Murray AW (1996) GFP tagging of budding yeast chromosomes reveals that protein-protein interactions can mediate sister chromatid cohesion. Curr Biol 6: 1599–1608 [DOI] [PubMed] [Google Scholar]
- Taddei A, Hediger F, Neumann FR, Gasser SM (2004) The function of nuclear architecture: a genetic approach. Annu Rev Genet 38: 305–345 [DOI] [PubMed] [Google Scholar]
- Tubo RA, Berezney R (1987) Identification of 100 and 150 S DNA polymerase alpha-primase megacomplexes solubilized from the nuclear matrix of regenerating rat liver. J Biol Chem 262: 5857–5865 [PubMed] [Google Scholar]
- Zappulla DC, Sternglanz R, Leatherwood J (2002) Control of replication timing by a transcriptional silencer. Curr Biol 12: 869–875 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4