

Abstract
Background
The role of IKs, the slow delayed rectifier K+ current, in cardiac ventricular repolarization has been a subject of debate.
Methods and Results
We develop a detailed Markov model of IKs and its α-subunit KCNQ1 and examine their kinetic properties during the cardiac ventricular action potential at different rates. We observe that interaction between KCNQ1 and KCNE1 (the β-subunit) confers kinetic properties on IKs that make it suitable for participation in action potential repolarization and its adaptation to rate changes; in particular, the channel develops an available reserve of closed states near the open state that can open rapidly on demand.
Conclusions
Because of its ability to form an available reserve, IKs can function as a repolarization reserve when IKr, the rapid delayed rectifier, is reduced by disease or drug and can prevent excessive action potential prolongation and development of arrhythmogenic early afterdepolarizations.
Keywords: action potentials, electrophysiology, ion channels
Mutations to the cardiac potassium channel gene KCNQ1 (KvLQT1) have been linked to the long QT syndrome LQT1, which predisposes patients to arrhythmia during exercise and emotional stress, conditions that involve high levels of β-adrenergic stimulation. KCNQ1 is a 6-transmembrane domain protein that can form functional homomeric potassium channels and can also coassemble with the single transmembrane domain protein KCNE1 (MinK).1 Together these gene products reconstitute the IKs channel. Mutations to KCNE1 have also been linked to LQT (LQT5).2 In addition, transmural heterogeneity of IKs expression in ventricular myocardium gives rise to mid-myocardial cells (M cells) with a longer action potential (AP) duration (APD) and greater APD rate adaptation than epicardial or endocardial cells in many species.3 IKs is also augmented by β-adrenergic stimulation,4 suggesting its important role in mediating cardiac electrophysiological response.
These findings suggest that IKs is important for AP repolarization and APD adaptation to changes in rate, as demonstrated in guinea pig.5–7 However, IKs density has been reported to be much lower in larger mammals, specifically in canine and human ventricle.8 In canine myocytes, L-type calcium current, an inward depolarizing current, has been shown to mediate APD rate adaptation under control conditions without β-adrenergic effects.7 However, AP repolarization requires a sufficient outward repolarizing current during phases 2 and 3 of the AP. Such current is carried by IKr (the rapid delayed rectifier) and IKs, with IKr playing a primary role in large mammals under normal physiological conditions and in the absence of β-adrenergic stimulation. Because phases 2 and 3 depend on a delicate balance between inward and outward currents, one cannot rule out a priori an important role for IKs. Moreover, the arrhythmic consequences of LQT1 and LQT52 mutations, the existence of only 2 repolarizing currents (IKr and IKs) that constitute the delayed rectifier, and incorporation of the β-adrenergic signaling molecules into the IKs channel complex4 strongly suggest an important role for IKs in human heart electrophysiology under various conditions. It has been hypothesized that in large mammals IKs constitutes a “repolarization reserve” (RR) that compensates for reductions in other repolarizing currents, in particular IKr, caused by mutations (hereditary LQT2) or drugs (acquired LQT syndrome).9,10 There is growing consensus that in the absence of RR, certain drugs such as sotalol (antiarrhythmic), erythromycin (anti-infective), chlorpromazine (antipsychotic), and methadone can trigger a life-threatening arrhythmia.9 The possibility of IKs generating this reserve and the dependence of IKs participation on its kinetics remain to be elucidated.
In the present study we examine the hypothesis that IKs can participate in AP repolarization because of kinetic properties conferred by interaction between its KCNQ1 and KCNE1 subunits. We present detailed, experimentally based Markov models of KCNQ1 and IKs and examine their kinetic behavior during the AP at slow and fast rates. By comparing KCNQ1 behavior with IKs, we isolate the effect of the modulatory KCNE1 subunit on the AP. Results show that because of its kinetic properties, IKs can create an available reserve (AR) of channels at fast rates that can open and generate a larger repolarizing current. In the presence of IKr block, this AR prevents excessive APD prolongation and the formation of arrhythmogenic early afterdepolarizations (EADs). Such properties are not present in homomeric KCNQ1 channels and therefore require interaction with KCNE1. The AR concept relates to reserve within a single channel, thereby extending the RR concept that involves compensation for one repolarizing current by another.
Methods
Markov models of KCNQ1 and IKs are derived from experimental data and published K+ channel models.11,12 Koren et al11 described the K+ delayed rectifier RCK1 with a Markov model of 4 independent voltage sensor transitions and 1 cooperative voltage-independent transition before the open state. Zagotta et al12 expanded this model to study Shaker K+ channels by assuming that each voltage sensor undergoes 2 conformational changes before channel opening and successfully reproduced delayed activation (sigmoidal activation). A delay of several milliseconds has also been observed for KCNQ113 and IKs activation,14 suggesting that at least 2 voltage sensor transitions occur before channel opening. Experiments (J. Cui, PhD, personal communication, November 2004) also suggest a voltage-independent transition immediately before the first IKs open state, as proposed by Koren et al11 for RCK1.
The Markov schemes we developed for KCNQ1 and IKs are shown in Figure 1A and 1B. Two closed-state zones are shown; green represents states in which at least 1 voltage sensor must still make a first transition, and blue represents states in which only faster second transitions are necessary for opening. Model derivation is described in the online-only Data Supplement. Criteria for fitting parameters are described below.
Figure 1.
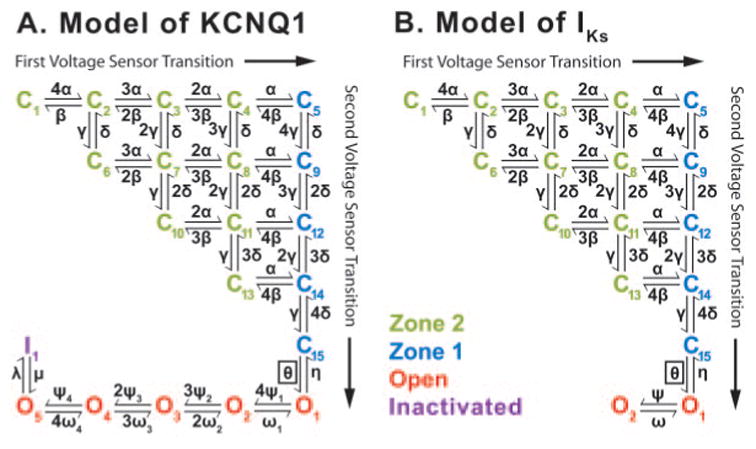
KCNQ1 and IKs Markov models. A, Model of KCNQ1 contains 15 closed (C1 to C15) states to account for 2 transitions of each of the 4 voltage sensors before channel opening. Green closed states represent channels in zone 2 that have not completed the first transition for all 4 channel subunits. Blue closed states represent channels in zone 1 that have completed 4 first voltage sensor transitions. The boxed transition (θ) to the open state is voltage independent. There are 5 open states (O1 to O5, red) and an inactivation state (I, purple). B, Model of IKs contains 2 open states with no inactivation.
The KCNQ1 model is based on frog oocyte recordings13 (Figure 2). Simulated activation to various potentials (Figure 2A), inactivation measured by a triple-pulse protocol (Figure 2C), and dependence of deactivation time constant on prepulse duration (Figure 2E) were included in the fitting procedure (details are in the online-only Data Supplement). The reversal potential was −90 mV and temperature 23°C, as dictated by experimental conditions. (Sufficient KCNQ1 data are not available at 37°C). Model comparisons with experimental data are shown in Figure 2B, 2D, and 2F.
Figure 2.
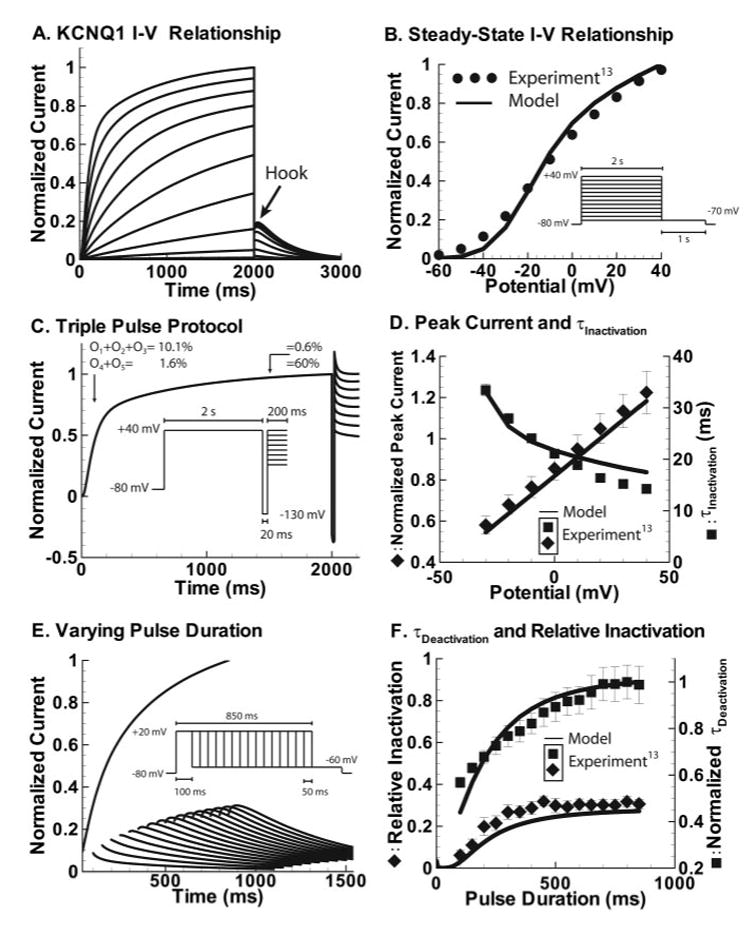
KCNQ1. A, Simulated current resulting from steps from −80 mV to various potentials (−70 to 40 mV) followed by a step to −70 mV (protocol shown in B). After stepping down to −70 mV, a hook is observed in the tail current indicating presence of inactivation, as observed experimentally.13 B, Simulated steady state current-voltage (I–V) curve (solid line) obtained from the protocol in A is compared with experiment (circles). C, Simulated current resulting from triple-pulse protocol (inset). Current resulting from the second depolarizing pulse shows no reactivation, indicating a delay before deactivation. The delay is due to accumulation in O4 and O5, as can be seen by the percentage (shown above tracing) of channels in these states late during the pulse. D, Simulated peak current for the second depolarizing pulse in C (solid line) normalized to the maximum current at 40 mV is compared with experiment (diamonds). Time constant of inactivation (from protocol in C) is also compared with experiment (squares). E, Simulated tail currents after various prepulse durations (protocol in inset). F, Relative inactivation and deactivation time constant with varying pre-pulse duration from the protocol in E. Simulated time constant of deactivation is normalized to 739 ms; experimental time constants (squares) are normalized to 365 ms. Relative inactivation is also compared with experiment (diamonds). B, D, and F were adapted from reference 13 with permission from Blackwell Publishing.
Lu et al15 have recorded E-4031 insensitive current (IKs) in guinea pig ventricular myocytes at 37°C for [K+]o=5.4 mmol/L and [Na+]o=143 mmol/L (Figure 3A). Data were digitized from the published experimental figure, and ion concentrations were set as reported. Channels were assumed to be closed at −50 mV on the basis of the current-voltage relationship that shows no activation below −10 mV (Figure 3B). Therefore, instantaneous current (possibly due to leakage) was subtracted. Current tracings were normalized according to the current-voltage relationship curve so that the steady state value for each tracing would match the experimental average. Deactivation at −50 mV was fit directly to published data.15 Rate of deactivation at resting Vm and current accumulation at fast pacing rates were simulated with IKs inserted in the whole-cell model (see below); comparison with experimental data15 is shown in Figure 3B. Additionally, mean current increase at fast rates and dynamic conductance (corresponding to APs and IKs simulated in Figure 4) were compared with recordings from AP clamp16 (Figure 5). A previously developed formulation of the reversal potential that accounts for sodium conductance through IKs channels was used (ratio of Na+:K+ permeability of 0.01833).6
Figure 3.
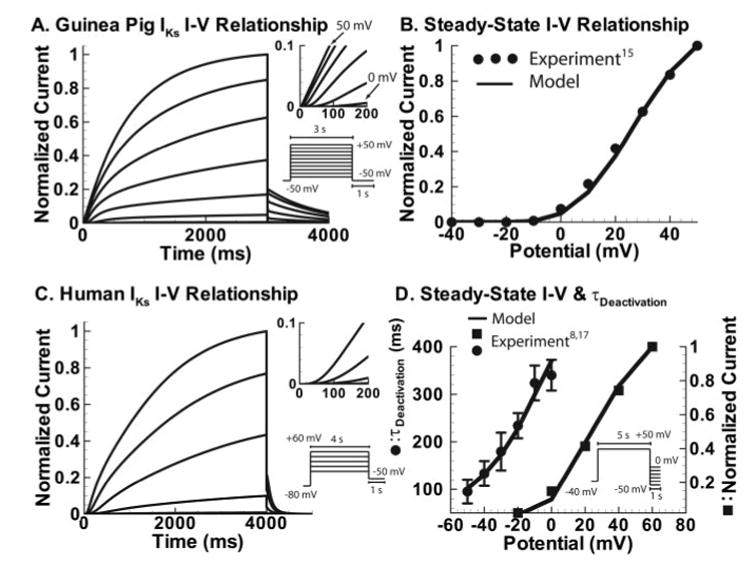
Guinea pig and human IKs. A, Simulated guinea pig IKs current resulting from protocol in inset. A delay of several milliseconds is observed before depolarization at voltages <20 mV and is inversely proportional to voltage (inset enlarges the initial portion of the current tracings). Tail currents show no hook, indicating absence of inactivation, as in experiment. B, Simulated steady state guinea pig current-voltage (I–V) relationship from the protocol in A is compared with experiment (circles). Adapted from reference 15 with permission from Lippincott Williams and Wilkins. C, Human IKs current resulting from the protocol in inset. Activation is slower than guinea pig IKs with a longer (20 ms) delay, and tail currents deactivate faster, as observe experimentally. D, Simulated steady state I–V relationship for human IKs compared with experiment17 (squares, protocol in C) and time constant of deactivation compared with experiment8 (circles, protocol in D inset). Adapted from references 8 and 17, copyright 2001 and 2002, with permission from The European Society of Cardiology.
Figure 4.
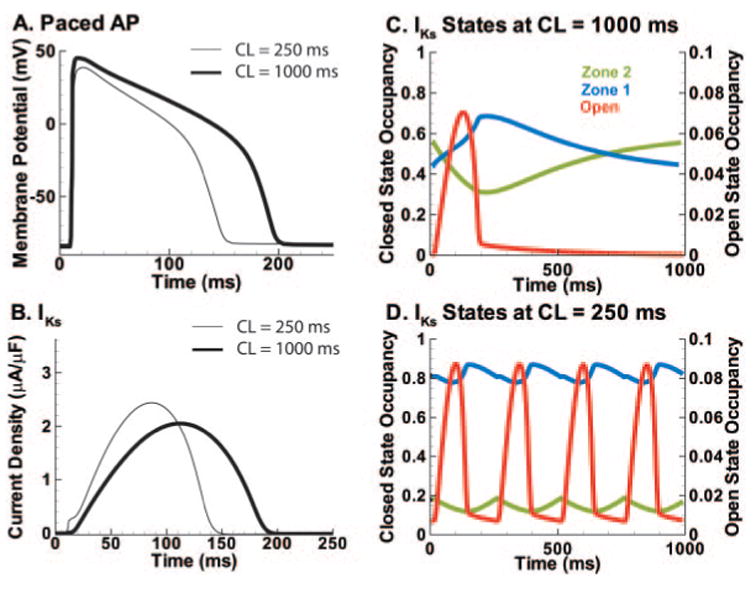
IKs role in APD adaptation. A, Fortieth AP at CL=250 ms (thin line) and CL=1000 ms (thick line) under control conditions. B, Guinea pig IKs during the APs in A. C, State occupancy during the 40th AP at slow rate (1000 ms). Green shows occupancy in zone 2 (Figure 1). Blue shows occupancy in zone 1. Red shows open channel occupancy (O1+O2). At AP initiation, most channels reside in zone 2. D, Occupancies at fast rate (CL=250 ms); same format as C. At the AP initiation, most channels reside in zone 1. Accumulation in zone 1 facilitates channel opening and larger IKs during the AP (B), causing APD shortening (A). Buildup of zone 1 reserve underlies the participation of IKs in APD rate adaptation. Open-state accumulation is minimal.
Figure 5.
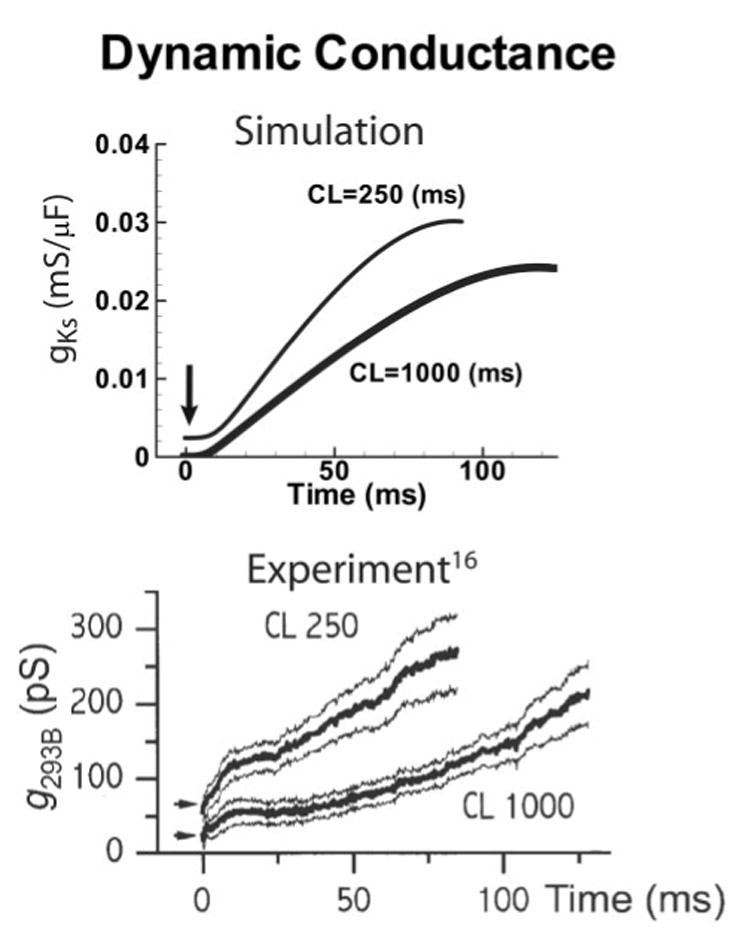
Dynamic guinea pig IKs conductance during AP clamp. Bottom, Experimentally measured IKs conductance (Chromanol 293B–sensitive current, g293B) at fast (CL=250 ms) and slow rates (CL=1000 ms) (thick and thin lines are mean and confidence limits, respectively). Arrows indicate open-state occupancy at AP outset. Reprinted from reference 16 with permission from Blackwell Publishing. Top, Simulation also shows little open-state accumulation (arrow) even at fast rates; instead, in both simulation and experiment an increase in IKs activation rate during fast pacing is responsible for greater IKs during the AP.
Human IKs activation kinetics (Figure 3C) were fit to data from Kupershmidt et al,17 who recorded expressed human IKs in HEK cells at 37°C. [Na+]o was 145 mmol/L, and [K+]o was 4 mmol/L. Expressed currents were fit because they display activation kinetics more clearly than currents recorded from native cells while still displaying activation at ≈−10 mV and a delay before slow activation, as in native cells.8 Deactivation was constrained by time constants measured in human ventricular myocytes at 37°C for [Na+]o=144 mmol/L and [K+]o=4 mmol/L to ensure that open-state accumulation was not overestimated.8 Reversal potential was calculated as for guinea pig IKs. Comparisons with experimental data are shown in Figure 3D.
Markov models for INa, IKr (updated version), and IKs were inserted in the Luo-Rudy (LRd) model of the guinea pig ventricular cell. AP simulation conditions are discussed in the online-only Data Supplement.
Results
KCNQ1 Model Validation and Current Properties
The KCNQ1 model (Figure 1A) was optimized to reproduce experimental protocols designed to isolate the essential channel features.13 Sigmoidal activation is reproduced with multiple closed-state transitions before opening. This allows for minimal initial current followed by a steep rise after ≈3 ms, as observed experimentally13 (Figure 2A). Two time constants of activation (fast followed by slow) are evident, with a slow rise in current observed even at 2-second depolarization, the last time point for which the current-voltage relationship is experimentally measured (Figure 2A, 2B).
Next, the triple-pulse protocol (Figure 2C, 2D), which measures channel inactivation at different potentials, is simulated. A short hyperpolarizing pulse to −130 mV allows most channels to recover from inactivation while preventing significant deactivation (Figure 2C). In the model, 5 open states allow channels to occupy open states far from the closed state (O4 and O5, Figure 2C), which delays deactivation during the hyperpolarizing pulse. However, transitions toward the closed state are rapid enough to reproduce the measured time constant of deactivation at −70 mV (Figure 2A; simulated τdeact=377 ms, measured τdeact 348 ms). Peak current and time constant of inactivation during the following (third) pulse are consistent with experimental results13 (Figure 2D), demonstrating rapid and voltage-dependent inactivation.
Finally, the dependence of deactivation time constant and relative inactivation (defined in the online-only Data Supplement) on pulse duration are simulated. KCNQ1 deactivation rate varies with pulse duration. As channels enter open states that are farther from the closed states, the rate of entry back into the closed states (τdeact) is slowed (Figure 2F). τdeact is greater than estimated experimentally from the dependence of deactivation on pulse duration (Figure 2E inset). However, a different experimental protocol (voltage dependence of deactivation) in the same study13 estimates τdeact close to the model-determined value at −60 mV (502±27 ms for experiment; 594 ms for model) with the same protocol. The different τdeact from the 2 experimental protocols may be explained by the need to compensate for the inactivation hook in the pulse-duration protocol (Figure 2E inset) when fitting an exponential, a procedure that can have a major effect on the estimate of τdeact. In the model such correction is not necessary because deactivation is measured directly as the decay of occupancy in the open and inactivated states.
In contrast to rapid inactivation during the third pulse (above), a single depolarizing pulse from −80 mV to 20 mV results in slow onset of inactivation (>200 ms pulse duration needed to elicit hook in tail current; Figure 2E) because of multiple transitions through open states. These transitions also govern relative inactivation (protocol not included in optimization), which shows an experimentally observed 75-ms delay13 (Figure 2F).
IKs Model Validation and Current Properties
The IKs model resembles the KCNQ1 model closely, with the number of open-state transitions truncated and inactivation removed (Figure 1). Significant differences are introduced by changes in the transition rates. For example, increased KCNE1 mRNA in frog oocytes results in greater delay before activation.14 This subunit effect is simulated by decreasing the transition rate from the first (resting) voltage sensor position (α) in the heteromeric guinea pig IKs channel to half the homomeric channel rate, confining more channels to zone 2 (Figure 3A, 3C insets). Once channels transition out of this zone, they activate rapidly. However, some channels remain in zone 2, causing a slow rise in current that continues even after several seconds (Figure 3A).
The first voltage sensor position is more stable for human IKs than KCNQ1 (C1 occupancy is ≈9 times greater at −80 mV), resulting in slower activation and a continuous current increase even after a 5-second pulse to 60 mV. Once channels transition into O1, they rapidly enter O2 (at 40 mV, the rate into O2 is 6.6 times faster than C15 to O1). The presence of 2 open states allows for simultaneous reproduction of fast activation, steady state current-voltage relationship, and slow deactivation by facilitating slow deactivation without requiring slow activation to reproduce the steady state current.
IKs Role in Rate Adaptation of APD
Simulated whole-cell APs computed with the guinea pig IKs Markov model are shown at fast and slow pacing rates (Figure 4A). At the fast rate, peak IKs increases moderately compared with the slow rate (Figure 4B); however, the more rapid rise of IKs at the fast rate leads to greater repolarizing current earlier during the AP. This earlier IKs rise causes a 19.7% increase in mean IKs during the AP, from 1.27 μA/μF at cycle length (CL)= 1000 ms to 1.52 μA/μF at CL=250 ms. This is a conservative value compared with AP clamp experiments that measured a 28% increase at these rates.16 The IKs increase during the AP accelerates repolarization to cause APD shortening at fast rates (adaptation).
IKs mediation of adaptation is a result of its closed-state transitions rather than its open-state accumulation. Figure 4C and 4D show total occupancy in zone 2 (green), zone 1 (blue), and the open states O1 and O2 (red) (Figure 1). During slow pacing (Figure 4C), most IKs channels reside in zone 2 before AP depolarization and must make a slow transition into zone 1 before opening. In contrast, at fast rates (Figure 4D) channels accumulate in zone 1 between APs, allowing IKs to activate rapidly via the second transition. There is not sufficient time between beats for the channels to transition back to zone 2 before the next AP, as at slow rates. Although there is some open-state accumulation due to slow deactivation, its effect on the current is opposed by the lower driving force resulting from a lower peak membrane potential at fast rates. Consequently, the primary mechanism for adaptation is the accumulation of closed states in zone 1, where a fast transition to the open states generates a rapid IKs rise during phase 1. Thus, at fast rates an AR is built ready to open very rapidly on demand. The overall low probability of channels residing in open states at slow and fast rates (Figure 4C, 4D) indicates that many channels are closed during the AP, creating a large IKs reserve.
AP clamp experiments have measured IKs conductance (gKs) during the AP at fast and slow rates, showing that open-state accumulation is minimal (Figure 5, bottom panel, arrows). Rather, IKs activates more rapidly at a fast rate than at a slow rate, as also observed in our simulations (Figure 5, top panel). The faster activation at fast pacing in experiments can be explained by the accumulation in closed states near to the open state that are readily available to open at the fast rate, as simulated by the model (Figure 4). Thus, the experiments support the concept of AR as a mechanism for IKs participation in repolarization and its dependence on rate.
Human IKs activates more slowly than guinea pig IKs and deactivates more rapidly (Figure 3C; compare with Figure 3A). The guinea pig cell provides a natural model for testing IKs in an environment in which IKr plays only a secondary role in repolarization. To evaluate the capability of human IKs to serve as a RR and to compensate for reduced IKr, we inserted a model of human IKs in the guinea pig LRd model (Figure 6). As can be seen in Figure 6F (solid line), with increased maximum conductance, human IKs can generate APD adaptation (gKs ≈4 times greater than guinea pig to obtain 200 ms APD). In contrast, when homomeric KCNQ1 channels are inserted, there is less APD adaptation at fast rates (Figure 6F, dashed line). This attenuation of APD adaptation is surprising because the deactivation kinetics of KCNQ1 are slower than human IKs, allowing for open-state accumulation (Figure 6D). However, IKs channels accumulate in zone 1 (Figure 6E, black), which is composed of closed states that only require rapid transitions to open, allowing for rapid activation that leads to large current during the repolarization phase of the AP (Figure 6C, thin line). This large IKs current during repolarization proves more important for shortening APD than the instantaneous current generated by KCNQ1. As shown earlier (Figure 4), zone 1 accumulation rather than open-state accumulation underlies the increase of IKs at fast rates. Such AR closed-state accumulation in zone 1 does not occur in KCNQ1 channels (Figure 6E, gray). Instead, a large percentage of channels activate even at slow rates (open-state occupancy at CL=10 000 ms is 30% for KCNQ1 versus 7% for IKs), preventing an increase at fast rates.
Figure 6.
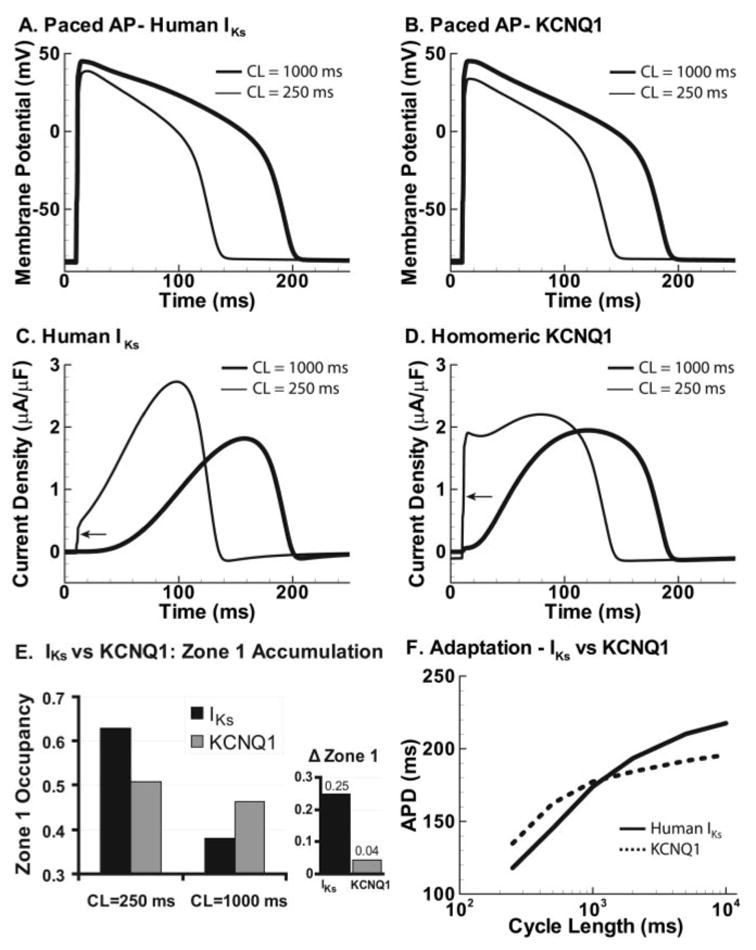
Adaptation with human IKs vs human KCNQ1. A, Fortieth AP shown at CL=250 ms (thin line) and at 1000 ms (thick line). B, Same as A with KCNQ1. C, Human IKs during the AP at fast and slow rates. Some open-state accumulation at fast rate causes a small early jump (arrow), whereas closed-state accumulation in zone 1 creates a reserve that allows the current to increase to a late peak that shortens APD (E). D, HKCNQ1 during the AP at fast and slow rates. Slow kinetics of deactivation cause large open-state accumulation at fast rates and large instantaneous current on depolarization (arrow). Note that in the absence of zone 1 reserve (E), the current stays constant during the AP, lacking the late, repolarizing peak of IKs (C). E, Human IKs (black) and KCNQ1 (gray) zone 1 occupancy at CL=250 ms and 1000 ms. Fast-rate accumulation in zone 1 allows IKs to participate in adaptation. In contrast, little accumulation is seen in zone 1 for KCNQ1. ΔZone 1 is difference in occupancy between CL=250 ms and 1000 ms. F, APD adaptation curves for an AP with human IKs (solid line) and KCNQ1 (dashed line). Lack of KCNQ1 accumulation in zone 1 results in less APD shortening at fast rates compared with IKs.
IKs and KCNQ1 During IKr Block
Figure 7A shows mean IKs (black) compared with KCNQ1 current (gray) during the first and 40th APs at CL=500 ms. IKs increases ≈50% from the first to the 40th AP, whereas KCNQ1 remains almost the same over 40 paced beats (inset shows current tracings for the first 9 beats). When IKr is blocked, greater IKs increase is observed over 40 paced beats (relative to control conditions), whereas only a limited increase is noted for KCNQ1. The difference between IKs and KCNQ1 behavior in the presence of IKr block is due to the nature of current accumulation. IKr block prolongs the AP plateau and elevates its potential, thus enhancing transitions toward the open states. KCNQ1 accumulation is mostly in the open state, which is at the expense of populating closed states that constitute the AR. Consequently, most channels open on every beat, allowing for only limited accumulation between beats. In contrast, IKs shows minimal open-state accumulation, and IKr block facilitates transitions into closed states that constitute the AR, allowing for greater IKs accumulation over many beats.
Figure 7.
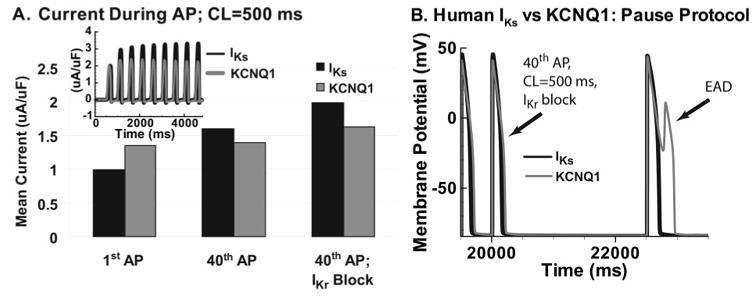
IKs participation in RR. A, IKs accumulation compared with KCNQ1 at CL=500 ms. Mean current during the first AP (left) compared with the 40th AP (middle) shows significant increase for IKs, whereas KCNQ1 shows only a small increase (time dependence over the first 9 APs is shown in inset). When IKr is blocked (right), IKs increases further (doubling compared with first control AP), and KCNQ1 increase is small. B, In presence of IKr block, pause after 40 APs at CL=500 ms is simulated with IKs or KCNQ1. Postpause AP with IKs shows normal repolarization; AP with KCNQ1 develops a pause-induced EAD.
When a pause is simulated after 40 beats in the presence of IKr block (Figure 7B), the postpause AP with KCNQ1 develops an EAD as a consequence of insufficient KCNQ1 current late during the AP. In contrast, the AP with IKs repolarizes normally because of the superior ability of IKs to activate during phase 3 of a prolonged AP.
Discussion
We show that interaction of KCNQ1 with the β-subunit KCNE1 to form IKs alters kinetics so that an AR is created at fast pacing rates. In contrast to KCNQ1, which does not generate a significant AR, IKs causes greater APD adaptation and protects against EADs when IKr is reduced.
The AR concept can be examined experimentally. For example, a gapped double-pulse protocol with variation in the gap width could be used to characterize the AR in both guinea pig and human ventricular myocytes. In this protocol, a depolarizing pulse to 40 mV for 2 seconds would activate the channel. This step would then be followed by a repolarizing step of variable duration (from 10 ms to 1 second) to −80 mV so that the channel could partially deactivate. A second depolarizing pulse to 40 mV would be used to measure the rate of activation, providing a measure of the AR.
Our simulations also predict that IKs will increase more at fast rates when IKr is blocked (Figure 7), a manifestation of AR. This prediction could be experimentally tested by blocking IKr and measuring the increase in IKs during an AP clamp at slow and fast rates. Another protocol to test interaction between IKr and IKs would be to reduce IKs and measure the change in APD from control during pacing, and then to apply IKr reduction to control and block IKs to the same degree as in the previous protocol. Our simulations predict that the same IKs reduction will have a much greater effect on APD when IKr is also reduced.
At fast rates, the ability of guinea pig IKs to accumulate between APs allows it to participate in APD shortening. However, in human myocytes IKs deactivates rapidly, bringing into question its ability to accumulate and mediate APD adaptation. Our simulations show that open-state accumulation is not necessary for IKs to increase at fast rates. Because of the complex nature of potassium channel activation, channels can accumulate in closed states near the open states (zone 1) to form an AR that opens rapidly and generates large IKs current during AP repolarization. This effect is confirmed by AP clamp experiments16 that show little accumulation in the open state but rapid activation at fast rates (Figure 5) that leads to greater IKs during the AP. In canine myocytes, IKs accumulation is also observed in the presence of β-agonist.18 Here the mechanism of IKs accumulation is identical for both human and guinea pig IKs, indicating a common mechanism for IKs increase across species.
This novel mechanism of adaptation contrasts with the behavior of homomeric KCNQ1 channels that accumulate in the open states because of slow deactivation but show no significant AR. Consequently, the KCNQ1 AP does not adapt as much as the IKs AP (Figure 6). The fundamental difference between IKs and KCNQ1 channels is the degree of participation of the first voltage sensor transition in channel activation. Because of the stabilization of the voltage sensor first position by KCNE1, only a small fraction of IKs channels activate at slow rates, creating a reserve of channels that can activate at fast rates to accelerate repolarization. In contrast, a large percentage of KCNQ1 channels activate even at slow rates, preventing buildup of such a reserve. As shown in Figure 6C, the initial jump (arrow) of IKs current that reflects open-state accumulation at fast rates is small and is followed by a steady increase to a peak of 3.4 μA/μF late during the AP. At this phase of the AP, IKs has a maximal effect on repolarization and APD. In contrast (Figure 6D), KCNQ1 current displays a large initial jump (arrow) due to large open-state accumulation but does not increase during the AP in the absence of an AR; its magnitude stays at ≈2 μA/μF and lacks the late peak that is important for repolarization. Thus, interaction with the KCNE1 subunit strongly influences the IKs profile during the AP; it augments AP shortening and is essential to normal IKs function and its participation in APD adaptation. Other differences between KCNQ1 and IKs, such as open-state behavior and flickery block, are discussed in the online-only Data Supplement.
As stated earlier in this report, IKs is not likely to participate in rate adaptation in large mammals under control conditions.7 However, when IKr is reduced, IKs is the only remaining major repolarizing current. The guinea pig myocyte provides a natural electrophysiological environment in which IKr is reduced. Under these conditions, we show that human IKs can mediate rate adaptation when the maximum conductance of the current is increased (Figure 6). β-Agonists, which are present in the normal physiological environment even at basal heart rates, can readily confer such an increase in IKs conductance.
In the case of pathologically reduced IKr (by a mutation or a drug), outward currents carried by other channels (RR) can prevent excessive APD prolongation, EADs, and triggered activity. It is hypothesized that IKr reduction in conjunction with a compromised RR is a precursor to arrhythmia, especially after a pause.9 To test the ability of IKs to participate in the RR, we compared IKs with KCNQ1 accumulation under control conditions and with IKr block. During pacing, IKs displays greater accumulation than KCNQ1 and increases further when IKr is reduced (Figure 7A). When the pause protocol is simulated with blocked IKr, increased IKs results in normal repolarization, whereas the postpause AP with KCNQ1 develops an EAD (Figure 7B). These simulations indicate that the ability of IKs to form an AR, which leads to accumulation at fast rates, makes it an ideal candidate to play a critical role in RR. The accepted RR concept involves compensation for one repolarizing current by another. This study demonstrates the existence of a reserve (AR) within a single channel (IKs), thereby extending the RR concept to include single-current reserve.
We describe IKs with 2 transitions, a slow transition (to zone 1) followed by a fast transition (to open), which implies that a 2-closed-state model (rather than 15 states) representing lumped voltage sensor transitions could adequately describe IKs activation. However, this model would not reproduce the experimentally observed delay before activation,14,19 which has important consequences during the AP. To reproduce these kinetics, a semi-Markov 2-closed-state model could be used that would introduce a memory property to the channel via time-dependent transition rates. Such models have been proposed20 but introduce another level of complexity with the addition of memory. The present model was chosen because of its correlation to the tetrameric symmetry of K+ channels and the simplicity of 4 transitions without memory that describe IKs activation. This detailed description of activation, in particular the delay before activation, is a channel feature that has not previously been incorporated into an IKs model.
Model parameters were determined with the use of nonlinear optimization. Although we incorporated a large set of experiments, there may be different parameters that also reproduce channel properties. Our conclusions depend on the participation of numerous closed states in IKs activation. The necessity of these states for generating a delay before activation has been rigorously documented.19 Thus, any model of this topology (Figure 1) that fits IKs activation, regardless of the parameter set, would generate an AR and cause more adaptation at fast rates (see online-only Data Supplement).
Not all data were obtained from native myocytes. For KCNQ1, we used frog oocyte experiments13 conducted at room temperature, in which transition rates differ from body temperature. Still, KCNQ1 activation in mammalian cells (37°C)1 and in oocytes (25°C) is quite similar, with 2 time constants of activation and similar current-voltage relationships.
Future work should incorporate β-adrenergic modulation of IKs and study its effects on whole-cell electrophysiological function. To accomplish this, a β-adrenergic model that simulates its effects on many cellular processes is necessary; these processes include sarcoplasmic reticulum calcium handling, ICa,L, the transient outward Cl− current (Ito,2), INa, INaCa, IKs, and IK1. To date there have been attempts to create such a model,21,22 but a sufficiently complete model that allows accurate study of AP dynamics in the context of β-adrenergic stimulation awaits development.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health/National Heart, Lung, and Blood Institute grants R01-HL49054 and R37-HL33343 (Dr Rudy) and F31-HL68318 (Dr Silva). Dr Rudy is the Fred Saigh Distinguished Professor of Engineering. We thank J. Cui, T. Hund, G. Faber, K. Decker, and T. O’Hara for helpful discussions and M. Sanguinetti for KCNQ1 data.
References
- 1.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 2.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 3.Antzelevitch C, Dumaine R. Electrical heterogeneity in the heart: physiological, pharmacological and clinical implications. In: Page E, Fozzard H, Solaro J, editors. Handbook of Physiology, Section 2: The Cardiovascular System, Volume I: The Heart. New York, NY: Oxford University Press; 2002. pp. 654–692. [Google Scholar]
- 4.Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 5.Sicouri S, Quist M, Antzelevitch C. Evidence for the presence of M cells in the guinea pig ventricle. J Cardiovasc Electrophysiol. 1996;7:503–11. doi: 10.1111/j.1540-8167.1996.tb00557.x. [DOI] [PubMed] [Google Scholar]
- 6.Viswanathan PC, Shaw RM, Rudy Y. Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation. 1999;99:2466–2474. doi: 10.1161/01.cir.99.18.2466. [DOI] [PubMed] [Google Scholar]
- 7.Hund TJ, Rudy Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation. 2004;110:3168–3174. doi: 10.1161/01.CIR.0000147231.69595.D3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Virag L, Iost N, Opincariu M, Szolnoky J, Szecsi J, Bogats G, Szenohradszky P, Varro A, Papp JG. The slow component of the delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc Res. 2001;49:790–797. doi: 10.1016/s0008-6363(00)00306-0. [DOI] [PubMed] [Google Scholar]
- 9.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 10.Volders PG, Stengl M, van Opstal JM, Gerlach U, Spatjens RL, Beekman JD, Sipido KR, Vos MA. Probing the contribution of IKs to canine ventricular repolarization: key role for beta-adrenergic receptor stimulation. Circulation. 2003;107:2753–2760. doi: 10.1161/01.CIR.0000068344.54010.B3. [DOI] [PubMed] [Google Scholar]
- 11.Koren G, Liman ER, Logothetis DE, Nadal-Ginard B, Hess P. Gating mechanism of a cloned potassium channel expressed in frog oocytes and mammalian cells. Neuron. 1990;4:39–51. doi: 10.1016/0896-6273(90)90442-i. [DOI] [PubMed] [Google Scholar]
- 12.Zagotta WN, Hoshi T, Aldrich RW. Shaker potassium channel gating, III: evaluation of kinetic models for activation. J Gen Physiol. 1994;103:321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tristani-Firouzi M, Sanguinetti MC. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J Physiol. 1998;510(pt 1):37–45. doi: 10.1111/j.1469-7793.1998.037bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui J, Kline RP, Pennefather P, Cohen IS. Gating of IsK expressed in Xenopus oocytes depends on the amount of mRNA injected. J Gen Physiol. 1994;104:87–105. doi: 10.1085/jgp.104.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Z, Kamiya K, Opthof T, Yasui K, Kodama I. Density and kinetics of I(Kr) and I(Ks) in guinea pig and rabbit ventricular myocytes explain different efficacy of I(Ks) blockade at high heart rate in guinea pig and rabbit: implications for arrhythmogenesis in humans. Circulation. 2001;104:951–956. doi: 10.1161/hc3401.093151. [DOI] [PubMed] [Google Scholar]
- 16.Rocchetti M, Besana A, Gurrola GB, Possani LD, Zaza A. Rate dependency of delayed rectifier currents during the guinea-pig ventricular action potential. J Physiol. 2001;534:721–732. doi: 10.1111/j.1469-7793.2001.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kupershmidt S, Yang IC, Sutherland M, Wells KS, Yang T, Yang P, Balser JR, Roden DM. Cardiac-enriched LIM domain protein fhl2 is required to generate I(Ks) in a heterologous system. Cardiovasc Res. 2002;56:93–103. doi: 10.1016/s0008-6363(02)00498-4. [DOI] [PubMed] [Google Scholar]
- 18.Stengl M, Volders PG, Thomsen MB, Spatjens RL, Sipido KR, Vos MA. Accumulation of slowly activating delayed rectifier potassium current (IKs) in canine ventricular myocytes. J Physiol. 2003;551:777–786. doi: 10.1113/jphysiol.2003.044040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zagotta WN, Hoshi T, Dittman J, Aldrich RW. Shaker potassium channel gating, II: transitions in the activation pathway. J Gen Physiol. 1994;103:279–319. doi: 10.1085/jgp.103.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clay JR. A simple model of K+ channel activation in nerve membrane. J Theor Biol. 1995;175:257–262. doi: 10.1006/jtbi.1995.0138. [DOI] [PubMed] [Google Scholar]
- 21.Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saucerman JJ, Brunton LL, Michailova AP, McCulloch AD. Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J Biol Chem. 2003;278:47997–48003. doi: 10.1074/jbc.M308362200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.