

Abstract
The deafness (dn) and Beethoven (Bth) mutant mice are models for profound congenital deafness (DFNB7/B11) and progressive hearing loss (DFNA36), respectively, caused by recessive and dominant mutations of transmembrane cochlear-expressed gene 1 (TMC1), which encodes a transmembrane protein of unknown function. In the mouse cochlea Tmc1 is expressed in both outer (OHCs) and inner (IHCs) hair cells from early stages of development. Immature hair cells of mutant mice seem normal in appearance and biophysical properties. From around P8 for OHCs and P12 for IHCs, mutants fail to acquire (dn/dn) or show reduced expression (Bth/Bth and, to a lesser extent Bth/+) of the K+ currents which contribute to their normal functional maturation (the BK-type current IK,f in IHCs, and the delayed rectifier IK,n in both cell types). Moreover, the exocytotic machinery in mutant IHCs does not develop normally as judged by the persistence of immature features of the Ca2+ current and exocytosis into adulthood. Mutant mice exhibited progressive hair cell damage and loss. The compound action potential (CAP) thresholds of Bth/+ mice were raised and correlated with the degree of hair cell loss. Homozygous mutants (dn/dn and Bth/Bth) never showed CAP responses, even at ages where many hair cells were still present in the apex of the cochlea, suggesting their hair cells never function normally. We propose that Tmc1 is involved in trafficking of molecules to the plasma membrane or serves as an intracellular regulatory signal for differentiation of immature hair cells into fully functional auditory receptors.
Deafness (dn) is a spontaneous autosomal recessive mutation in mice that leads to a complete lack of cochlear responses (measured up to 20 days after birth) and is associated with sensory hair cell defects in homozygotes (Steel & Bock, 1980; Bock & Steel, 1983). Although spontaneous auditory nerve activity is absent in adult dn mice (Durham et al. 1989), functional connections in the central auditory pathway do occur (Bock et al. 1982). The semidominant Beethoven (Bth) mutation arose from an ENU (N-ethyl-N-nitrosourea) mutagenesis programme (Hrabé de Angelis et al. 2000) and leads to progressive hearing loss in heterozygote and early onset deafness in homozygote mice (Vreugde et al. 2002). Both mutations affect transmembrane cochlear-expressed gene 1 (Tmc1: Vreugde et al. 2002; TMC1: Kurima et al. 2002). Mutations in human TMC1 cause dominant progressive hearing loss (DFNA36) and recessive profound congenital deafness (DFNB7/B11) (Kurima et al. 2002). Tmc1 is a member of a novel gene family and the function of the transmembrane protein that it encodes has remained elusive (Kurima et al. 2002; Vreugde et al. 2002). Although in situ hybridization studies have shown that in the mouse cochlea Tmc1 is strongly expressed in hair cells from postnatal day 5 (P5) onwards (Kurima et al. 2002; Vreugde et al. 2002), the protein is first detected just before the onset of hearing at about P10 in the pericuticular necklace and the endoplasmic reticulum (Makishima et al. 2005).
Functional maturation of mouse cochlear hair cells is a complex process that starts just after terminal mitosis at embryonic day 14.5 (E14.5) and lasts for about three to four weeks (Pujol et al. 1998). During this period, numerous morphological (Pujol et al. 1998) and electrophysiological (Eatock & Hurley, 2003) changes occur within the cochlea. Some of these changes are important for turning functionally immature hair cells into highly specialized sensory cells. Inner hair cells (IHCs), the primary sensory receptors of the cochlea, can fire Ca2+ action potentials (Kros et al. 1998; Marcotti et al. 2003b) before the onset of hearing, at around P10–P12 in mice (Romand, 1983). However, slow action potentials are not suited for encoding the high-frequency sounds that are typically transduced by the adult mammalian cochlea. This problem is solved in IHCs with the expression at around P12 of the large and rapidly activating BK-current IK,f (Kros et al. 1998; Marcotti et al. 2004a) and the unusually negatively activating delayed rectifier K+ current IK,n (Marcotti et al. 2003a; Oliver et al. 2003). By reducing the cell membrane time constant, these K+ currents allow IHCs to respond to sound with fast, graded receptor potentials. The onset of maturation in outer hair cells (OHCs), which are responsible for the amplification and tuning of auditory responses, starts from around P8 with the acquisition of both IK,n (Marcotti & Kros, 1999) and the electromotile activity (He et al. 1994; Marcotti & Kros, 1999) that is mediated by the motor protein prestin (Zheng et al. 2000). Since the expression of the protein encoded by Tmc1 takes place during this period of major developmental biophysical changes in both hair cell types, we investigated whether, when mutated in Bth and dn mice, it would affect normal development of hair cell function. The relation between abnormal hair cell physiology, degeneration and hearing loss was also studied.
Methods
Mice
The Beethoven (Tmc1Bth, also referred to as Bth below) colony was maintained on a C3HeB/FeJ background. The deafness (Tmc1dn, also referred to as dn below) stock has been maintained on an undefined genetic background within a closed colony for over 20 years. All animal experiments were carried out in full compliance with UK Home Office regulations.
Genotyping
Primers to identify the Tmc1Bth mutation were designed from the Tmc1 genomic sequence. Mice from the Beethoven stock were genotyped using PCR to amplify a 166-bp product around the point mutation in exon 13. The primers were: 5′-GAA CAT GGT AAT GTC CCT CCT GGG CA-3′ and 5′-CTC ATC CAT CAA GGC GAG AAT GAA-3′. The primers were designed to introduce a second mutation in the PCR product, which created an NspI restriction site in the wild type; subsequent digestion gave 142 bp and 30 bp fragments from wild-type DNA and 166 bp fragments from the Bth allele.
The mutation in Tmc1 in deafness mice is a 1.6-kb deletion (Kurima et al. 2002). This deletion includes all of exon 14 and part of introns 13 and 14. Primers were designed from the genomic sequence with one forward primer and two reverse primers. The primer sequence for the forward primer (5′-ATG TTC TGT CCC ACC CTG TTT GA-3′) was designed from intron 13. The sequences for reverse primer 1 (5′-CCC ACT ACA CTG TAG CCC AC-3′) and reverse primer 2 (5′-ACT ACC CAC TAC ACC CTA-3′) were designed from the Tmc1 sequence published in Kurima et al. (2002). Reverse primer 2 is designed from the sequence that is created when the intron 13 and intron 14 sequences meet after the deletion has occurred. Therefore mice carrying the dn allele will give a PCR product of approximately 200 bp when using reverse primer 2 and the forward primer. The wild-type allele will not be amplified as the primer sequence for either intron 13 or intron 14, which is contained in reverse primer 2, will not be long enough to allow robust binding. To amplify the wild-type allele, a long-range PCR reaction is used with reverse primer 1, designed from the sequence in intron 14, and the forward primer. The amplification product is approximately 2 kb long.
Structural investigations
For scanning electron microscopy (SEM), inner ears were fixed in glutaraldehyde, the organ of Corti was exposed by dissection, and samples were processed by a modified OTOTO method (Hunter-Duvar, 1978). Cochleae were examined in Beethoven (number of mice at P15: 5 +/+, 6 Bth/+, 6 Bth/Bth; P30–35: 19 +/+, 21 Bth/+, 4 Bth/Bth; P60: 5 +/+, 5 Bth/+, 5 Bth/Bth) and deafness (P30: 5 +/dn, 5 dn/dn).
Hair cell degeneration was quantified by counting the number of normal cells and the number of degenerating cells remaining in the organ of Corti from montages of scanning electron micrographs. Three distinct areas of the cochlea were analysed: 10–20%, 40–50% and 80–90% of the total length from the base. The corresponding approximate best frequency ranges are: 47–67 kHz, 15–22 kHz and 1.3–3.2 kHz, respectively (from eqn 13 in Ehret, 1975). The areas counted were a minimum of 100 μm in length, and number of cells counted was expressed per 100 μm. Hair cells were counted from 4–7 mice at each location of each genotype at each age. Hair cells were counted as normal if their hair bundle showed the normal V- (OHCs) or crescent-shaped (IHCs) arrangement with distinct stereocilia. Hair cells were classed as degenerating or damaged if they showed any defects (viewed at a higher resolution than Figs 1A and 2), including missing, damaged or fused stereocilia. This method is likely to lead to an underestimation of the degenerating cells since the hair bundle structure might not be affected during early stages of cell deterioration. In fact, a previous investigation has shown that all hair cells from deafness mice exhibit subtler signs of degeneration in the cytoplasm and the synaptic region as early as P15 when viewed using transmission electron microscopy, together with evidence of delayed maturation of the spaces of Nuel and tunnel of Corti by about 10 days (Bock & Steel, 1983). SEM was used here to allow quantitative comparisons of a large number of hair cells in different cochlear locations of Beethoven and deafness mice.
Figure 1. Morphological and physiological properties of deafness cochleae.
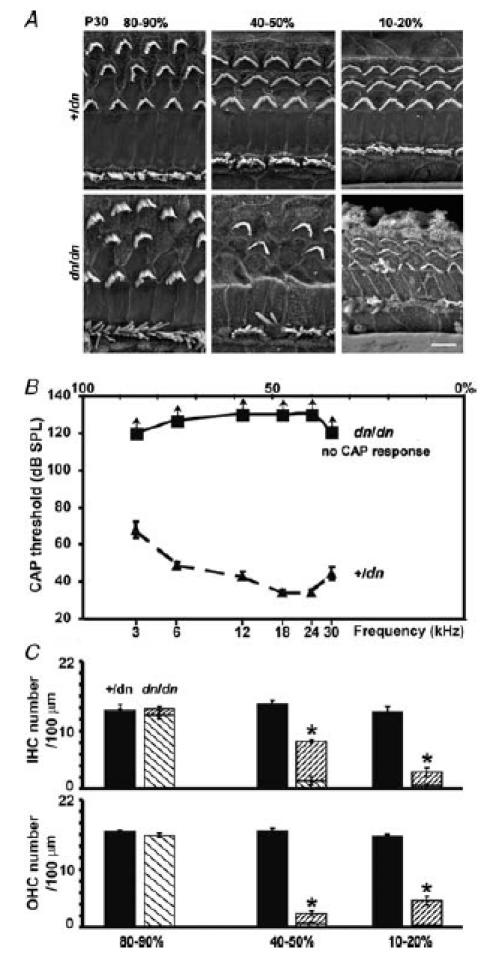
A, low-resolution scanning electron micrographs (SEM) from the apical (80–90%), middle (40–50%) and basal (10–20%) regions of P30 +/dn and dn/dn cochleae, showing the three rows of V-shaped hair bundles of OHCs and one row of crescent-shaped bundles of IHCs. Hair cell degeneration in dn/dn only appears to be present in the 40–50% and 10–20% regions. Scale bar represents 5 μm. B, mean compound action potential (CAP) thresholds in +/dn (▴) and dn/dn (▪) mice aged P30. The dn/dn mice showed no CAP response at the maximum sound intensities that could be used, which is represented with arrows pointing upwards. Sound frequency (lower axis) is scaled tonotopically as percentage distance (upper axis) from the base (calculated from Ehret, 1975). C, hair cell counts, using high-resolution SEM, per 100 μm in the same three relevant areas of P30 mouse cochleae described in A. Filled and diagonally striped bars (\) represent normal hair cells observed in the +/dn and dn/dn cochlea, respectively. The more densely striped bars (/) represent degenerating hair cells.
Figure 2. Morphological characteristics of Beethoven cochleae.
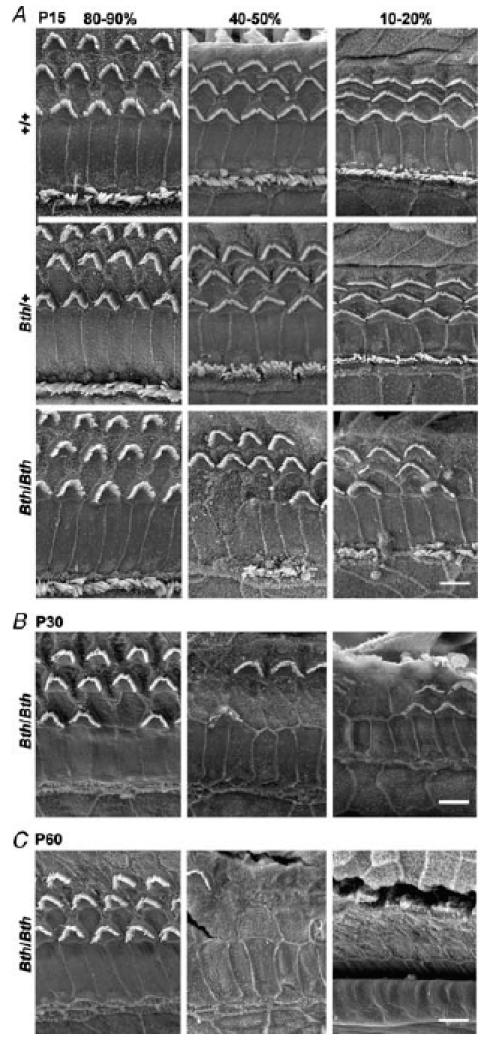
A, low-resolution SEM of +/+, Bth/+ and Bth/Bth at P15 from the same three cochlear regions as in Fig. 1A, B and C, SEM of Bth/Bth at P30 and P60, respectively. Bth/Bth cochleae show degeneration of both hair cell types in the 40–50% and 10–20% regions at all ages. No clear signs of degeneration were observed in the apical region (80–90%) of P15 Bth/Bth, or in any region of P15 Bth/+ cochleae. Scale bar represents 5 μm.
Cochlear electrophysiology
Mice were anaesthetized with urethane (i.p. injection using 2 mg (g body weight)−1 as a 20% solution), a recording electrode was placed on the round window of the cochlea, and thresholds for compound action potential (CAP: a measure of the average firing activity of the afferent auditory fibres) responses determined in 2 dB steps using a standard approach (Steel & Smith, 1992). The endocochlear potential, which is the transepithelial potential of the endolymph within the cochlear duct that increases the driving force for the transducer current, was recorded using a micropipette electrode inserted into the basal turn cochlear duct through the lateral wall (Steel & Smith, 1992). Both Beethoven (P15: 5 +/+, 8 Bth/+, 6 Bth/Bth; P30: 9 +/+, 12 Bth/+, 6 Bth/Bth; P60: 5 +/+, 5 Bth/+, 5 Bth/Bth) and deafness (P30: 5 +/dn, 5 dn/dn) mice were used for this analysis.
Single-hair cell electrophysiology
Tissue preparation
Apical-coil IHCs and OHCs of Beethoven (Bth) and deafness (dn) mutant mice and their phenotypically normal controls from the same strains were studied in acutely dissected organs of Corti (ages P6–P22 for Bth and P7–P58 for dn, where the day of birth is P0). Adult and immature mice were killed by cervical dislocation, in accordance with UK Home Office regulations. The cochleae were dissected in extracellular solution composed of (mM): 135 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 5.6 d-glucose, 10 Hepes-NaOH, 2 Na-pyruvate (pH 7.5, osmolality about 306 mmol kg−1). Amino acids and vitamins for Eagle's minimum essential medium (MEM) without l-glutamine were added from concentrates (Invitrogen, Paisley, UK). The organs of Corti were transferred to a microscope chamber containing extracellular solution, and immobilized under a nylon mesh. The chamber (volume 2 ml) was perfused at a flow rate of about 10 ml h−1 from a peristaltic pump, and mounted on the stage of an upright microscope (Zeiss ACM, Germany or Olympus, Japan). The organs of Corti were observed with Nomarski differential interference contrast optics (×40 water immersion objectives). The position of hair cells along the cochlea was calculated as percentage distance from the extreme base. In the immature cochlea, hair cells from which electrophysiological recordings were obtained were positioned at a distance of 76% to 84% from the base. Recordings in the mature cochlea were from cells positioned between 81% and 94% from the base, corresponding to a frequency range of approximately 0.8–3.0 kHz (Ehret, 1975). Tissue samples from all mice were kept for genotyping.
Electrical properties of the basolateral membrane
Hair cells (IHCs: 26 +/+, 27 Bth/+, 29 Bth/Bth, 16 +/dn, 28 dn/dn; OHCs: 13 +/+, 22 Bth/+, 30 Bth/Bth, 14 +/dn, 18 dn/dn) were whole-cell voltage clamped at room temperature (20–25°C) or near body temperature (35–37°C) using EPC-8 (HEKA, Lambrecht, Germany) or Optopatch (Cairn Research Ltd, Faversham, UK) amplifiers. Patch pipettes were pulled from soda glass capillaries (Harvard Apparatus Ltd, Edenbridge, UK) and electrode resistances in extracellular solution were 2–3 MΩ. In order to reduce the electrode capacitance, the shank of the electrode was coated with surf wax (Mr Zogs SexWax, Carpinteria, CA, USA). The normal pipette filling solution contained (mm): 131 KCl, 3 MgCl2, 1 EGTA-KOH, 5 Na2ATP, 5 Hepes-KOH, 10 Na-phosphocreatine (pH 7.3, 292 mmol kg−1). Calcium currents (ICa) were recorded using the following intracellular solution to minimize contamination due to the K+ currents (mm): 147 Cs-glutamate, 3 MgCl2, 5 Na2ATP, 0.3 Na2GTP, 1 EGTA-NaOH, 5 Hepes-CsOH (pH 7.3, 292 mmol kg−1). Data were acquired using pClamp software (Axon Instruments, Union City, CA, USA) with a LabMaster DMA or a Digidata 1320A data acquisition board. Data were filtered, depending on the protocols used, at 2.5, 5 or 10 kHz (8-pole Bessel), sampled at 5, 10 or 50 kHz and stored on computer. Offline data analysis was performed using Origin software (OriginLab, Northampton, MA, USA). For voltage-clamp experiments, current recordings were corrected offline for leak conductance (gleak). In most cases gleak was calculated between −84 mV and − 94 mV, as the main outward K+ currents activate positive to or around −80 mV. When the inward rectifier K+ current (IK1) was studied in immature IHCs gleak was calculated in response to 10 mV hyperpolarizing voltage steps from a holding potential of −64 mV (Marcotti et al. 1999). Values for gleak were 1.9 ± 0.1 nS (n = 63, P6–P8) for immature hair cells and 3.2 ± 0.3 nS (n = 62, P15–P58) for mature IHCs after the onset of hearing. In the experiments in which the negatively activating K+ current (IK,n) was studied in mature cells, gleak(IHCs: 1.1 ± 0.1 nS, n = 36; OHCs: 1.2 ± 0.1 nS, n = 49) was measured at very hyperpolarized potentials at which IK,n was deactivated (Marcotti & Kros, 1999; Marcotti et al. 2003a). Residual series resistance (Rs) after compensation (50–90%) was 2.5 ± 0.1 MΩ (range 0.4–9.8 MΩ, n = 223). Membrane potentials under voltage clamp were corrected for the voltage drop across the residual Rs at steady-state current level and for a liquid junction potential, measured between pipette and bath solutions, of −4 mV for the KCl-based and −11 mV for the Cs-glutamate-based intracellular solution. For current-clamp experiments, offline series resistance correction was applied only if the voltage drop exceeded 1 mV. For voltage-clamp recordings, the holding currents were plotted as zero current unless otherwise specified.
Mechano-electrical transducer currents
Mechano-electrical transducer currents were elicited in apical-coil OHCs (P6–P8) from Bth and dn mutant mice, using a fluid jet driven by a piezoelectric disc and recorded under whole-cell voltage clamp as previously described (Kros et al. 1992). Mechanical stimuli were applied using 45 Hz sinusoids with a driver-voltage amplitude of 35 V, resulting in saturating transducer currents (filtered at 1 kHz, 8-pole Bessel). Although bundle movements were not recorded for this study, the corresponding bundle displacements should be in the order of 150–200 nm (Géléoc et al. 1997; Marcotti et al. 2005). Data were acquired using Asyst software (Keithley Instruments, Rochester, NY, USA), filtered at 2.5 kHz, sampled at 5 kHz and stored on computer for off-line analysis using Origin software. For these experiments, no correction was made for the voltage drop across Rs, which was at most 4 mV at extreme potentials.
Membrane capacitance measurements
Real-time changes in membrane capacitance (ΔCm) were studied under voltage clamp at 37°C and measured using the track-in mode of the Optopatch (Johnson et al. 2002, 2005) in both Beethoven (P16–P17, n = 13) and deafness (P17–P18, n = 9) apical-coil IHCs. Simultaneous recordings of ΔCm and Ca2+ currents were performed using the same intracellular solutions used for ICa measurements (see above). A 2.5 kHz sine wave of 18.5 mV amplitude was applied to IHCs from a holding potential of −81 mV using the internal oscillator of the Optopatch. The sine wave was small enough not to activate any significant membrane current, since accurate membrane capacitance calculation requires a high and constant membrane resistance. The command sine wave was interrupted for the duration of the voltage steps so inward currents could be recorded. The capacitance signal from the Optopatch was amplified (×50), 2-pole filtered at 150 Hz with additional 8-pole Bessel filtering at 250 Hz, and sampled at 5 kHz. During multiple step protocols, the prestimulus Cm baseline for consecutive steps was set to zero during offline analysis. IHCs were stimulated using voltage steps of 100 ms duration from the holding potential of −81 mV to more positive potentials in 10 mV increments. Changes in membrane capacitance were calculated by subtracting the mean capacitance measured over a 300 ms period, starting at around 50 ms after the end of the voltage step, from the mean prepulse capacitance (averaged over a 200 ms period). We allowed approximately 5–10 s per stimulus for vesicle pool replenishment.
Extracellular superfusion
Acetylcholine (Sigma, Gillingham, UK), at a concentration of 100 μm, was used to assess the presence of the small-conductance Ca2+-activated K+ current ISK (Evans, 1996; He & Dallos, 1999; Glowatzki & Fuchs, 2000; Katz et al. 2004; Marcotti et al. 2004b) in mature OHCs and IHCs. Ca2+-free extracellular solutions contained 0.5 mm EGTA, and Mg2+ was increased to 3.9 mm to minimize changes in membrane charge screening. ΔCm and ICa were recorded during the superfusion of 30 mm TEA (Fluka, Gillingham, UK) to block most of the IHC K+ currents. In this solution, NaCl was reduced to keep the osmolality constant. Solutions were applied via a multibarrelled pipette positioned close to the hair cells, so that solution changes were almost instantaneous (dead space within the perfusion pipette is about 1 μl).
Statistical analysis
Statistical comparisons of means were made using Student's two-tailed t test or, for comparisons of multiple data sets, one-way ANOVA (followed by the Tukey post test). Two-way ANOVA, followed by the Bonferroni test, was used to compare CAP thresholds. For all statistical tests P < 0.05 was used as the criterion for statistical significance and mean values are quoted ± standard error (s.e.m.) in text and figures. In all figures, statistically significant difference is indicated by asterisks.
Results
Ultrastructural observations and cochlear function in deafness and Beethoven mutants
Deafness homozygotes (dn/dn) at P30 showed significant hair cell damage and loss in the middle (40–50%) and basal (10–20%) turns of the cochlea (Fig. 1A and C) compared to heterozygous controls (+/dn). In the apical region, many (80–90%) hair cells retained a normal, or near-normal, bundle appearance in dn/dn mice (Fig. 1A). Cells from a similar apical, low-frequency region were selected to study the developmental maturation of the biophysical properties of both IHCs and OHCs, to minimize the confounding factor of hair cell degeneration (see below). The physiological consequences of these hair cell defects were investigated in anaesthetized mice by measuring CAP thresholds. A previous investigation had shown that homozygous deafness mice lacked any cochlear responses (CAP, summating potentials and cochlear microphonics) to sound stimuli between the ages of P12 and P20 (Steel & Bock, 1980; Bock & Steel, 1983). In the present study we found that also at P30 no CAP responses could be elicited in homozygous deafness mice, even at the maximum sound intensity used over the entire frequency range tested (between 3 and 30 kHz), while heterozygotes showed normal thresholds (Fig. 1B). The absence of cochlear responses at low frequencies (3 and 6 kHz), despite the presence of apical-turn hair cells, suggests that these cells are not functioning normally even before they degenerate. Hence, the observed loss and profound degeneration of hair cells in the middle and basal region of the cochlea is likely to be a secondary consequence and not the primary cause of deafness in dn/dn mutants.
Beethoven homozygotes (Bth/Bth) showed hair cell damage and a significant reduction in the number of cells in the basal and middle turns by P15, but no significant hair cell loss in the apical region (Figs 2A and 3A, lower panel). The loss of hair cells in Beethoven homozygotes was even greater at P30 (Fig. 2B and Vreugde et al. 2002) and P60 (Figs 2C and 3B, lower panel), with an unusual pattern in that IHCs were being lost at an earlier age than OHCs. In the apical turns of homozygotes at these older ages, there were stretches with no IHCs remaining, but a complete complement of OHCs. This pattern of hair cell loss clearly differs from that in dn/dn mice, where IHCs and OHCs appear equally affected (Fig. 1C). In contrast to the complete lack of cochlear responses in dn/dn mice, Bth/Bth mice showed summating potential responses, both positive and negative, to sound (data not shown), suggesting some hair cell function in vivo. However, no CAP responses up to the maximum sound intensity used could be recorded in homozygous Beethoven mutant mice at P15 (Fig. 3A), P30 (not shown) and P60 (Fig. 3B). Similar to our findings in dn/dn mice (Fig. 1), the limited extent of hair cell degeneration in the apical turn of the cochlea in Bth/Bth mice did not seem to correlate with the absence of CAP responses already evident at P15, suggesting again that hair cell death alone is not sufficient to explain the lack of cochlear responsiveness.
Figure 3. Physiological properties and hair cell survival of Beethoven cochleae.
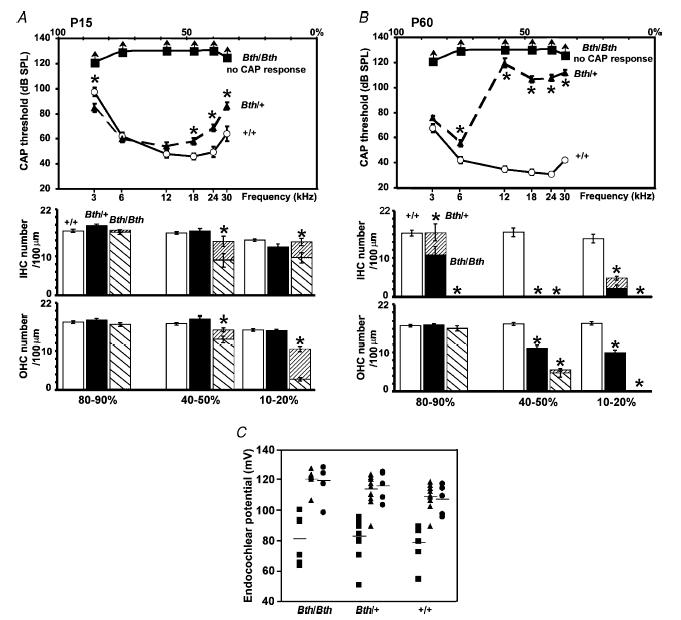
A and B, average CAP (top panels) and hair cell counts per 100 μm of the cochlear coil using high-resolution SEM (bottom panels) in +/+, Bth/+ and Bth/Bth mice at P15 and P60, respectively. The arrows shown for CAP responses in Bth/Bth mice indicate that the actual thresholds could not be measured because they must have been higher than the maximum sound intensities used (▪). The open bars shown in A and B (bottom panels) represent normal hair cells observed in the +/+ cochlea; filled bars are the normal cells in the Bth/+ cochlea, the striped bars (\) indicate normal cells in the Bth/Bth cochlea and the more densely striped bars (/) represent degenerating hair cells in all genotypes. C, endocochlear potential measured in Beethoven mice at P15 (▪), P30 (▴) and P60 (•). Horizontal lines represent the mean values of endocochlear potential at each age.
In the Beethoven heterozygous (Bth/+) mutant mice at P15, there was no obvious hair cell damage or loss in any cochlear turn (Figs 2A and 3A, lower panel), even though there was already a small but significant elevation of CAP thresholds compared to wild-type mice of the same genetic background (+/+) for frequencies of 18 kHz and higher (Fig. 3A, top panel). However, hair cell degeneration was evident in Bth/+ at P30 (Vreugde et al. 2002). A large increase in CAP threshold was observed in Bth/+ at P30 for frequencies of 12 kHz and above (previously reported by Vreugde et al. 2002) with a further worsening by P60 when the 6 kHz threshold was also raised (Fig. 3B, top panel). By contrast, wild-type mice from the Beethoven colony showed some improvement in CAP thresholds from P15 to P60 associated with maturation (Fig. 3A and B, top panels). Beethoven heterozygotes (Bth/+) had by P60 a similar pattern of hair cell loss as the Bth/Bth homozygotes, with IHC loss being more pronounced than OHC loss, but the process was delayed in heterozygotes compared with the homozygotes. Endocochlear potentials are present and entirely normal in Beethoven homozygotes and heterozygotes, increasing in size between P15 and P30 as the potential matures (Fig. 3C). Size and developmental time course of the endocochlear potential were also normal in dn/dn mice (Bock & Steel, 1983). Of the three types of mutant tested in this study, only Bth/+ showed a clear correlation between hair cell loss and raised CAP thresholds. Therefore in the Beethoven heterozygotes the hearing loss might be a consequence of hair cell loss, whereas in the Beethoven and deafness homozygotes a more profound functional problem might underlie the deafness. This hypothesis prompted us to investigate the physiological properties of auditory hair cells in the three mutants, since the two homozygotes might show abnormalities in their physiological development not detected in the Beethoven heterozygotes (Vreugde et al. 2002).
Deafness and Beethoven outer hair cells have large mechano-electrical transducer currents
The lack of cochlear microphonics (recorded between P12 and P20) in the presence of normal endocochlear potentials in dn/dn mice has prompted speculation that Tmc1 might be the mechano-electrical transducer channel (Steel & Bock, 1980; Kurima et al. 2002). Therefore we first sought to establish whether mechano-electrical transduction was functional in these mutants. OHCs only were studied because their transducer currents could be more conveniently recorded than those of IHCs, and because the contribution of IHCs to the cochlear microphonic is thought to be small (Dallos et al. 1972). Transducer currents in apical-coil OHCs (P6–P8) from both Beethoven and deafness control and mutant mice were elicited by alternating inhibitory and excitatory bundle displacements using sinusoidal force stimuli (Kros et al. 1992). When saturating excitatory stimuli were applied to the bundle, large transducer currents, up to about −1500 pA at −104 mV and in 1.3 mm extracellular Ca2+, could be recorded in both deafness (+/dn and dn/dn) and Beethoven (+/+, Bth/+ and Bth/Bth) OHCs. Figure 4A and B shows examples of mechano-electrical transducer currents recorded from heterozygous control (+/dn) and homozygous (dn/dn) deafness OHCs at P8, which in the mouse corresponds to their onset of functional maturation (Marcotti & Kros, 1999). At negative membrane potentials, large inward transducer currents were elicited upon moving the bundles towards the kinocilium in the excitatory direction. When the bundles were moved in the inhibitory direction any transducer channels open in the absence of mechanical stimulation (normally about 5–10%) were closed, resulting in a reduction of the inward current. Starting from −104 mV and stepping the membrane potential to more depolarized values, the transducer currents decreased in size at first, and then reversed near 0 mV in both +/dn (+4.6 ± 1.3 mV, n = 4) and dn/dn (+4.0 ± 0.5 mV, n = 6) OHCs, commensurate with the currents flowing through non-selective cation channels (Ohmori, 1985). For membrane potentials positive to this reversal potential, fluid jet stimuli in the excitatory direction now caused outward currents. Figure 4C and D shows the size of the transducer current recorded at −104 mV and +96 mV in both deafness (controls: +/dn; mutants: dn/dn) and Beethoven (controls: +/+; mutants: Bth/+ and Bth/Bth) OHCs, respectively, when saturating sinusoidal stimuli were applied to the bundle. The pooled average current sizes, recorded from OHCs at immature stages (P6–P7) and at their onset of maturation (P8), did not differ significantly between controls and mutants in both Beethoven (see also Vreugde et al. 2002) and deafness OHCs. The resting open probability of the transducer channels in +/dn OHCs increased from 5.6 ± 1.7% at −104 mV to 31.6 ± 6.1% at +96 mV (n = 4, P7–P8). Recordings from dn/dn cells yielded similar values (−104 mV: 5.0 ± 0.5%; +96 mV: 33.6 ± 4.0%, P7–P8, n = 6) Similar entirely normal shifts in resting open probability upon depolarization were found in Beethoven OHCs (P6–P8). The larger open probability at positive membrane potentials reflects an increased resting tension on the transducer channels due to the smaller driving force for Ca2+ influx reducing adaptation (Crawford et al. 1989).
Figure 4. Mechano-electrical transducer currents in deafness and Beethoven mutant mice.
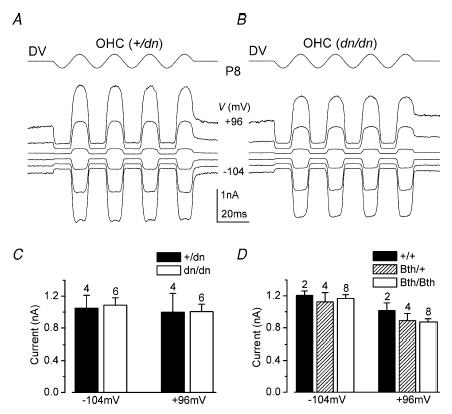
A and B, transducer currents recorded from a control +/dn (A) and a mutant dn/dn (B) OHC (P8, apical coil) by applying sinusoidal force stimuli of 45 Hz. Driver voltage signal (DV; amplitude 35 V) to the fluid jet is shown above the traces. Positive deflections of the DV are excitatory. The cells were held at −84 mV, and the membrane potential was stepped between −104 mV and +96 mV in 20 mV increments. For clarity, only responses to every other voltage step are shown. Current recordings in this and in the following figures are single traces unless otherwise stated. Traces in A and B are offset so that the zero-transducer current levels (responses to inhibitory stimuli) are equally spaced. +/dn: Cm 6.3 pF, Rs 1.2 MΩ. dn/dn: Cm 5.8 pF, Rs 2.5 MΩ. Bundle height for both cells 4 μm. C and D, absolute size of the transducer current, using saturating stimuli, at membrane potentials of −104 mV and + 96 mV in dn (C) and Bth (D) controls and mutant mice. All recordings were at room temperature.
OHCs from deafness mice fail to develop adult-type basolateral ion channels
Since the transducer current recorded from OHCs (+/dn and dn/dn) during either immature stages (P6–P7) or at their onset of functional maturation (P8) appeared unaffected by the mutation in Tmc1, we investigated whether basolateral membrane currents expressed by hair cells were altered. Typical examples of K+ currents recorded from +/dn and dn/dn immature (P7) OHCs are shown in Fig. 5A and B, respectively. Voltage steps positive to about −50 mV caused the activation of voltage-dependent outward currents (a delayed rectifier-type K+ current named IK,neo, Kros et al. 1998) in all OHCs investigated. The size of IK,neo, measured at 160 ms and at 0 mV, was not significantly different between control and mutant OHCs (Fig. 5C). The activation curve and the activation kinetics of IK,neo were also similar between mutant and control OHCs and to those found in normal age-matched CD-1 mice (Marcotti & Kros, 1999). Although we did not analyse ICa in immature OHCs in isolation, its presence was evident from the small inward currents preceding the much slower outward IK,neo in both +/dn and dn/dn cells (Fig. 5A and B, arrows). Moreover, dn/dn OHCs showed similar resting membrane potentials and linear leak conductance as +/dn controls, suggesting the absence of cell degeneration during immature stages of development. These results indicate that immature OHCs are unlikely to be affected by the mutation in Tmc1.
Figure 5. K+ currents in OHCs from deafness mutant mice.
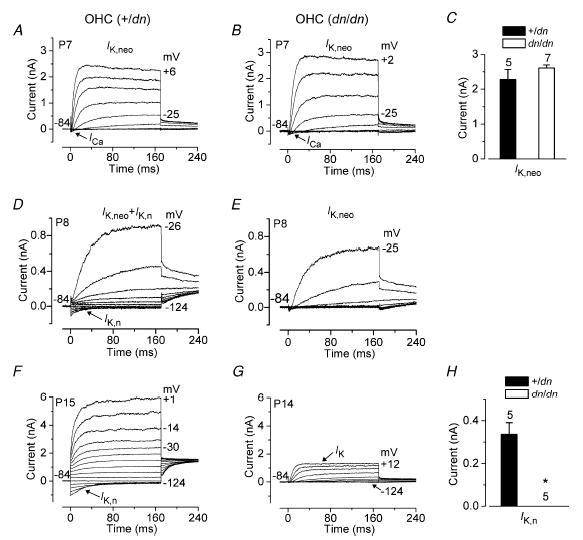
A and B, total current recorded from a control (A, +/dn) and a mutant (B, dn/dn) P7 apical-coil OHC. Membrane currents were elicited in response to depolarizing voltage steps (10 mV increments) from −104 mV to the various test potentials shown by some of the traces, starting from the holding potential of −84 mV. In this and in the following figure (panels A and B), arrows indicate the presence of the inward Ca2+ currents in immature IHCs. +/dn: Vm−68 mV; Cm 6.6 pF; Rs 4.1 MΩ; gleak 1.5 nS; dn/dn: Vm−69 mV; Cm 6.3 pF; Rs 1.5 MΩ; gleak 2.6 nS. C, size of IK,neo measured at 160 ms and at 0 mV in both +/dn and dn/dn apical OHCs. D and E, typical membrane currents recorded from +/dn (D) and dn/dn (E) P8 apical-coil OHCs. Currents were elicited by applying depolarizing voltage steps in 10 mV increments starting from −124 mV from the holding potential of −84 mV. Note the absence of the negatively activating K+ current IK,n in the dn/dn OHC (E). +/dn: Vm−72 mV; Cm 5.8 pF; Rs 1.8 MΩ; gleak 1.8 nS; dn/dn: Vm−66 mV; Cm 5.7 pF; Rs 1.4 MΩ; gleak 1.7 nS. F and G, membrane currents recorded from +/dn (P15) and dn/dn (P14) OHCs, respectively. Voltage protocol as in D and E. IK,n was absent in dn/dn OHCs, leaving only the small delayed rectifier IK. +/dn: Vm−79 mV; Cm 10.8 pF; Rs 2.5 MΩ; gleak 2.0 nS. dn/dn: Vm−66 mV; Cm 8.2 pF; Rs 2.8 MΩ; gleak 1.6 nS. H, size of IK,n measured as the difference between instantaneous and steady-state deactivating tail currents for voltage steps from −84 mV to −124 mV in +/dn (n = 5, P14–P15) and dn/dn (n = 5, P14) mice. All recordings were at room temperature.
The onset of functional maturation in mouse OHCs starts at P8 with the acquisition of both the slow delayed rectifier K+ current IK,n, which has a hyperpolarized activation range starting from −136 mV (calculated as 5% of the maximum activation), and electromotility (Marcotti & Kros, 1999). Figure 5D shows typical current recordings from a control heterozygous (+/dn) P8 OHC. Hyperpolarizing voltage steps from a holding potential of −84 mV elicited an inward K+ current that commenced instantaneously and decayed slowly to a steady-state level, indicating deactivation of channels that were open at the holding potential, similar to previously described results in OHCs for IK,n of normal CD-1 mice (Marcotti & Kros, 1999). Depolarizing voltage steps elicited IK,neo in addition to IK,n. When the same voltage protocol was applied to dn/dn P8 OHCs, only IK,neo was evident (Fig. 5E). The absence of IK,n in dn/dn cells was even more evident at later stages of development (Fig. 5G) when OHCs from +/dn (Fig. 5F) and normal CD-1 mice (Marcotti & Kros, 1999) expressed a large IK,n. The small remaining outward K+ current in dn/dn OHCs (IK) activates from a much more depolarized potential (about −48 mV: 5% of the maximum activation) than IK,n. This current was previously observed in normal CD-1 mice upon abolishing IK,n with a fully blocking concentration of linopirdine (Marcotti & Kros, 1999). It is possible that IK is simply a downregulated version of IK,neo, but this remains to be investigated in detail. The maximum size of IK,n in control and mutant cells at P14 (Fig. 5H) was measured as the deactivating current at −124 mV (difference between instantaneous and steady-state inward currents).
Since the expression of IK,n in OHCs from normal CD-1 mice coincides with the appearance of the cells' electromotile activity from about P8 onwards (Marcotti & Kros, 1999), we tried to investigate whether electromotility was also affected by the mutation. Despite the absence of IK,n, dn/dn OHCs (P19, n = 3) exhibited clear electromotile activity that was visualized under the experimental microscope when cells were depolarized from the holding potential of −64 mV (Marcotti & Kros, 1999). Non-linear capacitance was also evident in the current recordings. After reaching the whole-cell configuration, adult mutant OHCs appeared to be quite fragile, and usually only lasted long enough to record a few voltage-clamp protocols. This short lifetime prevented us from successfully measuring the displacements, so any modest reduction in electromotility may have gone undetected.
Adult-type basolateral ion channels are missing in IHCs from deafness mice
IHCs start to mature a few days later than OHCs, at around the onset of hearing at P12, with the acquisition of a rapidly activating large-conductance Ca2+-activated K+ current (IK,f, Kros et al. 1998; Marcotti et al. 2003a) and a smaller IK,n (Marcotti et al. 2003a; Oliver et al. 2003). Before P12, mouse IHCs express a variety of basolateral membrane currents, some of which are only transiently present during immature stages of development (Marcotti et al. 1999, 2003b, 2004b). Figure 6A and B shows typical outward K+ current (IK,neo) recordings from P7 +/dn and dn/dn IHCs. The activation kinetics, activation curve and size (Fig. 6C) of IK,neo were similar between control and mutant cells and to those found in normal age-matched CD-1 mice (Marcotti et al. 2003a). The size of the inward-rectifier K+ current (IK1), measured at −154 mV (recordings not shown), was also similar between mutant (−472 ± 72 pA, n = 3) and control (−441 ± 42 pA, n = 4) IHCs (Fig. 6C). The presence of ICa in immature IHCs was seen as a small, rapidly activating inward current preceding the much slower outward IK,neo in all +/dn and dn/dn cells investigated (Fig. 6A and B). The peak ICa, measured at room temperature and using 1.3 mm extracellular Ca2+, occurred at around −20 mV, and was similar between +/dn controls (−265 ± 63 pA, n = 4) and mutant dn/dn (−225 ± 24 pA, n = 7) IHCs. These results suggest that mutations in Tmc1 did not prevent the normal physiological development of prehearing IHCs.
Figure 6. K+ currents in IHCs from deafness mutant mice.

A and B, current recordings under voltage clamp from control (+/dn) and mutant (dn/dn) immature apical-coil IHCs (P7), respectively. Outward K+ currents (IK,neo) were elicited in response to depolarizing voltage steps (10 mV nominal increments) from −104 mV starting from the holding potential of −84 mV. +/dn: Vm−72 mV; Cm 7.0 pF; Rs 1.7 MΩ; gleak 4.0 nS; dn/dn: Vm−74 mV; Cm 8.3 pF; Rs 1.2 MΩ; gleak 1.3 nS. Recordings were at room temperature. C, size of IK,neo and the inward rectifier K+ current IK1 measured at 160 ms during voltage steps to 0 mV and −154 mV, respectively, in both +/dn and dn/dn IHCs. D and E, total outward K+ currents recorded from +/dn(IK,f+IK,s) and dn/dn (IK,s) apical IHCs (P58), respectively. Current recordings were elicited by applying depolarizing voltage steps in 10 mV nominal increments from −84 mV. The insets show the onset of the total current recorded from the same cells shown in A and B using shorter (20 ms) voltage steps of which the first 10 ms are shown. Note the slower current activation in dn/dn due to the absence of IK,f. +/dn: Vm−72 mV; Cm 7.2 pF; Rs 1.3 MΩ; gleak 12.0 nS. dn/dn: Vm−69 mV; Cm 6.4 pF; Rs 1.3 MΩ; gleak 1.0 nS. F, size of the total steady-state currents (IK,f+IK,s) and the isolated IK,f and IK,s measured at −25 mV in both +/dn (n = 8, P18–P58) and dn/dn (n = 14, P17–P58) IHCs. G and H, membrane currents recorded from +/dn and dn/dn IHCs (P58) in response to voltage steps in 10 mV increments from −124 mV to more depolarized values starting from the holding potential of −64 mV. Note that IK,f and IK,n(as for OHCs) were not present in the mutant cell. +/dn: Vm−76 mV; Cm 9.1 pF; Rs 1.5 MΩ; gleak 11.5 nS. dn/dn: Vm−64 mV; Cm 6.5 pF; Rs 1.9 MΩ; gleak 0.4 nS. I, size of IK,n measured as the difference between instantaneous and steady-state deactivating tail currents for voltage steps from −64 mV to −124 mV in +/dn (n = 5, P41–P58) and dn/dn (n = 14, P41–P58) mice. Recordings in D–I were at body temperature.
Figure 6D and E shows the total outward K+ current recorded in adult IHCs from control (+/dn) and mutant (dn/dn) mice, respectively. In +/dn IHCs (P18–P58, n = 8) depolarizing voltage steps caused the rapid activation of outward K+ currents similar to those recorded in adult CD-1 mice (Kros et al. 1998; Marcotti et al. 2004a). These currents are mainly composed of IK,f and a much slower delayed-rectifier current (IK,s) which develops from the immature IK,neo (Marcotti et al. 2003a). When the same voltage protocol was applied to dn/dn IHCs (P17–P58, n = 14), the total outward K+ current was smaller and activated more slowly (Fig. 6E), suggesting that IK,f is likely to be absent in these mutant cells. The different activation time course between +/dn and dn/dn IHCs is more evident when the current onsets are displayed on an expanded time scale (Fig 6D and E, insets). The size of the total steady-state current (IK,f+IK,s), measured at 160 ms and at −25 mV, was found to be significantly larger (P < 0.001) in +/dn than in dn/dn IHCs (Fig. 6F). To find out whether IK,f was indeed absent in dn/dn IHCs, we measured its size in isolation (at 1.5–3.0 ms from the start of the voltage steps) at a membrane potential of −25 mV. This time range was selected because the rapidly activating IK,f had nearly reached its steady-state, while the slower IK,s was only just beginning to activate (Kros et al. 1998; Marcotti et al. 2004a). Figure 6F shows that IK,f was absent in dn/dn cells, whereas IK,s, obtained by subtracting any IK,f from the total current, was present and of normal size.
The next step was to verify whether IK,n, the other K+ current characteristic of adult IHCs, was also absent in mutant IHCs, as was the case for dn/dn OHCs (Fig. 5E and G). In IHCs of +/dn mice, hyperpolarizing voltage steps from a holding potential of −64 mV elicited inward K+ currents that commenced instantaneously and decayed slowly to a steady-state level (Fig. 6G), indicating deactivation of IK,n that was similar to that found in IHCs of CD−1 mice (Marcotti et al. 2003a; Oliver et al. 2003). IK,n from IHCs was recorded from −64 mV instead of −84 mV as for OHCs (Fig. 5D and F), to allow comparisons with previous results recorded from IHCs of CD-1 mice (Marcotti et al. 2003a). Depolarizing voltage steps elicited IK,f and IK,s in addition to IK,n. When the same voltage protocol consisting of hyperpolarizing and depolarizing voltage steps was applied to dn/dn IHCs, only IK,s was evident (Fig. 6H). The maximum size of IK,n in control and mutant IHCs (Fig. 6I) was measured as the deactivating current at −124 mV from the holding potential of −64 mV. The size of IK,n in +/dn IHCs was similar to that recorded from CD-1 mice (Marcotti et al. 2003a; Oliver et al. 2003).
Adult-type ionic currents from Beethoven mice are reduced in size
Since deafness and Beethoven are mutations of the same Tmc1 gene, we investigated whether basolateral ion channels were also affected in hair cells from Beethoven mice. Earlier results had suggested that basolateral K+ and Ca2+ currents developed normally in Bth/+ IHCs and OHCs up to P15 (Vreugde et al. 2002). Immature hair cells from control (+/+) and mutant (Bth/+ and Bth/Bth) mice all had similar biophysical properties (data not shown), analogous to the results from immature dn mice. We also looked in immature Bth IHCs for the presence of the small-conductance Ca2+-activated K+ current (ISK) which was previously demonstrated in immature CD-1 IHCs (Marcotti et al. 2004b). The current was evident as a slowly developing outward current (4 s voltage steps, as in Fig. 2A of Marcotti et al. 2004b) that was reversibly abolished in Ca2+-free extracellular solution (Bth/Bth, P7, n = 4, data not shown). The apparently normal development of immature currents indicates that the mutations in Tmc1 are unlikely to affect these cells before their onset of mature function (P8 for OHCs and P12 for IHCs).
Hair cells from mature Bth mutants did have abnormal expression of basolateral membrane currents. Figure 7B, E and G shows that the size of the total outward K+ current from Bth/Bth IHCs and OHCs, recorded using voltage protocols similar to those described in Fig. 5 (for OHCs) and Fig. 6 (for IHCs), was considerably reduced when compared to that measured in controls (+/+, Fig. 7A, D and F, respectively). The average sizes of the total current (IK,f+IK,s) and the isolated IK,f and IK,s in control and mutant IHCs are shown in Fig. 7C. In contrast to deafness mice, IK,f was still present in Beethoven mutant IHCs, although its size was considerably reduced (P < 0.0001), more so in the homozygous mutants than in Bth/+. Post tests showed that IK,f of Bth/+ IHCs was both smaller than that of +/+ (P < 0.001) and larger than that of Bth/Bth (P < 0.01) IHCs. This corrects the earlier observation of normal development of basolateral currents in Bth/+ hair cells (Vreugde et al. 2002): the effect of the mutation on IK,f was probably missed because this current is still quite small just after the onset of hearing, and hair cell currents were only tested up to P15 in that study. Just as for dn mice, the size of IK,s did not seem to be affected by the mutation. In both cell types, IK,n could be observed in isolation during hyperpolarizing voltage steps as deactivating inward currents (Fig. 7D and F). It was again found to be reduced by the mutation in Tmc1 (IHCs: P < 0.0005; OHCs: P < 0.0001), but only significantly so in Bth/Bth and not in Bht/+ (Fig. 7H).
Figure 7. K+ currents in mature IHCs and OHCs from Beethoven mutant mice.
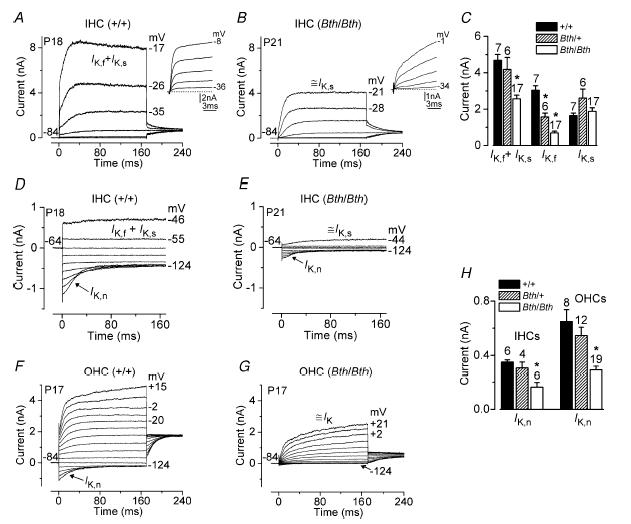
A and B, outward K+ currents in a control IHC (+/+, P18) and a homozygous mutant IHC (Bth/Bth, P21), respectively. In this figure, all current recordings were obtained using the same voltage protocols described in Fig. 5 (for OHCs) and Fig. 6 (for IHCs). In contrast to deafness mutants, some residual IK,f is still present in the Bth/Bth IHC (B). +/+ IHC: Vm−75 mV; Cm 10.0 pF; Rs 0.4 MΩ; gleak 6.0 nS. Bth/Bth IHC: Vm−76 mV; Cm 10.0 pF; Rs 1.6 MΩ; gleak 2.0 nS. C, size of the total current (IK,f+IK,s) and the isolated IK,f and IK,s measured at −25 mV in +/+ (n = 7, P19–P20), Bth/+ (n = 6, P16–P22) and Bth/Bth (n = 17, P16–P22) IHCs. D and E, membrane currents recorded from P18 +/+ (D) and P21 Bth/Bth (E) IHCs. Note the small residual IK,n present in the Bth/Bth cell. Both cells are the same as in A and B. F and G, current recordings from +/+ (F) and Bth/Bth (G) P17 OHCs. As for IHCs, IK,n was very small in the Bth/Bth OHC. +/+ OHC: Vm−77 mV; Cm 10.8 pF; Rs 1.9 MΩ; gleak 0.5 nS. Bth/Bth OHC: Vm−69 mV; Cm 10.0 pF; Rs 1.8 MΩ; gleak 1.0 nS. H, size of IK,n measured as the difference between instantaneous and steady-state deactivating tail currents for voltage steps from −64 mV for IHCs and −84 mV for OHCs to −124 mV in +/+ (IHCs: n = 6, P41–P58; OHCs: n = 8, P15–P17), Bth/+ (IHCs: n = 4, P41–P58; OHCs: n = 12, P14–P17) and Bth/Bth (IHCs: n = 7, P41–P58; OHCs: n = 19, P14). The symbol ≅ before IK,s and IK indicates that these currents are contaminated by residual currents not fully absent in Bth/Bth hair cells. All recordings were at room temperature.
We also investigated whether the ACh-activated current, which is normally expressed in adult OHCs (Evans, 1996; Dulon & Lenoir, 1996; He & Dallos, 1999) and in immature but not adult IHCs (Glowatzki & Fuchs, 2000; Katz et al. 2004; Marcotti et al. 2004b), was present in mutant cells. Currents were recorded from adult Bth/Bth hair cells during superfusion of 100 μm ACh as previously described (Marcotti et al. 2003b). As expected, the ACh-activated K+ current, isolated by subtracting control currents from currents recorded in the presence of ACh, was expressed in OHCs (P20–P24, n = 4) but not in IHCs (P22, n = 2), so the ACh responses appeared qualitatively normal. The response of OHCs to ACh was quantified by measuring the steady-state slope conductance of the ACh-activated current around the holding potential of −84 mV (Marcotti et al. 2004b). The slope conductance in Beethoven mutant OHCs (Bth/Bth, 6.9 ± 0.7 nS, n = 4) was found to be significantly (P < 0.05) reduced compared to that measured in controls (+/+, 13.5 ± 2.1 nS, n = 3).
The exocytotic machinery in mutant IHCs remains at an immature stage of development
In normal CD-1 mice the size of ICa in IHCs increases from embryonic stages of development up to about P6, and then gradually decreases to a constant maximum size of about −100 pA (1.3 mm extracellular Ca2+ and at around −20 mV) after the onset of hearing (Marcotti et al. 2003b; Johnson et al. 2005). Although the size of ICa decreases during development, its activation kinetics are not significantly different between pre- and posthearing IHCs (Johnson et al. 2005). Moreover, the observation that the Cav1.3 Ca2+ channel subunit is responsible for >90% of ICa in immature as well as mature IHCs (Platzer et al. 2000; Brandt et al. 2003), indicates that the decline of the current starting at P6 is due to a reduction in the number of Ca2+ channels rather than a change in channel composition. Immature IHCs (P7) from both control and mutant deafness mice express an ICa of similar size to that previously reported for CD-1 mice of the same age (Marcotti et al. 2003b; Johnson et al. 2005). By contrast, ICa in mature IHCs was affected by the mutation. Figure 8A and B shows ICa recorded from P18 +/dn and dn/dn IHCs, respectively. The size of ICa recorded in mutant (both dn and Bth) IHCs was always larger than that found in controls (see Fig. 8A, B and D for deafness), but similar to that measured in immature, prehearing cells. The different inactivation kinetics between the control (+/dn) and mutant (dn/dn) IHCs shown in Fig. 8A and B are not due to the mutation but represent variations in the effectiveness of the block of contaminating outward K+ currents seen also in IHCs from normal CD-1 mice (Johnson et al. 2005).
Figure 8. Ca2+ currents and ΔCm in mature deafness and Beethoven IHCs.
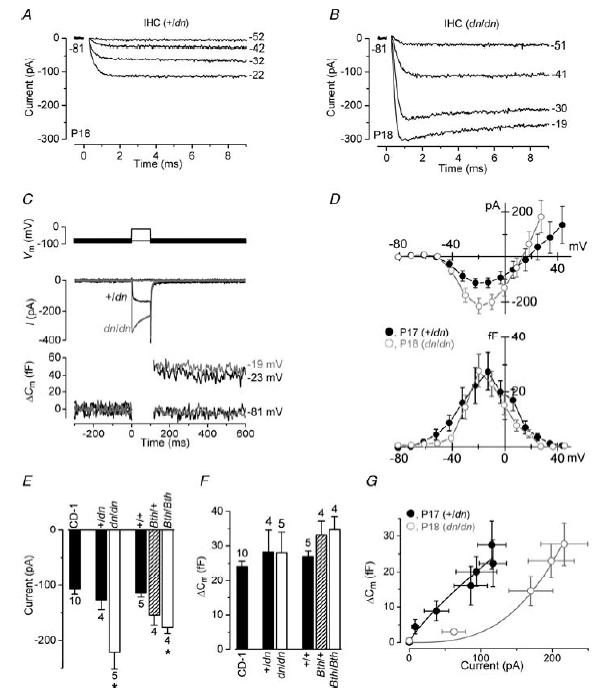
A and B, inward ICa recordings from +/dn and dn/dn IHCs (P18), respectively, in response to voltage steps from the holding potential of −81 mV to more depolarized levels in 10 mV increments. For clarity only some of the traces are shown and the membrane potentials (inmV) are indicated next to the traces. The current traces in A and B are averages from five and six repetitions, respectively. +/dn: Cm 8.3 pF; Rs 6.8 MΩ; gleak 4.0 nS. dn/dn: Cm 6.3 pF; Rs 7.9 MΩ; gleak 1.3 nS. C, ICa (middle panel) and ΔCm (bottom panel) responses in adult IHCs from +/dn and dn/dn mice elicited by applying 100 ms depolarizing voltage steps from the holding potential of −81 mV to near −21 mV. +/dn: Cm 7.5 pF; Rs 6.0 MΩ; gleak 3.2 nS; Peak ICa−145 pA elicited a ΔCm of 45 fF. dn/dn: Cm 6.3 pF; Rs 7.9 MΩ; gleak 0.4 nS; Peak ICa−336 pA elicited a ΔCm of 49 fF. The command protocol (top panel) consists of a sinusoidal waveform (used to track Cm) that appears as a thick black line, which is interrupted for the duration of the voltage steps. D, peak current–voltage (ICa–V, upper panel) and capacitance–voltage (ΔCm–V, lower panel) curves, measured at different membrane potentials from −81 mV to more depolarized potentials in nominal 10 mV increments, from the +/dn (n = 4, P17) and dn/dn (n = 5, P18) IHCs. E, average peak (near −21 mV) ICa recorded in mature IHCs from normal CD-1 (P16–P20), +/dn (P17), dn/dn (P18), +/+and Bth/+ (P16–P17) and Bth/Bth (P17). F, average ΔCm measured in response to the ICa recorded in E. G, average ΔCm responses plotted against the corresponding ICa size from the I–V and ΔCm–V curves shown in (D). Continuous lines are fits to data points according to eqn (1). All recordings were at body temperature.
Because of the larger ICa size in mature mutant IHCs, we investigated whether exocytosis, a Ca2+-dependent process that is considered to be a sign of neurotransmitter release from presynaptic cells (Parsons et al. 1994; Von Gersdorff et al. 1998; Moser & Beutner, 2000) was also affected. The degree of exocytosis, elicited by voltage steps, was estimated by measuring the change in cell membrane capacitance (ΔCm). Figure 8C shows ICa and the corresponding ΔCm recorded from mature +/dn (P17) and dn/dn (P18) IHCs using 100 ms voltage steps from −81 mV to near −21 mV, which elicited maximum ICa. Although the dn/dn IHC responded to the same voltage step with a significantly larger peak ICa, the size of the induced ΔCm was similar to that seen in the +/dn cell. The peak ICa–voltage (I–V) and ΔCm-voltage (ΔCm–V) curves for +/dn (n = 4) and dn/dn (n = 5) cells were obtained from responses to 100 ms depolarizing voltage steps in nominal 10 mV increments from the holding potential of −81 mV (Fig. 8D). ICa and ΔCm both followed a bell-shaped trend, but note the narrower voltage range for ΔCm in homozygous mutant cells. The average peak size of ICa recorded in both dn and Bth mutants and controls is shown in Fig. 8E. ICa in homozygous mutant IHCs was significantly larger than that recorded in controls (P < 0.05) and normal age-matched CD-1 mice (dn/dn: P < 0.001; Bth/Bth: P < 0.01). The size of ICa in Bth/+ was intermediate, and did not differ significantly from that recorded in either +/+ or Bth/Bth IHCs, although it was larger (P < 0.05) than that in CD-1 IHCs. The average size of ICa in mature IHCs of homozygous dn and Bth mutants closely matches that recorded at about P6 in immature IHCs of normal CD-1 mice (Marcotti et al. 2003b; Johnson et al. 2005). The maximum ΔCm was similar between all IHCs investigated, and also to that in adult CD-1 mice (Fig. 8F). In both mutant and control IHCs, ΔCm for the largest ICa measured (at around −21 mV) was about 30 fF (femtofarad) implying that mature IHCs, whether mutant or not, were capable of releasing about 800 synaptic vesicles in response to 100 ms voltage steps, using a conversion factor of 37 aF (attofarad) per vesicle (Lenzi et al. 1999). As their Ca2+ currents were larger, the Ca2+ efficiency of the mature mutant IHCs was thus reduced compared to normal mature IHCs (Johnson et al. 2005).
The relation between Ca2+ entry and exocytosis (Fig. 8G) for +/dn and dn/dn IHCs, estimated using a synaptic transfer function (Augustine et al. 1985; Johnson et al. 2005), was obtained by plotting ΔCm against peak ICa for 100 ms voltage steps over the range from −81 mV to near −11 mV. Data were approximated using a power function:
where N is the power. The average power obtained from the transfer functions of mature +/dn IHCs was 0.82 ± 0.10 (n = 4), suggesting a near-linear relation between Ca2+ entry and exocytosis as previously described for mature IHCs from normal CD-1 mice (Johnson et al. 2005). The transfer curve obtained from dn/dn IHCs was best approximated using a power of 2.99 ± 0.46 (n = 5), significantly larger (P < 0.005) than that found in the controls, indicating that each release event requires around three Ca2+-binding steps to occur (Dodge & Rahamimoff, 1967). The Ca2+ dependence of exocytosis in Beethoven IHCs was also significantly steeper than in control cells (Bth/Bth: 2.48 ± 0.38 n = 3; +/+: 0.72 ± 0.08 n = 5; P < 0.05, post test), with intermediate values for Bth/+ (1.36 ± 0.61 n = 3). This high-order dependence of exocytosis on Ca2+ entry and the lower Ca2+ efficiency in adult mutant IHCs is reminiscent of that found in prehearing IHCs of normal CD-1 mice (power N≈ 3, Johnson et al. 2005), suggesting that Tmc1 is also likely to be involved in the maturation of the exocytotic machinery in these cells. Although the factors that lead to the developmental linearization in the Ca2+ dependence of exocytosis in normal IHCs are unknown, the expression of Tmc1 might ensure the normal maturation (downregulation) of ICa and/or changes in the Ca2+-sensing molecules of exocytosis from around the onset of hearing. These anomalies in exocytosis due to Tmc1 mutations may reflect the apparently abnormal development or maintenance of synaptic connections previously reported in deafness mutant hair cells (Bock & Steel, 1983).
Voltage responses of IHCs under current clamp
Immature IHCs from normal CD-1 mice are capable of generating spontaneous or evoked repetitive Ca2+ action potentials prior to the onset of hearing (Kros et al. 1998; Glowatzki & Fuchs, 2000; Beutner & Moser, 2001; Marcotti et al. 2003a; Marcotti et al. 2003b). Figure 9A and B shows that immature P8 IHCs from both +/dn and dn/dn fire action potentials upon current injection. The same results were also obtained in both homo- and heterozygous Beethoven IHCs (P7). In the latter a Ca2+-free extracellular solution was tested, which reversibly abolished the action potentials, demonstrating their Ca2+ dependence (data not shown). As in normal CD-1 mice, ICa expressed in heterozygous and homozygous deafness IHCs (see Fig. 6A and B, arrows) is likely to play the major role in the generation of action potentials. The mean resting membrane potential (Vm, mostly measured in current clamp but in some cases as zero-current potential) of immature IHCs was found not to be significantly different between control and mutant (dn/dn, Bth/+ and Bth/Bth) cells, and the values were pooled together (−74.1 ± 0.8 mV, n = 29, P6–P8). The same applied to the Vm of control and mutant immature OHCs (−67.2 ± 1.2 mV, n = 15, P6–P8). Since the disappearance of induced action potentials in IHCs at the onset of hearing was previously correlated with the expression of IK,f (Kros et al. 1998) and IK,n (Marcotti et al. 2003a), we investigated how the absence of these currents affected the voltage responses of adult mutant cells. In +/dn IHCs, small hyperpolarizing and depolarizing current injections produced fast, small and graded voltage responses (Fig. 9C) as previously described in IHCs of normal CD-1 mice (Kros et al. 1998; Marcotti et al. 2004a). In adult dn/dn IHCs, small hyperpolarizing current injections caused large and almost passive voltage responses (Fig. 9D), most likely due to the absence of both IK,n and IK,f, which in normal hair cells are both partially active around the resting membrane potential (Marcotti et al. 2003a, 2004a; Oliver et al. 2003). Voltage changes in response to depolarizing current injections were always much larger but slower in onset than in dn/dn IHCs (Fig. 9D) compared to those recorded from +/dn cells (Fig. 9C). Moreover, small depolarizing current injections generated slow oscillatory responses reminiscent of the small action potentials recorded from late embryonic IHCs (Marcotti et al. 2003a,b). Voltage oscillations were never observed in mature Bth/+ and Bth/Bth IHCs, but voltage responses to depolarizing and hyperpolarizing current steps appeared larger and slower than in controls, particularly in Bth/Bth IHCs. These voltage responses were not analysed in detail but resembled those observed when TEA was used to block IK,f (Kros & Crawford, 1990) or BAPTA to shift its operating range in the depolarizing direction (Marcotti et al. 2004a). Figure 9E and F shows the resting membrane potential (Vm) in adult IHCs (Fig. 9E) and OHCs (Fig. 9F) from mutants and controls. In homozygous mutant hair cells, Vm was found to be significantly depolarized (IHCs: dn/dn P < 0.0001; Bth/Bth P < 0.05; OHCs: dn/dn P < 0.005; Bth/Bth P < 0.001) relative to that measured in control cells from the same strains. In Bth/+, Vm was similar to the controls and more hyperpolarized than Bth/Bth in both cell types (P < 0.01).
Figure 9. Voltage responses of IHCs from deafness mice.
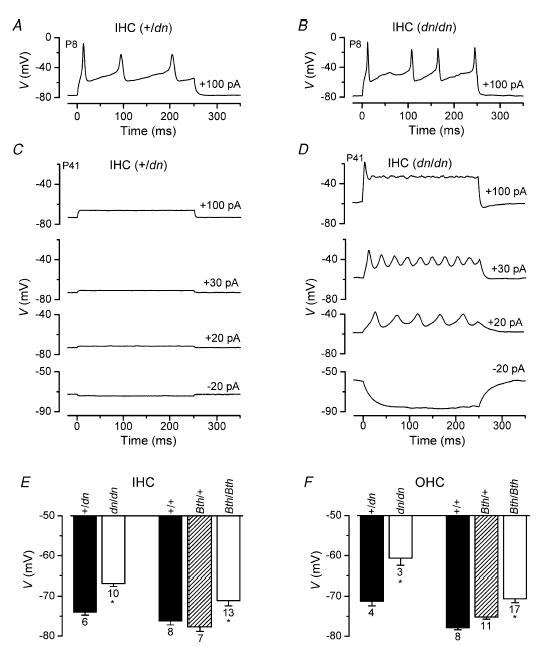
A and B, voltage responses under current clamp from +/dn (A) and dn/dn (B) IHCs at P8. Depolarizing current injections (+100 pA) triggered action potentials. The membrane potential (V) is plotted along the ordinates. +/dn: Vm−78 mV; Cm 9.4 pF; Rs 5.0 MΩ. dn/dn: Vm−79 mV; Cm 9.9 pF; Rs 6.2 MΩ. C, voltage responses from a P41 +/dn IHC. Vm−74 mV; Cm 8.2 pF; Rs 7.1 MΩ. D, voltage responses from a P41 dn/dn IHC exhibiting slow repetitive action potentials when small current injections were applied. Vm−64 mV; Cm 6.2 pF; Rs 6.2 MΩ. E and F, average resting membrane potentials (Vm) of IHCs and OHCs, respectively, from dn and Bth mutant mice. IHCs: +/dn, P18–P58; dn/dn, P17–P58; +/+, Bth/+ and Bth/Bth, P19–P20. OHCs: +/dn and dn/dn, P14; +/+ and Bth/+, P14–P17; Bth/Bth, P12–P17. All recordings were at body temperature.
Discussion
Both the dn and Bth mutations in mouse and the DFNA36 and DFNB7/B11 loci in humans lead to hearing impairment by affecting the Tmc1 (TMC1) gene (Vreugde et al. 2002; Kurima et al. 2002). Although the function of Tmc1 is unknown, its expression in both IHCs and OHCs in early postnatal development (from P5: Kurima et al. 2002; Vreugde et al. 2002) suggests that it might play a role in the normal maturation of these cells. Therefore, in the present study we examined the effects that mutations in Tmc1 have on the structural appearance and biophysical properties of immature and mature cochlear hair cells using the deafness and Beethoven mouse mutants.
Mutations in Tmc1 can impede the development of cochlear hair cells into fully functional sensory receptors
After terminal mitosis, at about embryonic day 14–15 in normal mice (Pujol et al. 1998), hair cell precursors start to differentiate electrophysiologically by acquiring a small delayed-rectifier K+ current (Marcotti et al. 2003a; Helyer et al. 2005). During the following two weeks, hair cells undergo extensive biophysical changes mainly due to the acquisition of the mechano-electrical transducer current and various basolateral membrane currents (Fig. 10A and B). Immature IHCs generate trains of spontaneous action potentials (Kros et al. 1998; Marcotti et al. 2003a 2003b) prior to the onset of sound-induced responses, thought to be involved in the refinement of synaptic connections within the immature cochlea by analogy with the visual system (Zhang & Poo, 2001). With the onset of functional maturation at P8 in OHCs (Marcotti & Kros, 1999) and P12 in IHCs (Kros et al. 1998; Marcotti et al. 2003a), most of the basolateral membrane currents expressed in immature hair cells are partially or completely downregulated (IHC: Fig. 10A and C; OHC: Fig. 10B and E). At the same time the acquisition of IK,f and IK,n in IHCs and IK,n, IK,(ACh) and electromotility in OHCs (Figs 10C and E) changes these cells into fully functional sensory receptors.
Figure 10. Main physiological differences between immature and mature hair cells from normal CD-1 mice and deafness mutant mice.
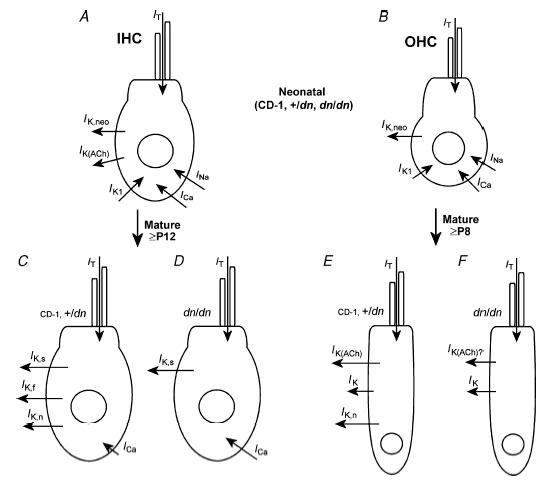
A and B, schematic representation showing the different types of membrane currents likely to be expressed in immature IHCs (A) and OHCs (B) from normal CD-1, +/dn and dn/dn mice. C and E, currents expressed by mature hair cells of +/dn and normal CD-1 mice. D and F, membrane currents expressed in mutant hair cells. For IK(ACh) there is some uncertainty as the data are only from Bth/Bth. IT is the mechano-electrical transducer current. The effects of the Tmc1 mutation on the various ionic currents are qualitatively similar but less severe in Bth/Bth and least severe in Bth/+ (see text for details).
Single-cell recordings from dn/dn, Bth/+ and Bth/Bth mice show that immature hair cells express basolateral membrane and mechano-electrical transducer currents that are indistinguishable from those found in controls or normal CD-1 mice, suggesting that prehearing cells are unlikely to be affected by mutations in Tmc1. The development of normal transducer currents in the mutant OHCs shows that Tmc1 does not encode a component of the transducer channel, and makes it likely that the inability to record cochlear microphonics from dn/dn mice is simply a consequence of the degeneration of large numbers of OHCs. The most striking effect of Tmc1 mutations is the absence (dn/dn) or the reduced (Bth/Bth and, to a more modest extent, Bth/+) expression of K+ currents that characterize the mature hair cell phenotype (Fig. 10D and F). Moreover, the size of ICa in mutant adult IHCs (dn/dn, Bth/+ and Bth/Bth) is larger than that recorded in age-matched control (+/dn and, for Bth, +/+) cells, but similar to that measured in immature IHCs from both controls and mutants (see Results for deafness) and normal CD-1 mice (Beutner & Moser, 2001; Marcotti et al. 2004b; Johnson et al. 2005), indicating that its normal maturation has also been altered. It is thus likely that Tmc1 mutations prevent the apparently normal downregulation of the Ca2+ current starting in the second postnatal week (Beutner & Moser, 2001; Marcotti et al. 2003b). Despite this large ICa, maximum Ca2+-induced exocytosis was similar between controls and mutant adult IHCs, indicative of a reduced Ca2+ efficiency in the mutants. Moreover, the Ca2+ dependence of exocytosis was steeper in mutant cells. Since a high-order Ca2+ dependence of exocytosis has only been found in prehearing IHCs (Johnson et al. 2005), it seems conceivable that Tmc1 also plays a role in the normal maturation of the exocytotic machinery. However, the presence of electromotile activity in adult mutant OHCs, and their responsiveness to ACh, together with the normal developmental downregulation of most of the immature-type currents in both cell types suggest that mutations in Tmc1 do not cause a general failure of hair cell maturation, but instead prevent the acquisition of specific adult phenotypes.
Physiological consequences of mutations in Tmc1 in the organ of Corti
Scanning electron microscopy of dn/dn and Bth/Bth mutants revealed progressive hair cell degeneration from P15 onwards. This degeneration is unlikely to be the direct cause for the raised thresholds for CAP responses (dn/dn: Bock & Steel, 1983; Bth/Bth: Fig. 3A and B), because many hair cells remain with hair bundles of normal appearance, especially in the apex (80–90% region, Figs 1 and 2 and Vreugde et al. 2002). The presence of apparently healthy apical cells could conceivably be explained by assuming that the expression of Tmc1 was restricted to the middle and basal regions of the cochlea, as recently shown for the chick basilar papilla (Mutai et al. 2005), the avian equivalent of the organ of Corti. However, our electrophysiological findings show that apical hair cell function is directly affected by the mutation, indicating that the electrically resonant low-frequency region of the basilar papilla may be a special case that is different from the mammalian cochlea and one in which Tmc1 has no functional role.
In mammals, the hearing impairment associated with Tmc1 mutations is most likely due to the absence (in dn) or reduced expression (in Bth) of adult-type currents, and the lack of maturation of the exocytotic machinery. IK,n is substantially (about 70%) activated at the resting membrane potentials of adult OHCs (Marcotti & Kros, 1999) and IHCs (Marcotti et al. 2003a; Oliver et al. 2003), providing an efficient exit route for K+ ions entering the cells through the transducer channels. In IHCs this role is aided by IK,f, since it contributes about half of the resting conductance (Marcotti et al. 2004a). The absence of IK,n in mutant OHCs and of both IK,n and IK,f in IHCs might lead to an accumulation of intracellular K+ that could be responsible for the progressive degeneration of these cells observed in mutant mice. The slower progression of hair cell degeneration in the apex could be due to the smaller transducer currents in this region (He et al. 2004). Slow degeneration of hair cells has been inferred for DFNA2, another form of non-syndromic dominant progressive hearing loss in humans starting at high frequencies, caused by mutation of the KCNQ4 gene (Kubisch et al. 1999; Kharkovets et al. 2006) that is likely to underlie IK,n in hair cells (Marcotti & Kros, 1999; Marcotti et al. 2003a; Oliver et al. 2003). The absence of IK,f in dn/dn IHCs, which is believed to be essential for high-frequency phase locking in the auditory nerve (Palmer & Russell, 1986; Kros & Crawford, 1990), allows these cells to fire slow Ca2+ action potentials even at adult stages (up to P58, the oldest stage investigated). These action potentials (Fig. 9D), together with the steeper Ca2+ dependence of exocytosis (Fig. 8G), which results in a more depolarized threshold for neurotransmitter release, would prevent adult IHCs of dn/dn from accurately encoding high-frequency sound stimuli (Johnson et al. 2005). Synchronous neural activity is required for generation of a CAP, so abnormal action potentials in mature mutant IHCs could lead to the absence of the CAPs in dn/dn (Bock & Steel, 1983) The absence of normal auditory nerve activity of cochlear origin has been suggested as the possible cause of the increased transmitter release probability (Oleskevich & Walmsley, 2002), altered inhibitory transmission (Leao et al. 2004a) and enhanced cell excitability (Leao et al. 2004b) observed in auditory brain stem neurons of dn/dn mutants studied in vitro. Action potentials were not observed in Bth/Bth IHCs, but their voltage responses were slower and larger than normal. Together with the more depolarized threshold for neurotransmitter release, this could partially but perhaps not fully explain the absence of CAP responses in Bth/Bth mice. In mature Bth/+ cochlear hair cells, the various K+ currents were only somewhat smaller than the controls, and the Ca2+ current was somewhat larger, but with the exception of IK,f in IHCs not significantly so. Exocytosis also appeared nearly normal. Perhaps with more experiments, statistical significance might have been achieved. It seems likely that because of these near-normal properties reasonable CAP responses could be recorded from Bth/+ mice over the full frequency range by P15, with the later deterioration at high frequencies at P30 and P60 following the progress of hair cell degeneration due to impaired K+ efflux from the basolateral membrane. Finally, the more depolarized membrane potential observed in homozygous mutant IHCs, due to the partial (Bth/Bth) or complete (dn/dn) absence of IK,n and IK,f (Figs 5–7), would of itself increase resting ICa in mature mutant IHCs, which could raise the intracellular Ca2+ levels (Oliver et al. 2003), possibly leading to cytotoxicity and cell degeneration (Orrenius et al. 2003).
Possible functional roles of Tmc1
Tmc1 encodes a transmembrane protein predicted to have 6–11 transmembrane domains with charged intracellular amino and carboxy termini (Kurima et al. 2002; Vreugde et al. 2002), reminiscent of voltage-gated channels (Hille, 2001). Our finding of relatively modest defects in hair cell physiology and slower degeneration of the organ of Corti in Bth/+ than in Bth/Bth mice, together with the absence of any obvious defects in +/dn mice, suggests that the Bth allele acts through a dominant negative mechanism rather than through haploinsufficiency, while the dn allele is likely to be a functional null mutation. This is compatible with the notion that the Tmc1 protein may be a multimer of which the function is disturbed by the incorporation of Bth subunits. Tmc1 protein appears to be localized in the area surrounding the hair cell cuticular plate and at the endoplasmic reticulum from P10 (Makishima et al. 2005). The presence of TMC1 in endoplasmic reticulum membrane was also demonstrated in Cos7 cells (Labay et al. 2005). Tmc1 is unlikely to encode an ion channel since when mutated it appears to interfere with the normal development of a number of different ion channel types (IK,f, IK,n, ICa and possibly IK(ACh)). Moreover, such mutations also prevent the maturation of the synaptic machinery. However, Tmc1 could be responsible for up- or downregulating various ion channels or exocytotic molecules during development.
Channel expression can be regulated by Ca2+ entry into the cell (Linsdell & Moody, 1995; Liu & Kaczmarek, 1998a; Muller et al. 1998; Xie & Black, 2001) and/or by neurotrophic factors via the activation of intracellular signalling pathways (Liu & Kaczmarek, 1998b; Dryer et al. 2003; Huang & Reichardt, 2003). In mature cochlear IHCs from mice lacking the Cav1.3 L-type Ca2+ channel, which contributes 90% of the total Ca2+ current in these cells (Platzer et al. 2000), IK,f is absent (Brandt et al. 2003), suggesting that ICa is necessary for the appearance of IK,f. The absence or reduced expression of IK,f in IHCs with Tmc1 mutations is not due to the lack of ICa, since the latter is in fact unusually large in these cells. The possibility that mutations in Tmc1 only cause a delay in the normal physiological maturation of cochlear hair cells, as previously described for mice lacking thyroid hormone receptors (Rüsch et al.1998, 2001), where the maturation of IK,f was delayed by about two weeks, was considered. Transmission electron microscopy has shown that the opening of the spaces of Nuel around the OHCs of homozygous deafness mice was delayed by about 10 days (Bock & Steel, 1983). This delay may well be a consequence of the finding presented here that IK,n never develops in dn/dn OHCs, hampering the K+ recycling pathway at this point (Steel & Kros, 2001). However, a general delay in hair cell maturation is unlikely as an explanation for our observations, since the effects of the Tmc1 mutations were still evident in IHCs from two-month-old dn/dn mice.
Tmc1 expression could be involved in the activation or modulation of intracellular signals responsible for the physiological differentiation of immature hair cells into fully functional sensory receptors. Alternatively, Tmc1 could be involved in intracellular trafficking, such as controlling the exit of various membrane proteins characteristic of mature hair cells from the endoplasmic reticulum (Ellgaard & Helenius, 2003; Vandenberghe & Bredt, 2004) before being inserted into the plasma membrane.
Acknowledgments
This work was supported by the MRC and Deafness Research UK. W.M. is a Royal Society University Research Fellow. We thank Jabulani Sithole for helping with statistical analysis, and Martin Hrabé de Angelis and Helmut Fuchs for the initial discovery of the Beethoven mutant.
References
- Augustine GJ, Charlton MP, Smith SJ. Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. J Physiol. 1985;367:163–181. doi: 10.1113/jphysiol.1985.sp015819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner D, Moser T. The presynaptic function of mouse cochlear inner hair cells during development of hearing. J Neurosci. 2001;21:4593–4599. doi: 10.1523/JNEUROSCI.21-13-04593.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock GR, Frank MP, Steel KP. Preservation of central auditory function in the deafness mouse. Brain Res. 1982;239:608–612. doi: 10.1016/0006-8993(82)90536-4. [DOI] [PubMed] [Google Scholar]
- Bock GR, Steel KP. Inner ear pathology in the deafness mutant mouse. Acta Otolaryngol. 1983;96:39–47. doi: 10.3109/00016488309132873. [DOI] [PubMed] [Google Scholar]
- Brandt A, Striessnig J, Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford AC, Evans MG, Fettiplace R. Activation and adaptation of transducer currents in turtle hair cells. J Physiol. 1989;419:405–434. doi: 10.1113/jphysiol.1989.sp017878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallos P, Billone MC, Durrant JD, Wang C, Raynor S. Cochlear inner and outer hair cells: functional differences. Science. 1972;177:356–358. doi: 10.1126/science.177.4046.356. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Rahamimoff R. Co-operative action of calcium ions in transmitter release at the muscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryer SE, Lhuillier L, Cameron JS, Martin-Caraballo M. Expression of K(Ca) channels in identified populations of developing vertebrate neurons: role of neurotrophic factors and activity. J Physiol. 2003;97:49–58. doi: 10.1016/j.jphysparis.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Dulon D, Lenoir M. Cholinergic responses in developing outer hair cells of the rat cochlea. Eur J Neurosci. 1996;8:1945–1952. doi: 10.1111/j.1460-9568.1996.tb01338.x. [DOI] [PubMed] [Google Scholar]
- Durham D, Rubel EW, Steel KP. Cochlear ablation in deafness mutant mice: 2-deoxyglucose analysis suggests no spontaneous activity of cochlear origin. Hear Res. 1989;43:39–46. doi: 10.1016/0378-5955(89)90057-9. [DOI] [PubMed] [Google Scholar]
- Eatock RA, Hurley KM. Functional development of hair cells. Curr Topics Dev Biol. 2003;57:389–448. doi: 10.1016/s0070-2153(03)57013-2. [DOI] [PubMed] [Google Scholar]
- Ehret G. Masked auditory thresholds, critical ratios, and scales of the basilar membrane of the housemouse (Mus musculus) J Comp Physiol. 1975;103:329–341. [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Evans MG. Acetylcholine activates two currents in guinea-pig outer hair cells. J Physiol. 1996;491:563–578. doi: 10.1113/jphysiol.1996.sp021240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géléoc GSG, Lennan GWT, Richardson GP, Kros CJ. A quantitative comparison of mechanoelectrical transduction in vestibular and auditory hair cells of neonatal mice. Proc R Soc Lond B. 1997;264:611–621. doi: 10.1098/rspb.1997.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA. Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science. 2000;288:2366–2368. doi: 10.1126/science.288.5475.2366. [DOI] [PubMed] [Google Scholar]
- He DZ, Dallos P. Development of acetylcholine-induced responses in neonatal gerbil outer hair cells. J Neurophysiol. 1999;81:1162–1170. doi: 10.1152/jn.1999.81.3.1162. [DOI] [PubMed] [Google Scholar]
- He DZ, Evans BN, Dallos P. First appearance and development of electromotility in neonatal gerbil outer hair cells. Hear Res. 1994;78:77–90. doi: 10.1016/0378-5955(94)90046-9. [DOI] [PubMed] [Google Scholar]
- He DZ, Jia S, Dallos P. Mechanoelectrical transduction of adult outer hair cells studied in a gerbil hemicochlea. Nature. 2004;429:766–770. doi: 10.1038/nature02591. [DOI] [PubMed] [Google Scholar]
- Helyer RJ, Kennedy HJ, Davies D, Holley MC, Kros CJ. Development of outward potassium currents in inner and outer hair cells from the embryonic mouse cochlea. Audiol Neurootol. 2005;10:22–34. doi: 10.1159/000081545. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. 3. Sunderland, MA: Sinauer Associates; 2001. [Google Scholar]
- Hrabé de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, Marschall S, Heffner S, Pargent W, Wuensch K, Jung M, Reis A, Richter T, Alessandrini F, Jakob T, Fuchs E, Kolb H, Kremmer E, Schaeble K, Rollinski B, Roscher A, Peters C, Meitinger T, Strom T, Steckler T, Holsboer F, Klopstock T, Gekeler F, Schindewolf C, Jung T, Avraham K, Behrendt H, Ring J, Zimmer A, Schughart K, Pfeffer K, Wolf E, Balling R. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet. 2000;25:444–447. doi: 10.1038/78146. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Hunter-Duvar IM. A technique for preparation of cochlear specimens for assessment with the scanning electron microscope. Acta Otolaryngol Suppl. 1978;351:3–23. doi: 10.3109/00016487809122718. [DOI] [PubMed] [Google Scholar]
- Johnson SL, Marcotti W, Kros CJ. Increase in efficiency and reduction in Ca2+ dependence of exocytosis during development of mouse inner hair cells. J Physiol. 2005;563:177–191. doi: 10.1113/jphysiol.2004.074740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Thomas MV, Kros CJ. Membrane capacitance measurement using patch clamp with integrated self-balancing lock-in amplifier. Pflugers Arch. 2002;443:653–663. doi: 10.1007/s00424-001-0763-z. [DOI] [PubMed] [Google Scholar]
- Katz E, Elgoyhen AB, Gomez-Casati ME, Knipper M, Vetter DE, Fuchs PA, Glowatzki E. Developmental regulation of nicotinic synapses on cochlear inner hair cells. J Neurosci. 2004;24:7814–7820. doi: 10.1523/JNEUROSCI.2102-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 2006;25:642–652. doi: 10.1038/sj.emboj.7600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kros CJ, Crawford AC. Potassium currents in inner hair cells isolated from the guinea-pig cochlea. J Physiol. 1990;421:263–291. doi: 10.1113/jphysiol.1990.sp017944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kros CJ, Ruppersberg JP, Rüsch A. Expression of a potassium current in inner hair cells during development of hearing in mice. Nature. 1998;394:281–284. doi: 10.1038/28401. [DOI] [PubMed] [Google Scholar]
- Kros CJ, Rüsch A, Richardson GP. Mechano-electrical transducer currents in hair cells of the cultured neonatal mouse cochlea. Proc R Soc Lond B. 1992;249:185–193. doi: 10.1098/rspb.1992.0102. [DOI] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lütjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, Arnaud D, Drury S, Mo J, Makishima T, Ghosh M, Menon PS, Deshmukh D, Oddoux C, Ostrer H, Khan S, Riazuddin S, Deininger PL, Hampton LL, Sullivan SL, Battey JF, Jr, Keats BJ, Wilcox ER, Friedman TB, Griffith AJ. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet. 2002;30:277–284. doi: 10.1038/ng842. [DOI] [PubMed] [Google Scholar]
- Labay V, Makishima T, Griffith A. Transmembrane topologic organization of TMC1. Assoc Res Otolaryngol Abstract. 2005;795 [Google Scholar]
- Leao RN, Berntson A, Forsythe ID, Walmsley B. Reduced low-voltage activated K+ conductances and enhanced central excitability in a congenitally deaf (dn/dn) mouse. J Physiol. 2004b;559:25–33. doi: 10.1113/jphysiol.2004.067421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao RN, Oleskevich S, Sun H, Bautista M, Fyffe RE, Walmsley B. Differences in glycinergic mIPSCs in the auditory brain stem of normal and congenitally deaf neonatal mice. J Neurophysiol. 2004a;91:1006–1012. doi: 10.1152/jn.00771.2003. [DOI] [PubMed] [Google Scholar]
- Lenzi D, Runyeon JW, Crum J, Ellisman MH, Roberts WM. Synaptic vesicle populations in saccular hair cells reconstructed by electron tomography. J Neurosci. 1999;19:119–132. doi: 10.1523/JNEUROSCI.19-01-00119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell P, Moody WJ. Electrical activity and calcium influx regulate ion channel development in embryonic Xenopus skeletal muscle. J Neurosci. 1995;15:4507–4514. doi: 10.1523/JNEUROSCI.15-06-04507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SQ, Kaczmarek LK. The expression of two splice variants of the Kv3.1 potassium channel gene is regulated by different signaling pathways. J Neurosci. 1998a;18:2881–2890. doi: 10.1523/JNEUROSCI.18-08-02881.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SQ, Kaczmarek LK. Depolarization selectively increases the expression of the Kv3.1 potassium channel in developing inferior colliculus neurons. J Neurosci. 1998b;18:8758–8769. doi: 10.1523/JNEUROSCI.18-21-08758.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima T, Macnamare R, Kurima K, Griffith A. Expression and localization studies of the TMC1 protein. Assoc Res Otolaryngol Abstract. 2005;625 [Google Scholar]
- Marcotti W, Géléoc GSG, Lennan GWT, Kros CJ. Developmental expression of an inwardly rectifying potassium conductance in inner and outer hair cells along the mouse cochlea. Pflugers Arch. 1999;439:113–122. doi: 10.1007/s004249900157. [DOI] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Holley MC, Kros CJ. Developmental changes in the expression of potassium currents of embryonic, neonatal and mature mouse inner hair cells. J Physiol. 2003a;548:383–400. doi: 10.1113/jphysiol.2002.034801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Kros CJ. Effects of intracellular stores and extracellular Ca2+ on Ca2+-activated K+ currents in mature mouse inner hair cells. J Physiol. 2004a;557:613–633. doi: 10.1113/jphysiol.2003.060137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Kros CJ. A transiently expressed SK current sustains and modulates action potential activity in immature mouse inner hair cells. J Physiol. 2004b;560:691–708. doi: 10.1113/jphysiol.2004.072868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Rüsch A, Kros CJ. Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J Physiol. 2003b;552:743–761. doi: 10.1113/jphysiol.2003.043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Kros CJ. Developmental expression of the potassium current IK,N contributes to maturation of the mouse outer hair cells. J Physiol. 1999;520:653–660. doi: 10.1111/j.1469-7793.1999.00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, van Netten SM, Kros CJ. The aminoglycoside antibiotic dihydrostreptomycin rapidly enters hair cells through the mechano-electrical transducer channel. J Physiol. 2005;567:505–521. doi: 10.1113/jphysiol.2005.085951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T, Beutner D. Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci U S A. 2000;97:883–888. doi: 10.1073/pnas.97.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller YL, Reitstetter R, Yool AJ. Regulation of Ca2+-dependent K+ channel expression in rat cerebellum during postnatal development. J Neurosci. 1998;18:16–25. doi: 10.1523/JNEUROSCI.18-01-00016.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutai H, Mann S, Heller S. Identification of chicken transmembrane channel-like (TMC) genes: expression analysis in the cochlea. Neurosci. 2005;132:1115–1122. doi: 10.1016/j.neuroscience.2005.01.046. [DOI] [PubMed] [Google Scholar]
- Ohmori H. Mechano-electrical transduction currents in isolated vestibular hair cells of the chick. J Physiol. 1985;359:189–217. doi: 10.1113/jphysiol.1985.sp015581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleskevich S, Walmsley B. Synaptic transmission in the auditory brainstem of normal and congenitally deaf mice. J Physiol. 2002;540:447–455. doi: 10.1113/jphysiol.2001.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Knipper M, Derst C, Fakler B. Resting potential and submembrane calcium concentration of inner hair cells in the isolated mouse cochlea are set by KCNQ-type potassium channels. J Neurosci. 2003;23:2141–2149. doi: 10.1523/JNEUROSCI.23-06-02141.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Palmer AR, Russell IJ. Phase-locking in the cochlear nerve of the guinea-pig and its relation to the receptor potential of inner hair-cells. Hear Res. 1986;24:1–15. doi: 10.1016/0378-5955(86)90002-x. [DOI] [PubMed] [Google Scholar]
- Parsons TD, Lenzi D, Almers W, Roberts WM. Calcium-triggered exocytosis and endocytosis in an isolated presynaptic cell: capacitance measurements in saccular hair cells. Neuron. 1994;13:875–883. doi: 10.1016/0896-6273(94)90253-4. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Pujol R, Lavigne-Rebillard M, Lenoir M. Development of sensory and neural structures in the mammalian cochlea. In: Rubel EW, Popper AN, Fay RR, editors. Development of the Auditory System. New York: Springer-Verlag; 1998. pp. 146–192. [Google Scholar]
- Romand R. Development of the cochlea. In: Romand R, editor. Development of Auditory and Vestibular Systems. New York: Academic Press; 1983. pp. 47–88. [Google Scholar]
- Rüsch A, Erway LC, Oliver D, Vennstrom B, Forrest D. Thyroid hormone receptor beta-dependent expression of a potassium conductance in inner hair cells at the onset of hearing. Proc Natl Acad Sci U S A. 1998;95:15758–15762. doi: 10.1073/pnas.95.26.15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüsch A, Ng L, Goodyear R, Oliver D, Lisoukov I, Vennstrom B, Richardson G, Kelley MW, Forrest D. Retardation of cochlear maturation and impaired hair cell function caused by deletion of all known thyroid hormone receptors. J Neurosci. 2001;21:9792–9800. doi: 10.1523/JNEUROSCI.21-24-09792.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel KP, Bock GR. The nature of inherited deafness in deafness mice. Nature. 1980;288:159–161. doi: 10.1038/288159a0. [DOI] [PubMed] [Google Scholar]
- Steel KP, Kros CJ. A genetic approach to understanding auditory function. Nat Genet. 2001;27:143–149. doi: 10.1038/84758. [DOI] [PubMed] [Google Scholar]
- Steel KP, Smith RJ. Normal hearing in Splotch (Sp/+), the mouse homologue of Waardenburg syndrome type 1. Nat Genet. 1992;2:75–79. doi: 10.1038/ng0992-75. [DOI] [PubMed] [Google Scholar]
- Vandenberghe W, Bredt DS. Early events in glutamate receptor trafficking. Curr Opin Cell Biol. 2004;16:137–139. doi: 10.1016/j.ceb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Sakaba T, Berglund K, Tachibana M. Submillisecond kinetics of glutamate release from a sensory synapse. Neuron. 1998;21:1177–1188. doi: 10.1016/s0896-6273(00)80634-0. [DOI] [PubMed] [Google Scholar]
- Vreugde S, Erven A, Kros CJ, Marcotti W, Fuchs H, Kurima K, Wilcox ER, Friedman TB, Griffith AJ, Balling R, Hrabe De Angelis M, Avraham KB, Steel KP. Beethoven, a mouse model for dominant, progressive hearing loss DFNA36. Nat Genet. 2002;30:257–258. doi: 10.1038/ng848. [DOI] [PubMed] [Google Scholar]
- Xie J, Black DL. A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature. 2001;410:936–939. doi: 10.1038/35073593. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Poo M. Electrical activity and development of neural circuits. Nat Neurosci. 2001;4:1207–1214. doi: 10.1038/nn753. [DOI] [PubMed] [Google Scholar]
- Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature. 2000;405:149–155. doi: 10.1038/35012009. [DOI] [PubMed] [Google Scholar]