

Abstract
The monoclonal antibody (mAb) rituximab produces objective clinical responses in patients with B-cell non-Hodgkin's lymphoma and antibody-based autoimmune diseases. Mechanisms mediating B-cell depletion by rituximab are not completely understood and may include direct effects of signalling via the target antigen CD20. Like most but not all CD20 mAbs, rituximab induces a sharp change in the solubility of the CD20 protein in the non-ionic detergent Triton-X-100, reflecting a dramatic increase in the innate affinity of CD20 for membrane raft signalling domains. Apoptosis induced by rituximab hypercrosslinking has been shown to require src family kinases (SFK), which are enriched in rafts. In this report we provide experimental evidence that SFK-dependent apoptotic signals induced by rituximab are raft dependent. Cholesterol depletion prevented the association of hypercrosslinked CD20 with detergent-insoluble rafts, and attenuated both calcium mobilization and apoptosis induced with rituximab. CD20 cocapped with the raft-associated transmembrane adaptor LAB/NTAL after hypercrosslinking with CD20 mAbs, regardless of their ability to induce a change in the affinity of CD20 for rafts. Taken together, the data demonstrate that CD20 hypercrosslinking via rituximab activates SFKs and downstream signalling events by clustering membrane rafts in which antibody-bound CD20 is localized in a high-affinity configuration.
Keywords: B cells, apoptosis, calcium, leukaemia/lymphoma/myeloma
Introduction
Therapeutic depletion of neoplastic, autoimmune or alloreactive B cells has been made possible in recent years through the development and application of monoclonal antibodies (mAb) directed against the CD20 antigen.1 Although CD20 antibodies are clinically effective, resistance occurs or develops in a significant fraction of patients, emphasizing the importance of defining the mechanisms that mediate in vivo depletion by these reagents.2 Studies from numerous laboratories have collectively yielded complex and sometimes conflicting results, probably indicating the involvement of multiple mechanisms of depletion operating at variable potency under different conditions.2,3 The situation is complicated further by the diversity of effects elicited by mAbs directed against different CD20 epitopes.4,5 In the case of rituximab, a human immunoglobulin G1 (IgG1)-mouse chimeric anti-CD20 mAb, considerable evidence supports a major role for complement-mediated cytotoxicity6–12 yet a requirement for IgG Fc receptors13,14 indicates the additional involvement of antibody-dependent cellular mechanisms and possibly direct (antiproliferative and apoptotic) effects of crosslinking the target antigen. Crosslinking cell-bound rituximab in vitro with either a secondary antibody or with fibroblasts ectopically expressing FcRs activates intracellular signalling events that can lead to apoptosis.15–21 Apoptosis occurring in vivo has also been described22,23 and may be more significant than currently appreciated. It would likely be mediated by hypercrosslinking via FcR-bearing cells within tissues, and therefore difficult to detect.
A remarkable property of most CD20 antibodies is their ability to induce a profound change in the solubility of the CD20 protein in the non-ionic detergent Triton-X-100.4,11,24 As we recently described, this change in Triton-solubility does not require cell signalling or crosslinking, and probably reflects a sudden conformational shift in CD20 that dramatically increases its innate affinity for detergent-resistant membrane rafts.25 Rafts are membrane signalling domains enriched in dually acylated signalling proteins like src-family tyrosine kinases (SFK) and it is known that CD20 hypercrosslinking activates SFKs upstream of a calcium-dependent signalling pathway leading to apoptosis.16,17 Importantly, there is a correlation between the ability of antibodies to elicit CD20's translocation to Triton-insoluble rafts and their ability to initiate SFK-dependent calcium mobilization upon hypercrosslinking (4, 11, 16, 26 and data in this report). These observations predict that CD20 hypercrosslinking with rituximab delivers apoptotic signals that are raft-dependent.27 Here, we provide experimental evidence in support of this conclusion.
The integrity of rafts is compromised by depletion of membrane cholesterol; therefore, we examined the effect of cholesterol depletion on calcium mobilization and apoptosis induced by rituximab hypercrosslinking. We first confirmed the basic observations that underpin this study, namely that calcium signalling and apoptosis induced by CD20 hypercrosslinking are SFK dependent. Then we show that rituximab induced calcium mobilization is inhibited by cholesterol depletion, indicating a requirement for the integrity of rafts in initiating SFK-dependent intracellular signals. Using annexin V and propidium iodide staining for phosphatidylserine exposure and cell viability, respectively, we show that cholesterol depletion significantly reduces apoptosis induced by hypercrosslinking CD20 with rituximab. CD20 cocapped with the raft-associated transmembrane adaptor LAB/NTAL after hypercrosslinking with CD20 antibodies, regardless of their ability to induce a change in the affinity of CD20 for rafts. These findings are consistent with the conclusion that activation of calcium mobilization and apoptotic signalling downstream of hypercrosslinked CD20 is a consequence of clustering SFK-containing rafts in which antibody-bound CD20 is localized in a high-affinity configuration.
Materials and methods
Cells and antibodies
Ramos Burkitt's lymphoma B cells were maintained in 7·5% fetal bovine serum (FBS)/RPMI-1640. BJAB B cells stably expressing transfected human LAB-GFP, established in this laboratory, were maintained in 10% FBS/RPMI with 1 mg/ml geneticin (Life Technologies, Gaithersburg, MD). Hybridoma SFM medium (Grand Island, NY) was used in apoptotic experiments requiring serum free culture.
CD20 mAbs 2H7 (IgG2b) and 1F5 (IgG2a) were provided by Dr J. Ledbetter (Bristol-Myers Squibb, Seattle, WA). Rituximab (hIgG1) was obtained from IDEC Pharmaceuticals (San Francisco, CA), B1 (IgG2a) from Coulter (Hialeah, FL), FMC7 (IgM) from Serotec (Raleigh, NC), human IgG1 and fluoroscein isothiocyanate (FITC)-conjugated antimouse IgM from Caltag (Burlingame, AL), anti-CD45 from Transduction Laboratories (Lexington, KY), and IgG2b and IgM isotype control mAbs from Sigma (St. Louis, MO). Goat anti-human F(ab′)2 IgG Fcγ-specific (GAH), rabbit anti-mouse IgG (RAM), goat anti-human F(ab′)2 IgM Fcµ-specific, goat anti-human F(ab′)2 IgGCy3 and goat antimouse F(ab′)2 IgGCy3 were from Jackson Immunoresearch (West Grove, PA). Anti-CD20 rabbit serum was generated as described.28 Horseradish peroxidase conjugates of Protein A and rabbit anti-mouse IgG were purchased from Bio-Rad (Hercules, CA) and Southern Biotechnology Associates (Birmingham, AL), respectively.
Generation of LAB-GFP construct
LAB fused to the amino-terminus of green fluorescent protein (GFP) was expressed using the pEGFPN1 expression vector from Clonetech. The entire human LAB sequence (including 5′ UTR and excluding stop codon) was cloned from BJAB cDNA using the forward primer AGTCAGATCTTCACCAGGCCACGCATCACAAG and reverse primer GACTAAGCTTGGCTTCTGTGGCTGCCACCTC. The forward primer led to the incorporation of a BglII restriction site and the reverse primer led to the incorporation of a HindIII restriction site that allowed the amplified sequence to be inserted into the multicloning site of pEGFPN1. BJAB cells expressing LAB-GFP were generated by electroporation at 340 V and 950 µF (Gene Pulser II; Bio-Rad) with 20 µg of DNA. GFP-positive cells were sorted by flow cytometry (FACStar cytometer; BD Biosciences).
Calcium mobilization
Cells were incubated in RPMI containing 20 µm fluo-3 AM (Molecular Probes, Eugene, OR) for 30 min at room temperature, then further incubated for 15 min with rituximab, 2H7, 1F5 or B1 mAbs (1 µg/106 cells). Cells (2 × 106) were resuspended in 2 ml buffer (20 mm HEPES, pH 7·4, containing 150 mm NaCl, 1·5 mm CaCl2, 3 mm KCL, 10 mm glucose, and 250 µm sulphinpyranone). Cells were excited at 480 nm and emission was measured at 530 nm. A baseline was obtained for 60 s before stimulation with the relevant secondary antibody at concentrations indicated in the figure legends. The data were acquired using a Becton Dickinson FACScan and analysed using the FlowJo program (Tree Star Inc, San Carlo, CA).
Induction and analysis of apoptosis
1·5 × 106 cells/well were loaded into a 24-well plate with 2 ml 7·5% FBS/RPMI or Hybridoma SFM medium. Cells were incubated at 37° for the times indicated with either 25 µg 2H7 or IgG2b and 50 µg RAM, or with 2 µg rituximab or hIgG and 20 µg GAH. At the end of the assay, cells were washed once with RPMI, stained with annexin V (Molecular Probes) and propidium iodide (PI; ICN, Irvine CA), and analysed on a Becton Dickinson FACScan cytometer. Cells staining positive for annexin V and negative for PI were considered apoptotic. Groups of data were analysed using a Student's paired t-test with Bonferroni's correction for multiple comparisons. All values are mean ± SEM. Statistical significance was set at P < 0·05.
Pharmacological inhibitors
For inhibition of SFKs, Ramos cells were pretreated with either 10 µm PP2 (Calbiochem, La Jolla CA) or 2·5 µm SU6656 (Calbiochem), for 30 min at 37° before the addition of antibodies. PP3, an inactive analog of PP2, was used as a control. For inhibition of PLCγ, Ramos cells were pretreated with 5 µm U73122 (Calbiochem), for 30 min at room temperature before the addition of antibodies. All inhibitors were maintained throughout the experiments.
Cholesterol modulation
For cholesterol depletion, cells were treated in RPMI containing 10 mm methyl-β-cyclodextrin (MβCD; Sigma, Oakville, ON) for the indicated times at room temperature.
For cholesterol repletion, cholesterol-depleted cells were washed and treated in RPMI containing MβCD/cholesterol complex (1·4 mm cholesterol in 10 mm MβCD) (Sigma) for the indicated times at room temperature, followed by washing.
Isolation of lipid rafts
Lipid rafts were isolated as low density, detergent-resistant membranes (DRMs) by sucrose density gradient centrifugation as described29 using 1% Triton-X-100. Briefly, 108 cells were lysed for 15 min in ice-cold lysis buffer (25 mm morpholineethanesulphonic acid (MES), 150 mm NaCl, 1 µg/ml leupeptin, 1 µg/ml aprotinin, 1 mm phenylmethylsulphonyl fluoride, 1 mm ethylenediaminetetra-acetic acid, 1 mm Na2MoO4, 1 mm Na3VO4, and 1% Triton-X-100). Lysates were mixed with an equal volume of 80% sucrose in MBS (25 mm MES and 150 mm NaCl), overlaid with 5 ml of 30% sucrose and 5 ml of 5% sucrose in MBS, and centrifuged at 200 000 g for 17 hr in a SW41 Ti rotor in a Beckman XL-70 ultracentrifuge (Fullerton, CA). Eight fractions (1·5 ml each) were collected from the top to the bottom of the gradients and an aliquot of each fraction was mixed with sodium dodecyl sulphate (SDS) sample buffer. The pellets were solubilized in SDS sample buffer by repeated freeze–thaw cycles. Samples were resolved by SDS-polyacrylamide gel electrophoresis and processed for immunoblotting using the antibodies indicated. Images were acquired using a Fluor-S MAX MultiImager (Bio-Rad).
Immunofluorescence microscopy and digital deconvolution
BJAB LAB-GFP cells were incubated either with 2H7, rituximab, or B1 mAbs (1 µg/106 cells) at 37° for 20 min in RPMI. CD20 was crosslinked and labelled by the addition of GAMCy3 for B1 and 2H7, and GAHCy3 for rituximab, at 37° for 30 min (1 µg/106 cells) and washed. Cells were fixed by incubation for 15 min in 4% paraformaldehye (Electron Microscopy Sciences Hatfield, PA), at times indicated in the figure legend. The cells were washed and mounted on microscope slides coated with poly L-lysine (Sigma-Aldrich) with no. 1·5 coverslips (Fisher), and visualized using a DeltaVision Restoration Microscopy System (Applied Precision, Issaquah, WA). Stacks of optical sections were acquired using an Olympus 60×/1·40 NA oil immersion objective with sequential recording of fluorescent and DIC images. GFP was visualized using the standard FITC filter set (Ex 490/20; Em 528/38) and Cy3 was visualized using the standard RD filter set (Ex 555/28; Em 617/73). Single-labelled control samples were imaged separately to confirm minimal bleed-through of fluorophores by non-specific filter sets (data not shown). Digital deconvolution was performed on the stacks of fluorescent optical sections using SoftWoRx constrained iterative deconvolution (Applied Precision).
Results
Signaling and apoptosis induced by CD20 hypercrosslinking are SFK dependent
Dramatic differences are found in the ability of different anti-CD20 mAbs to induce insolubility of CD20 in Triton-X-100. At opposite ends of the spectrum, the 2H7 and B1 mAbs, respectively, induce almost complete or no translocation of CD20 from the soluble to the insoluble fraction, and a 3rd mAb, 1F5, falls between the two extremes.4,24,25 As shown in Fig. 1(a), this property of CD20 mAbs correlates well with their ability to elicit calcium mobilization upon hypercrosslinking with a secondary antibody. Ramos B cells were loaded with fluo-3 and incubated with 2H7, 1F5, or B1. No calcium flux was induced by ligation of CD20 in the absence of hypercrosslinking (not shown), but the addition of RAM elicited a robust calcium flux in 2H7-bound cells, a small flux with 1F5 and none with B1. The correlation with the ability to induce CD20 insolubility in Triton, which reflects high affinity association with SFK-containing rafts25 suggests that SFKs are involved in activation of the signalling pathway leading to calcium mobilization. To test this, cells were treated with the SFK inhibitor PP2 or its inactive analogue, PP3, prior to and during CD20 crosslinking with 2H7/RAM. PP2, but not PP3, completely abolished the calcium flux, indicating that crosslinking CD20 with 2H7/RAM activates SFK-dependent calcium signals (Fig. 1b). Cells were then incubated with 2H7/RAM for 24 hr in the presence or absence of PP2 or PP3, and apoptosis was measured using annexin V/PI staining. Apoptosis was reproducibly detected after 2H7 hypercrosslinking and was attenuated by PP2 (60 ± 3%), whereas PP3 had no significant effect (Fig. 1c).
Figure 1.

Calcium mobilization and apoptosis induced by 2H7 hypercrosslinking is SFK-dependent. (a) Ramos B cells were loaded with fluo-3AM and then preincubated with 2H7, 1F5, or B1 as indicated. After establishing the baseline cells were stimulated with 50 µg RAM (arrow). (b) As in (a) except that cells were untreated or treated with PP2 or PP3 before incubation with 2H7. Data in (a) and (b) are representative of three independent experiments. The scales on the y-axes show arbitrary units assigned by the FlowJo analysis program and are only comparable for samples run in the same experiment. (c) Ramos B cells were untreated or preincubated with PP2 or PP3. Cells were plated and either received no further treatment or were incubated with 2H7 or IgG2b and RAM for 24 hr at 37°, then labelled with AnnV and PI. Data shown represent the average and standard error of three independent experiments. Significant differences between mean results are indicated (**P < 0·0178).
Rituximab, like 2H7, strongly induces CD20 insolubility4 and was therefore predicted to elicit a robust calcium flux upon hypercrosslinking. Indeed, the kinetics and amplitude of the response to rituximab were slightly faster and higher than that elicited by 2H7 (Fig. 2a). The calcium response obtained by stimulating the B-cell receptor (BCR) with F(ab′)2 anti-IgM is shown for comparison. To further investigate the calcium flux induced by rituximab, the concentration of the secondary antibody used for crosslinking, GAH, was titrated from 50 µg down to 0·625 µg. Figure 2(b) shows that the highest concentrations of GAH induced the most rapid mobilization of Ca2+, but as the concentration was decreased the amplitude of the Ca2+ flux increased, peaking at 5 µg. This concentration of GAH (5 µg) was therefore used for subsequent experiments.
Figure 2.

Rituximab hypercrosslinking induces SFK-dependent calcium mobilization. (a) Ramos B cells were loaded with fluo-3AM and then incubated with rituximab or 2H7. After establishing the baseline, samples were stimulated with 50 µg GAH or GAM (arrow). Unligated cell samples were stimulated with anti-IgM for comparison (solid line). (b) Cells incubated with rituximab were stimulated with concentrations of GAH ranging from 0·625 to 50 µg, as indicated. (c) Cells were untreated or treated with 10 µm PP2 or PP3, then incubated with rituximab. After establishing the baseline, cells were stimulated with 5 µg of GAH. Data are representative of at least three independent experiments. The scales on the y-axes show arbitrary units assigned by the FlowJo analysis program and are only comparable for samples run in the same experiment.
As with 2H7, PP2 but not PP3 completely abolished the Ca2+ flux induced by rituximab crosslinking (Fig. 2c). To further examine the SFK-dependence of the calcium response we used another inhibitor, SU6656, which was reported to inhibit SFK with over sixfold greater selectivity relative to many other kinases tested.30 We found that SU6656 inhibited the rituximab-induced calcium response at concentrations as low as 2·5 µm (supplementary Fig. 1a). Calcium release from intracellular stores is typically a downstream consequence of activated phospholipase C (PLC) and we have previously shown that hypercrosslinking CD20 activates tyrosine phosphorylation of PLC-γ.26 As shown here, the PLC inhibitor, U73122, completely inhibited calcium mobilization by rituximab crosslinking (supplementary ); U73122 had no effect on thapsigargin-mediated calcium release, indicating the integrity of the internal calcium stores (supplementary ). Together, these data indicate that rituximab-mediated CD20 hypercrosslinking activates a SFK-dependent PLCγ signalling pathway leading to release of calcium from intracellular stores.
Rituximab crosslinking clusters stable CD20 rafts
Recently, we showed that CD20 is constitutively associated with lipid rafts even though the affinity of the association does not withstand lysis in 1% Triton-X-100.25 Unligated CD20 is found in the soluble fraction of Triton lysates but moves into the buoyant insoluble fraction upon ligation with 2H7, due to an in situ increase in its affinity for the raft environment. However, 2H7 or rituximab alone is insufficient to induce SFK activation leading to calcium mobilization, which requires further crosslinking with secondary antibodies. The effect of hypercrosslinking on CD20-raft affinity was therefore examined in Ramos cells treated with the indicated antibodies before lysis and sucrose density gradient centrifugation. The gradient fractions were probed by blotting for CD20 and the non-raft marker CD45 (Fig. 3a). In lysates from untreated cells, CD20 was found in the high density soluble fractions (fractions 7 and 8). Ligation with rituximab induced redistribution of CD20 to the buoyant (raft) fractions, as expected. Hypercrosslinking with GAH had no significant effect on the rituximab-induced shift to the buoyant fractions. Importantly, cholesterol depletion using methyl-β-cyclodextrin (MβCD) caused the majority of CD20 to remain in the soluble fraction, whether it was hypercrosslinked or not. Replenishment of cholesterol with MβCD/cholesterol after cholesterol depletion restored the buoyancy of CD20 on the gradients. To monitor changes in membrane cholesterol following treatment with MβCD and/or MβCD/cholesterol, we took advantage of the FMC7 mAb, which detects an epitope on CD20 that is exquisitely sensitive to the level of cholesterol in the plasma membrane.31 FMC7 expression fell dramatically after MβCD treatment, but was fully recovered by repletion of cholesterol (Fig. 3b). In conclusion, the data shown in Fig. 3 indicate that CD20 hypercrosslinked with rituximab/GAH retains strong raft association that is sensitive to cholesterol depletion.
Figure 3.

Association of hypercrosslinked CD20 with Triton-insoluble rafts is cholesterol sensitive. (a) Cells were untreated or incubated with rituximab alone or with rituximab/GAH; where indicated, cells were pretreated with MβCD or with MβCD followed by MβCD/cholesterol before incubation with rituximab. Cells were lysed in 1% Triton X-100 and fractionated on sucrose density gradients. Fractions 1–8 were collected from the top to the bottom of the gradients; P represents the solubilized pellets. Proteins in each fraction were probed by immunoblot for CD20 and CD45 as indicated. (b) Cells were labelled with FMC7 mAb and FITC-conjugated anti-IgM, before and after treatment with MβCD and MβCD/cholesterol. The dashed line, indicating FMC7 staining after MβCD treatment, was coincident with the isotype control.
To examine the distribution of CD20 at the cell surface in relation to lipid rafts we used the raft-localized transmembrane adaptor LAB/NTAL32,33 fused to GFP. LAB-GFP was expressed in BJAB cells and was observed predominantly at the plasma membrane with a relatively uniform distribution (Fig. 4). In the absence of hypercrosslinking, incubation with 2H7 or rituximab induced no obvious change in the distribution of either CD20 or LAB-GFP (Fig. 4, compare panel 1 to panel 2 and panel 4 to panel 5), consistent with our previous data using the GPI-linked protein CD59 as a marker of rafts;25 this result supports our conclusion that antibody-induced Triton-insolubility reflects a sharp in situ increase in CD20-raft affinity rather than clustering of rafts. In contrast, hypercrosslinking caused clustering of CD20 and coclustering with LAB-GFP, regardless of the primary antibody used (Fig. 4, panels 3, 6 and 9). Even though B1 does not induce a change in the affinity of CD20 for rafts, hypercrosslinking with B1 induced CD20 coclustering with LAB-GFP, consistent with the constitutive raft localization of CD20. Taken together, the data in Figs 1–4 suggest that in order to activate SFK and induce calcium mobilization, an anti-CD20 mAb must be able to induce both increased CD20-raft affinity and clustering of the rafts; neither alone is sufficient.
Figure 4.
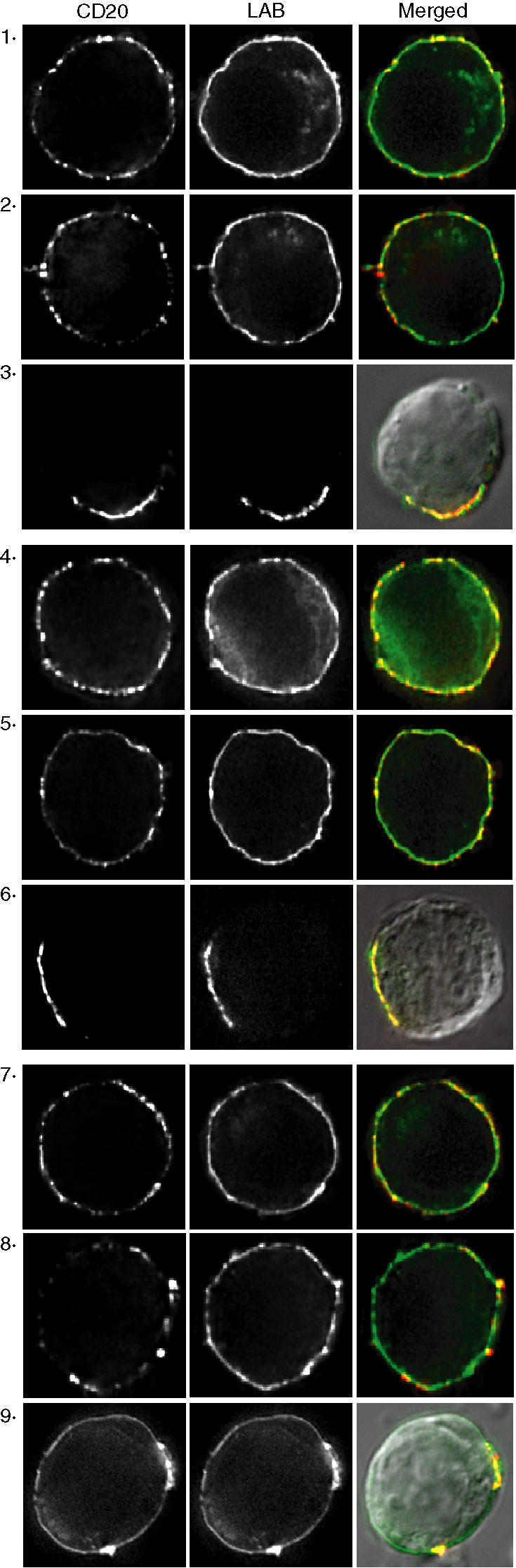
Hypercrosslinking induces cocapping of CD20 with LAB/NTAL. BJAB cells expressing LAB-GFP were incubated with rituximab (panels 1–3), 2H7 (panels 4–6), or B1 (panels 7–9) and bound mAb was detected with Cy3-conjugated F(ab′)2 anti-mouse or -human IgG. Cells were fixed before labelling (panels 1, 4, and 7), or after incubation with the primary antibody (panels 2, 5, and 8), or after incubation with the Cy3-conjugated F(ab′)2 anti-mouse IgG (panels 3, 6, 9). In panels 3, 6, and 9 the merged image is overlaid with a DIC image. Images shown are representative of at least 12 cells analysed in three independent experiments.
Calcium mobilization and apoptosis induced by CD20 hypercrosslinking is cholesterol dependent
To determine if raft localization is necessary for calcium mobilization, cells were incubated in the absence or presence of MβCD before CD20 ligation. MβCD attenuated the calcium flux induced with either rituximab/GAH or 2H7/RAM, and cholesterol repletion restored the response (Fig. 5, top and middle panels). As previously reported29,34 MβCD did not inhibit the initial peak of calcium flux induced by crosslinking the BCR with F(ab′)2 anti-IgM (Fig. 5, lower panel), demonstrating that signalling pathways were intact in MβCD-treated cells. Together with the data shown in Figs 1–3, these results demonstrate the cholesterol-dependence of SFK-activated calcium mobilization when specifically induced by crosslinking CD20 with rituximab or 2H7.
Figure 5.

Calcium mobilization by CD20 hypercrosslinking is cholesterol-dependent. Ramos B cells were loaded with fluo-3 and then incubated in the presence or absence of MβCD; some MβCD treated samples were further incubated with MβCD/cholesterol. Cells were then incubated with rituximab or 2H7. After establishing the baseline, cells were stimulated with either 5 µg GAH or 50 µg RAM (top and middle panels). Bottom panel, cells were stimulated with 10 µg F(ab′)2 anti-human IgM. Data shown are representative of at least three independent experiments.
Calcium mobilization has previously been shown to be essential for apoptosis induced by 2H7/GAM,16,17 and disruption of lipid rafts reduces the Ca2+ flux (Fig. 4); therefore, it was predicted that MβCD treatment would also attenuate apoptosis. Because extended incubation of MβCD-pretreated cells in cholesterol-free culture medium is incompatible with cell viability, we needed to first establish appropriate conditions for these experiments. First, a time-course experiment in standard conditions determined that the minimum incubation time for clear and reproducible detection of apoptosis, as measured by annexin V/PI staining, was 15 hr. Second, we identified a serum-free culture medium that supported Ramos cell viability (see Materials and methods). Incubation for 15 hr in serum-free medium still compromised cell viability when the cells were pretreated with MβCD. Therefore, serum was added at varying times after the initiation of culture to support the MβCD-treated cells after a serum-free period. We found that MβCD-treated cells were fully viable after 15 hr if serum was added to the cultures after 4 hr in serum-free medium (data not shown); these conditions were therefore used for subsequent experiments.
To test the effect of cholesterol depletion on the apoptotic response, Ramos cells were incubated in the absence or presence of MβCD, with some samples further incubated with MβCD/cholesterol. The cells were cultured in serum-free medium in the presence of rituximab/GAH or 2H7/RAM for 4 hr and then FBS was added for the remaining 11 hr. MβCD pretreatment attenuated apoptosis as determined using annexin V and PI; apoptosis was inhibited by 41 ± 8% in cells treated with 2H7/RAM (Fig. 6a) and by 47 ± 7% in cells treated with rituximab/GAH (Fig. 6b). MβCD-treated samples that were cholesterol-replenished exhibited normal amounts of apoptosis.
Figure 6.

Apoptosis induced by CD20 crosslinking is cholesterol-dependent. Ramos B cells were incubated in the absence or presence of MβCD; some MβCD treated samples were further treated with MβCD/cholesterol. Cells were washed and incubated in serum-free medium with the antibodies indicated. After 4 hr FBS was added to each well and incubation continued for an additional 11 hr. Cells were stimulated with (a) 2H7 or (b) rituximab and stained with AnnV and PI. Data shown are the average and standard error of at least three independent experiments. Significant differences between mean results are indicated (**P < 0·0049; *P < 0·0241).
Discussion
In this study we investigated a potential role for lipid rafts in rituximab-induced intracellular signalling and apoptosis. CD20 is constitutively associated with membrane rafts at an affinity that is too low to withstand lysis in 1% Triton-X-100, but most CD20 mAbs, including 2H7 and rituximab, induce translocation of CD20 into Triton-insoluble rafts.4,24,25 Here, we show that CD20 translocation into Triton-insoluble rafts is essential to activate SFKs and subsequent calcium mobilization and apoptosis. However, translocation is not sufficient to activate SFKs and a second essential step involves hypercrosslinking. These conclusions are supported by several observations:
The B1 mAb, which has higher affinity for CD20 than rituximab35 does not translocate CD20, or activate calcium mobilization even though hypercrosslinking with B1 clusters lipid rafts. This indicates that translocation is essential.
Rituximab and the 2H7 mAb efficiently translocate CD20 and also activate SFK-dependent calcium mobilization and apoptosis; however, SFK-dependent calcium mobilization and apoptosis both additionally require hypercrosslinking.
Calcium mobilization and apoptosis induced by rituximab or 2H7 were both significantly and specifically reduced by cholesterol depletion, demonstrating a requirement for the integrity of lipid rafts.
Because the induction of calcium signalling by rituximab hypercrosslinking is abolished by SFK (and PLC) inhibition, it is probably unrelated to the normal function of CD20 in store-operated calcium entry34 but is rather a serendipitous consequence of its localization to membrane rafts. Supporting this conclusion is that crosslinking other raft-associated proteins, such as GPI-linked proteins CD24 and CD48, can also induce intracellular signalling leading to apoptosis.36
A pressing question is whether CD20 hypercrosslinking occurs in vivo. Although there is currently no clear answer, it is possible that the Fc regions of cell-bound rituximab are cross-linked by binding to Fc receptors on myeloid lineage cells. Accordingly, there is in vitro evidence that crosslinking can be mediated by FcR-bearing cells15 and that apoptosis does occur in vivo after rituximab infusion.22,23
It is not known precisely why mAbs like rituximab and 2H7 induce CD20 translocation, while others, like B1, do not. Translocation appears to be an epitope-dependent phenomenon that can occur independently of crosslinking (highly purified Fab fragments of 2H7 are effective), signalling, cytoskeletal reorganization and microscopic clustering.25 B1 recognizes an epitope expressed on monomeric CD20 whereas the epitope detected by 2H7 requires quaternary interactions.4 Ligation of mAbs like 2H7 might induce altered conformation or re-organization of CD20 molecules within an oligomeric complex, in such a way that increased affinity for cholesterol-rich raft domains is induced. The existence of a cholesterol-dependent epitope on CD20, detected by the FMC7 mAb31 indicates that such structural changes are likely. Unfortunately, it was not possible to test whether accessibility of this epitope is altered after CD20 translocation since FMC7 binding is blocked by prior ligation of other CD20 mAbs. One scenario that could account for the findings reported here would place unligated CD20 in peripheral regions of rafts that are more accessible to Triton and therefore solublized more readily. If core regions of rafts are less accessible to Triton and also concentrate SFKs, as previously suggested37 this could offer a possible explanation for both antibody-induced Triton-insolubility of CD20 and the requirement for translocation of CD20 to the core of rafts in order to activate signalling by rituximab and 2H7. Hypercrosslinking rituximab would induce transactivation of SFKs by first moving CD20 into the SFK-rich cores of rafts, whereas hypercrosslinking with an antibody like B1 would cluster the rafts without inducing CD20 association with SFK-rich raft cores.
Cragg et al. reached a different conclusion regarding the role of rafts in CD20-induced apoptosis;38 however, there are essential differences between the two studies. First, their observations were primarily related to the B1 mAb, which does not induce CD20 translocation and, interestingly, induces apoptosis in the absence of further crosslinking.35,38 We had previously identified a short membrane-proximal cytoplasmic region in CD20 (residues 219–225) that reduced antibody-induced translocation by 75% when expressed in the Molt-4 T-cell line.39 Cragg et al. expressed a similar deletion mutant (residues 216–226) in 293T cells and found that it did not affect apoptosis induced by B1.38 Although it is possible that there is sufficient raft association retained by the 216–226 deletion mutant to mediate raft-dependent signals, their conclusion that B1-induced apoptosis is raft-independent is consistent with the lack of translocation induced by B1 and the lack of any requirement for hypercrosslinking. Second, their experiments with rituximab did not involve hypercrosslinking. A low level of apoptosis can be induced by rituximab in some B cell lines without additional crosslinking, and this was also shown to be raft-independent using the 216–226 deletion mutant in 293T cells.38 New evidence for signalling mediated by rituximab without additional crosslinking was recently provided by Cittera et al. using gene expression profiling.40 The membrane-proximal, raft-independent signalling events activated by B1, or rituximab in the absence of hypercrosslinking, have not been defined but are expected to be independent of SFK activation.
We attempted to determine the effect of the 219–225 deletion on apoptosis induced by rituximab hypercrosslinking. Apoptosis was efficiently induced in Molt-4 cells expressing wild-type CD20 but not the 219–225 mutant; however, the mutant was expressed at a lower level than wild-type CD20. We generated a new cell line expressing wild-type CD20 at a lower level to match that of 219–225, but found that apoptosis could no longer be detected. Thus, this line of investigation was inconclusive.
Rituximab-induced CD20 translocation has been reported to alter the organization of raft components without affecting the overall protein or lipid composition of rafts.41 Functional consequences of this appear to include decreased lyn kinase activity in the presence of the high concentrations of Csk binding protein/phosphoprotein associated with glycolipid enriched membranes (Cbp/PAG) found in Raji cells, and increased susceptibility to phospholipase-mediated cleavage of the GPI-linked complement defence protein, CD55. Rituximab is more effective than B1 at initiating the complement cascade and this also appears to be related to differential effects of mAbs on CD20 translocation.11 An interesting explanation for this unexpected finding is that early components of the complement cascade may insert preferentially into raft domains, although it may also be due to a particular organization of antibody clusters accompanying translocation.11 Rituximab-induced CD20 translocation has now also been linked to sphingomyelinase activation with ceramide production in rafts, raising interesting new avenues for investigation into direct effects of antibody ligation.42
While this manuscript was under revision a paper reporting similar effects of MβCD on rituximab-induced calcium flux and apoptosis was published.43 The only substantial difference from our study is that activation of intracellular signalling pathways was not examined. These authors conclude that the calcium flux is caused by opening of the calcium channel; however, there is no experimental evidence to support this. On the contrary, we have previously shown that CD20 hypercrosslinking activates calcium mobilization in the absence of extracellular calcium26 and several studies, including those reported here, show that CD20 ligation activates intracellular signalling.
In summary, we have shown here that both translocation and hypercrosslinking of CD20 are necessary for rituximab to activate SFK-dependent signalling and apoptosis. These effects are probably unrelated to CD20's function in store-operated calcium entry but instead are a consequence of its association with cholesterol-dependent membrane rafts.
Acknowledgments
These studies were supported by an operating grant from the Alberta Cancer Board to J. P. D. and by personnel awards from the Canadian Institutes of Health Research (CIHR) Training Program in Immunology, Immunopathogenesis and Inflammation at the University of Calgary (T. L. U.), the Alberta Heritage Foundation for Medical Research (J. P. D. and C. M. M.), and the Natural Sciences and Engineering Research Council of Canada (C. M. M.). The Live Cell Imaging Facility at the University of Calgary is supported by CIHR. The authors gratefully acknowledge the expert assistance of Laurie Robertson and Fusun Turesin, Flow Cytometry Facility, and Dr Pina Colarusso, Live Cell Imaging Facility.
References
- 1.Boye J, Elter T, Engert A. An overview of the current clinical use of the anti-CD20 monoclonal antibody rituximab. Ann Oncol. 2003;14:520–35. doi: 10.1093/annonc/mdg175. [DOI] [PubMed] [Google Scholar]
- 2.Cartron G, Watier H, Golay J, Solal-Celigny P. From the bench to the bedside: ways to improve rituximab efficacy. Blood. 2004;104:2635–42. doi: 10.1182/blood-2004-03-1110. [DOI] [PubMed] [Google Scholar]
- 3.Johnson P, Glennie M. The mechanisms of action of rituximab in the elimination of tumor cells. Semin Oncol. 2003;30(Suppl. 2):3–8. doi: 10.1053/sonc.2003.50025. [DOI] [PubMed] [Google Scholar]
- 4.Polyak MJ, Deans JP. Alanine-170 and proline-172 are critical determinants for extracellular CD20 epitopes; heterogeneity in the fine specificity of CD20 monoclonal antibodies is defined by additional requirements imposed by both amino acid sequence and quaternary structure. Blood. 2002;99:3256–62. doi: 10.1182/blood.v99.9.3256. [DOI] [PubMed] [Google Scholar]
- 5.Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2004;103:2738–43. doi: 10.1182/blood-2003-06-2031. [DOI] [PubMed] [Google Scholar]
- 6.Anderson DR, Grillo-Lopez A, Varns C, Chambers KS, Hanna N. Targeted anti-cancer therapy using rituximab, a chimaeric anti-CD20 antibody (IDEC-C2B8) in the treatment of non-Hodgkin's B-cell lymphoma. Biochem Soc Trans. 1997;25:705–8. doi: 10.1042/bst0250705. [DOI] [PubMed] [Google Scholar]
- 7.Golay J, Zaffaroni L, Vaccari T, et al. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000;95:3900–8. [PubMed] [Google Scholar]
- 8.van der Kolk LE, Grillo-Lopez AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol. 2001;115:807–11. doi: 10.1046/j.1365-2141.2001.03166.x. [DOI] [PubMed] [Google Scholar]
- 9.Di Gaetano N, Cittera E, Nota R, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581–7. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 10.Kennedy AD, Beum PV, Solga MD, et al. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol. 2004;172:3280–8. doi: 10.4049/jimmunol.172.5.3280. [DOI] [PubMed] [Google Scholar]
- 11.Cragg MS, Morgan SM, Chan HT, Morgan BP, Filatov AV, Johnson PW, French RR, Glennie MJ. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid ‘rafts’. Blood. 2002;101:1045–52. doi: 10.1182/blood-2002-06-1761. [DOI] [PubMed] [Google Scholar]
- 12.Manches O, Lui G, Chaperot L, et al. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood. 2003;101:949–54. doi: 10.1182/blood-2002-02-0469. [DOI] [PubMed] [Google Scholar]
- 13.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443–6. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 14.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 15.Shan D, Ledbetter JA, Press OW. Apoptosis of maligant human B cells by ligation of CD20 with monoclonal antibodies. Blood. 1998;91:1644–52. [PubMed] [Google Scholar]
- 16.Shan D, Ledbetter JA, Press OW. Signaling events involved in anti-CD20-induced apoptosis of malignant human B cells. Cancer Immunol Immunother. 2000;48:673–83. doi: 10.1007/s002620050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofmeister JK, Cooney D, Coggeshall KM. Clustered CD20 induced apoptosis. src-family kinase, the proximal regulator of tyrosine phosphorylation, calcium influx, and caspase 3-dependent apoptosis. Blood Cells Mol Dis. 2000;26:133–43. doi: 10.1006/bcmd.2000.0287. [DOI] [PubMed] [Google Scholar]
- 18.Mathas S, Rickers A, Bommert K, Dorken B, Mapara MY. Anti-CD20- and B-cell receptor-mediated apoptosis: evidence for shared intracellular signaling pathways. Cancer Res. 2000;60:7170–6. [PubMed] [Google Scholar]
- 19.Taji H, Kagami Y, Okada Y, Andou M, Nishi Y, Saito H, Seto M, Morishima Y. Growth inhibition of CD20-positive B lymphoma cell lines by IDEC-C2B8 anti-CD20 monoclonal antibody. Jpn J Cancer Res. 1998;89:748–56. doi: 10.1111/j.1349-7006.1998.tb03280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghetie MA, Bright H, Vitetta ES. Homodimers but not monomers of Rituxan (chimeric anti-CD20) induce apoptosis in human B-lymphoma cells and synergize with a chemotherapeutic agent and an immunotoxin. Blood. 2001;97:1392–8. doi: 10.1182/blood.v97.5.1392. [DOI] [PubMed] [Google Scholar]
- 21.Pedersen IM, Buhl AM, Klausen P, Geisler CH, Jurlander J. The chimeric anti-CD20 antibody rituximab induces apoptosis in B-cell chronic lymphocytic leukemia cells through a p38 mitogen activated protein-kinase-dependent mechanism. Blood. 2002;99:1314–9. doi: 10.1182/blood.v99.4.1314. [DOI] [PubMed] [Google Scholar]
- 22.Byrd JC, Kitada S, Flinn IW, Aron JL, Pearson M, Lucas D, Reed JC. The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction. Blood. 2002;99:1038–43. doi: 10.1182/blood.v99.3.1038. [DOI] [PubMed] [Google Scholar]
- 23.Ramanarayanan J, Hernandez-llizaliturri FJ, Chanan-Khan A, Czuczman MS. Pro-apoptotic therapy with the oligonucleotide Genasense (oblimersen sodium) targeting Bcl-2 protein expression enhances the biological anti-tumour activity of rituximab. Br J Haematol. 2004;127:519–30. doi: 10.1111/j.1365-2141.2004.05239.x. [DOI] [PubMed] [Google Scholar]
- 24.Deans JP, Robbins SM, Polyak MJ, Savage JA. Rapid redistribution of CD20 to a low density detergent-insoluble membrane compartment. J Biol Chem. 1998;273:344–8. doi: 10.1074/jbc.273.1.344. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Ayer LM, Polyak MJ, et al. The CD20 calcium channel is localized to microvilli and constitutively associated with membrane rafts: antibody binding increases the affinity of the association through an epitope-dependent cross-linking-independent mechanism. J Biol Chem. 2004;279:19893–901. doi: 10.1074/jbc.M400525200. [DOI] [PubMed] [Google Scholar]
- 26.Deans JP, Schieven GL, Shu GL, Valentine MA, Gilliland LA, Aruffo A, Clark EA, Ledbetter JA. Association of tyrosine and serine kinases with the B cell surface antigen CD20. Induction via CD20 of tyrosine phosphorylation and activation of phospholipase C-gamma 1 and PLC phospholipase C-gamma 2. J Immunol. 1993;151:4494–504. [PubMed] [Google Scholar]
- 27.Deans JP, Li H, Polyak MJ. CD20-mediated apoptosis: signalling through lipid rafts. Immunology. 2002;107:176–82. doi: 10.1046/j.1365-2567.2002.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrie RJ, Deans JP. Colocalization of the B cell receptor and CD20 followed by activation-dependent dissociation in distinct lipid rafts. J Immunol. 2002;169:2886–91. doi: 10.4049/jimmunol.169.6.2886. [DOI] [PubMed] [Google Scholar]
- 29.Petrie RJ, Schnetkamp PP, Patel KD, Awasthi-Kalia M, Deans JP. Transient translocation of the B cell receptor and Src homology 2 domain-containing inositol phosphatase to lipid rafts: evidence toward a role in calcium regulation. J Immunol. 2000;165:1220–7. doi: 10.4049/jimmunol.165.3.1220. [DOI] [PubMed] [Google Scholar]
- 30.Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20:9018–27. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polyak MJ, Ayer LM, Szczepek AJ, Deans JP. A cholesterol-dependent CD20 epitope detected by the FMC7 antibody. Leukemia. 2003;17:1384–9. doi: 10.1038/sj.leu.2402978. [DOI] [PubMed] [Google Scholar]
- 32.Janssen E, Zhu M, Zhang W, Koonpaew S, Zhang W. LAB. A new membrane-associated adaptor molecule in B cell activation. Nat Immunol. 2003;4:117–23. doi: 10.1038/ni882. [DOI] [PubMed] [Google Scholar]
- 33.Brdicka T, Imrich M, Angelisova P, et al. Non-T cell activation linker (NTAL): a transmembrane adaptor protein involved in immunoreceptor signaling. J Exp Med. 2002;196:1617–26. doi: 10.1084/jem.20021405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Ayer LM, Lytton J, Deans JP. Store-operated cation entry mediated by CD20 in membrane rafts. J Biol Chem. 2003;278:42427–34. doi: 10.1074/jbc.M308802200. [DOI] [PubMed] [Google Scholar]
- 35.Cardarelli PM, Quinn M, Buckman D, Fang Y, Colcher D, King DJ, Bebbington C, Yarranton G. Binding to CD20 by anti-B1 antibody or F (ab′) (2) is sufficient for induction of apoptosis in B-cell lines. Cancer Immunol Immunother. 2002;51:15–24. doi: 10.1007/s00262-001-0247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki T, Kiyokawa N, Taguchi T, Sekino T, Katagiri YU, Fujimoto J. CD24 induces apoptosis in human B cells via the glycolipid-enriched membrane domains/rafts-mediated signaling system. J Immunol. 2001;166:5567–77. doi: 10.4049/jimmunol.166.9.5567. [DOI] [PubMed] [Google Scholar]
- 37.McCabe JB, Berthiaume LG. N-terminal protein acylation confers localization to cholesterol, sphingolipid-enriched membranes but not to lipid rafts/caveolae. Mol Biol Cell. 2001;12:3601–17. doi: 10.1091/mbc.12.11.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan HT, Hughes D, French RR, Tutt AL, Walshe CA, Teeling JL, Glennie MJ, Cragg MS. CD20-induced lymphoma cell death is independent of both caspases and its redistribution into triton X-100 insoluble membrane rafts. Cancer Res. 2003;63:5480–9. [PubMed] [Google Scholar]
- 39.Polyak MJ, Tailor SH, Deans JP. Identification of a cytoplasmic region of CD20 required for its redistribution to a detergent-insoluble membrane compartment. J Immunol. 1998;161(7):3242–8. [PubMed] [Google Scholar]
- 40.Cittera E, Onofri C, D'Apolito M, et al. Rituximab induces different but overlapping sets of genes in human B-lymphoma cell lines. Cancer Immunol Immunother. 2004;54:273–86. doi: 10.1007/s00262-004-0599-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semac I, Palomba C, Kulangara K, Klages N, Echten-Deckert G, Borisch B, Hoessli DC. Anti-CD20 therapeutic antibody rituximab modifies the functional organization of rafts/microdomains of B lymphoma cells. Cancer Res. 2003;63:534–40. [PubMed] [Google Scholar]
- 42.Bezombes C, Grazide S, Garret C, Fabre C, Quillet-Mary A, Muller S, Jaffrezou JP, Laurent G. Rituximab antiproliferative effect in B-lymphoma cells is associated with acid-sphingomyelinase activation in raft microdomains. Blood. 2004;104:1166–73. doi: 10.1182/blood-2004-01-0277. [DOI] [PubMed] [Google Scholar]
- 43.Janas E, Priest R, Wilde JI, White JH, Malhotra R. Rituxan (anti-CD20 antibody)-induced translocation of CD20 into lipid rafts is crucial for calcium influx and apoptosis. Clin Exp Immunol. 2005;139:439–46. doi: 10.1111/j.1365-2249.2005.02720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]