

Abstract
The majority of intracellular signalling cascades in higher eukaryotes are initiated by GPCRs (G-protein-coupled receptors). Hundreds of GPCRs signal through a handful of trimeric G-proteins, raising the issue of signal specificity. In the present paper, we illustrate a simple kinetic model of G-protein signalling. This model shows that stable production of significant amounts of free GαGTP (GTP-bound Gα subunit) and βγ is only one of multiple modes of behaviour of the G-protein system upon activation. Other modes, previously uncharacterized, are sustained production of βγ without significant levels of GαGTP and transient production of GαGTP with sustained βγ. The system can flip between different modes upon changes in conditions. This model demonstrates further that the negative feedback of receptor uncoupling or internalization, when combined with a positive feedback within the G-protein cycle, under a broad range of conditions results not in termination of the response but in relaxed oscillations in GPCR signalling. This variety of G-protein responses may serve to encode signal specificity in GPCR signal transduction.
Keywords: G-protein-coupled receptor (GPCR), kinetics, modelling, oscillation, transient response, trimeric G-protein
Abbreviations: GAP, GTPase-activating protein; GEF, guanine-nucleotide-exchange factor; GPCR, G-protein-coupled receptor; GRK, GPCR kinase
INTRODUCTION
GPCRs (G-protein-coupled receptors) constitute one of the biggest protein families in animals: more than 800 GPCR genes have been identified in the human genome, far outnumbering genes for other receptor types [1]. GPCRs recognize a vast array of extracellular signals, ranging from quanta of light to small nucleotide molecules and peptides to huge glycoproteins. They govern a multitude of cellular and organismal responses, such as receiving of sensory information, neurotransmission, immunity and development. More than half of all marketed therapeutic agents target GPCRs [2]. Our understanding of the mechanisms of GPCR functioning is crucial for biology and medicine.
GPCRs signal through activation of trimeric G-proteins. These proteins in the resting state exist as heterotrimers of α, β and γ subunits, where the α-subunit is bound to GDP. The GαGDPβγ complex can associate with GPCRs. Biochemical analysis provides a wealth of information about the G-protein cycle [3–5]. Activated GPCRs function as GEFs (guanine-nucleotide-exchange factors) for trimeric G-proteins, catalysing the exchange of GDP on Gα for GTP. This leads to dissociation of the complex into GαGTP and βγ, which can signal independently to various downstream effectors. Over time, GTP on Gα is hydrolysed back to GDP by the intrinsic GTPase activity of Gα, with the aid of GAPs (GTPase-activating proteins). GAP activity can be exerted by some effectors, such as PLCβ (phospholipase Cβ), or by specialized RGS (regulator of G-protein signalling) proteins [5]. Upon conversion into the GαGDP state, Gα can bind βγ and re-constitute the original complex, ready for a new round of signalling.
A generally accepted view of GPCR signalling is that receptor activation leads to a large increase in steady-state concentrations of free GαGTP and βγ, proportional to the ‘strength’ of GPCR activation. High concentrations of free GαGTP and βγ trigger intracellular signalling cascades, and fall back as a result of removal of the ligand from the extracellular space (signal termination), removal of the receptor from the cell surface (internalization) or covalent uncoupling of the receptor from G-proteins. This view is based on biochemical observations, such as the stable rise of GTP incorporation into and hydrolysis by G-proteins upon receptor stimulation [3], as well as stable receptor-induced dissociation of βγ from Gα in cell populations, terminated only by ligand washout [6] or receptor internalization [7]. These observations have been reproduced using kinetic simulations [7–11].
The uniformity in G-protein activation by GPCRs raises the issue of specificity in GPCR signalling. Indeed, hundreds of GPCRs have to signal through a limited set of G-proteins: only 16 genes for Gα subunits exist in humans [12] (six in Drosophila [13]). This issue is aggravated further by the promiscuity in the GPCR signalling: receptors and effectors usually do not discriminate between different βγ subunits [4]; moreover, receptors can activate G-proteins containing different Gα subunits [14], and genetic ablation of a Gα subunit often results in no or limited phenotypes, as other Gα subunits step up to fulfil the disrupted function [12].
In the present paper, we describe a novel kinetic model of trimeric G-protein signalling. Its simplicity is combined with careful examination of the reactions governing the G-protein cycle, as well as extensive parameter estimation from experimental data. This model challenges the conventional view of trimeric G-protein signalling and predicts a large variety of G-protein kinetic responses to receptor activation.
MATERIALS AND METHODS
Modelling equations
The kinetic model of the trimeric G-protein cycle (Figure 1) contains as variables concentrations of the trimeric G-protein complex ([GαGDPβγ]) and its derivatives ([βγ], [GαGTP] and [GαGDP]), and as constants concentrations of the activated GPCR ([Rc*]) and the GAP ([GAP]).
Figure 1. Trimeric G-protein cycle.

The cycle contains four dependent variables (GαGDPβγ, GαGTP, βγ and GαGDP) and two independent variables [Rc* (activated receptor) and GAP]. Three reactions (labelled ➀–➂ respectively) govern the cycle: association of βγ and GαGDP into the trimeric complex, Rc*-catalysed dissociation of the trimeric complex into βγ and GαGTP, and GAP-catalysed hydrolysis of GTP on GαGTP with formation of GαGDP. The rates of these reactions, V1, V2 and V3, are described in eqns (2)–(4) in the Materials and methods section.
Changes in molar concentrations of the components of the cycle are described through rates of production and destruction of the respective components; it becomes apparent that only three rates (designated as V1, V2 and V3) govern the behaviour of this simple cycle:
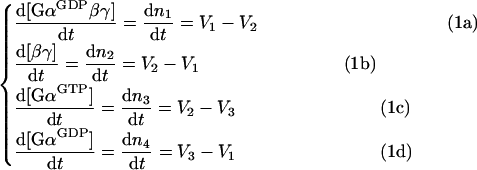 |
where V1 is the rate of re-association of the trimeric G-protein complex, V2 is the rate of dissociation of the trimeric complex, and V3 is the rate of hydrolysis of GTP on Gα. The respective reactions are depicted as ➀, ➁ and ➂ in Figure 1.
As one Gα subunit and one βγ heterodimer make one trimeric complex, and their association is not enzyme-catalysed, the rate V1 is described simply as:
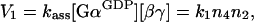 |
2 |
where kass (≡k1) is the association rate constant. In contrast, reactions described by rates V2 and V3 are enzymatic, with activated receptor (Rc*) catalysing dissociation of the trimeric complex, and GAP catalysing the GTP hydrolysis by Gα (Figure 1). Thus the Michaelis–Menten rate equations have to be used:
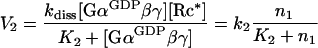 |
3 |
where kdiss is the dissociation rate constant, K2 is the Michaelis–Menten constant for the receptor-catalysed dissociation of the trimeric complex, and k2≡kdiss[Rc*]. Similarly:
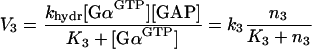 |
4 |
where khydr is the hydrolysis rate constant, K3 is the Michaelis–Menten constant for the GAP-accelerated GTP hydrolysis, and k3≡khydr[GAP].
Mass conservation requires that:
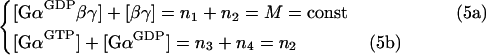 |
where M is the total (trimeric-complexed plus free) concentration of the G-protein.
Thus the four dependent differential equations (eqns 1a–1d) can be rewritten as a system of two independent differential equations:
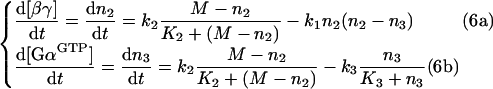 |
having [GαGTP] and [βγ] as the two variables. After solving eqns (6a) and (6b), concentrations of GαGDPβγ and GαGDP are determined using eqns (5a) and (5b).
Alternative modelling equations (resulting in the same characteristics of the trimeric G-protein system) are given in the Supplementary Materials section at http://www.BiochemJ.org/bj/401/bj4010485add.htm.
Obtaining parameters for kinetic modelling
Published data (summarized in Supplementary Tables 2–5 at http://www.BiochemJ.org/bj/401/bj4010485add.htm) were used to obtain the rate constants and the Michaelis–Menten constants, as well as to find the range of trimeric G-protein, receptor and GAP intracellular concentrations. To convert the published data on the number of molecules per cell, cell volume estimates were used as shown in Supplementary Table 1 (http://www.BiochemJ.org/bj/401/bj4010485add.htm).
The obtained values (see Supplementary Table 2) show that the intracellular concentrations of trimeric G-proteins vary in the range 200 nM–3 μM, those of GPCRs vary in the range 1–500 nM, and those of GAPs vary in the range 10–300 nM.
It should be noted that local plasma membrane concentrations of the components of the trimeric G-protein cycle may be much higher than the total intracellular concentrations shown above. However, kinetic modelling at much higher concentrations reproduces the phenomena presented in the Results section, as with increased concentrations of the components of the cycle all three reactions speed up.
The kass value for re-association of the trimeric complex from GαGDP and βγ varied in different experiments (see Supplementary Table 3): kass for the trimeric G-protein Gi using two different methods was found to be 4×104 M−1·s−1 [15] or 0.7×106 M−1·s−1 [16], with respective dissociation constant, Kd=100 or 3 nM. Later Kd measurements [17] yielded even lower value of 0.2 nM for Gi, and 17–27 nM for the trimeric G-proteins Go and Gs. It is thus likely that the kass value for Gi is fairly high (approx. 106 M−1·s−1), whereas other G-proteins such as Go and Gs have an kass value of approx. 105 M−1·s−1 or lower.
Published kdiss values for receptor-driven trimeric G-protein dissociation vary widely (see Supplementary Table 4), from approx. 1 to 300 s−1. The low values of kdiss were obtained in reconstituted systems and are likely to be underestimations, while the extremely high values obtained for rhodopsin/transducin may not be transferable to other receptor/G-protein systems. Likely in vivo kdiss values were estimated to be 20 s−1 [18] and were varied in 5–25 s−1 intervals for the present analysis.
From a general biochemical standpoint, the Michaelis–Menten constant for the receptor-driven trimeric G-protein dissociation (K2 in eqn 3) is likely to be in the range of physiological G-protein concentration [19], which is indeed the case for transducin activation by rhodopsin [20]. Thus, although direct measurements for the Michaelis–Menten constant for other G-proteins are unavailable, we set K2 at 500 nM.
GAPs were found to accelerate the GTP hydrolysis by orders of magnitude with a low Michaelis–Menten constant (K3 in eqn 4) of approx. 2 nM [21]. Specific khydr values differ depending on the Gα–GAP pair (see Supplementary Table 5) and were varied in the 1–20 s−1 interval in the present modelling.
Steady-state analysis
Numerical solutions to the system of non-linear equations (eqn 6a and 6b) are shown in the Results section. Here we present approximate solutions allowing us to estimate analytically the steady-state concentrations of the trimeric G-protein components.
The trimeric G-protein system reaches the steady-state when the derivatives in eqns (6a) and (6b) become zero and the rates V1, V2 and V3 become equal. Attempts to solve eqns (6a) and (6b) directly produce cumbersome equations of the fourth order. However, the Michaelis–Menten equations for the rates V2 and V3 eqns (3) and (4) approach constants at substrate concentrations greatly exceeding the Michaelis–Menten constant (K2 and K3 in eqns 3 and 4). This is easily the case for V3, as K3 is low (see above). So, we separately analysed two different steady-state situations: with high steady-state [GαGTP] (>K3), and low steady-state [GαGTP] (<K3). Under conditions of high [GαGTP], V3 becomes:
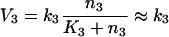 |
7 |
Eqns (6a) and (6b) for the steady-state can now be rewritten as:
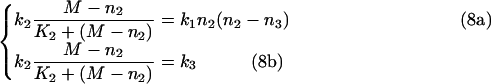 |
Solving eqns (8a) and (8b) produces:
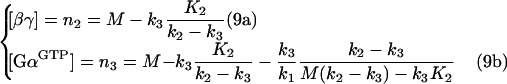 |
Eqns (9a) and (9b) have been obtained by approximating K3=0. This approximation is only valid for the set of parameters resulting in high steady-state [GαGTP]. In a different set of parameters, one or both solutions of eqns (9a) and (9b) can become ‘non-biological’, i.e. <0 or >M. It appears that if n2>0 and n3>0, the conditions n2<M and n3<M will apply. As a result, the following inequalities follow from eqns (9a) and (9b):
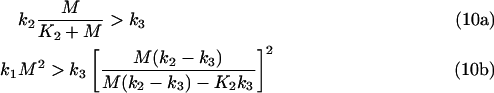 |
Inequalities (eqns (10a) and (10b)) describe relationships between the maximal values of the rates V1, V2 and V3, as:
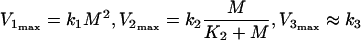 |
11 |
From the consideration that K2≈M (see above), it follows that V2max≈k2/2, and the inequalities (eqns (10a) and (10b) become:
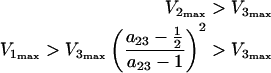 |
12 |
where a23=V2max/V3max. Inequalities (eqns 10a, 10b, 12a and 12b) describe the relationships between the Vmax values required to obtain high steady-state [GαGTP]. Violation of these inequalities results in low steady-state [GαGTP].
If the steady-state [GαGTP] is low (<K3), V3 is no longer constant, but can be rewritten in a linear form. We decided to simplify V2 into the linear form as well, as in the steady-state situation of trimeric G-protein activation, the concentration of the undissociated trimeric G-protein complex, GαGDPβγ, is low (<M≈K2). Thus V2 and V3 become:
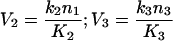 |
13 |
Rewriting eqns (6a) and (6b) and solving them for the steady-state gives the following solutions:
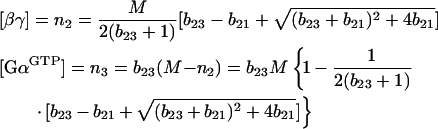 |
14 |
where bij≡Wi/Wj, such that W1=k1M2=V1max, W2=k2M/K2, and W3=k3M/K3. Here W2 and W3 are maximal rates for V2 and V3 written in the linear form (eqn 13). In eqn (14), solutions with a negative square root are discarded as being ‘non-biological’.
The steady-state analysis should be started with analysis of inequalities (eqns 12a and 12b). If they are fulfilled, the steady-state concentrations can be estimated using eqns (9a) and (9b). If not, eqn (14) should be used.
Modelling feedback reactions in the trimeric G-protein cycle
To model the positive feedback of GαGTP-enhanced GPCR activity, the V2 rate equation (eqn 3) can be rewritten in the following form, after [22]:
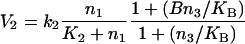 |
15 |
where B and KB are kinetic constants. In Figure 7, B is set as 100, and KB is set as 500 nM.
Figure 7. Feedback loops can produce oscillations in the trimeric G-protein cycle.

Mode 2 trimeric G-protein signalling (A) becomes transient upon incorporation of the negative-feedback loop of GαGTP-stimulated receptor internalization (B). Further addition of the positive-feedback loop of GαGTP-mediated stimulation of receptor activity induces oscillations in the trimeric G-protein cycle (D, E). The frequency and shape of oscillations depend on properties of the system: lower receptor/GAP activity ratio produces lower frequency (D) than does the higher receptor/GAP activity ratio (E). Strong off-balance activity of one enzymatic component prevents oscillations (C). Curve presentation is as in Figure 3. Kinetic parameters, other than in Figure 3, are: kass=0.01 nM−1·s−1; kdiss=20 s−1 (in A–C and E) and 18 s−1 (in D); khydr=3 s−1 (in A–C and E) and 6 s−1 (in D); [Rc*]=50 nM (in A–C and E) and 20 nM (in D); [GAP]=40 nM (in A, B, D and E) and 100 nM (in C). Time scale is 15 s in (A) and (B), and 300 s in (C–E).
To model the negative feedback of GαGTP-induced receptor internalization (removal from the cell surface), [Rc*] was set as a variable rather than as a constant:
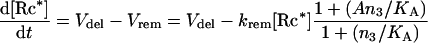 |
16 |
where Vdel is the rate of receptor delivery to the cell surface, krem is the rate constant for receptor removal, and A and KA are kinetic constants (set as 0.3 nM−1·s−1, 0.01 s−1, 180 and 400 nM respectively in Figure 7).
Computer modelling
Kinetic modelling was performed with the PLAS (Power Law Analysis and Simulation) software [19] (http://www.dqb.fc.ul.pt/docentes/aferreira/plas.html) using two numerical solvers: the BDF stiff integrator for rate-law systems and Taylor series method for power-law systems.
RESULTS
Description of the four kinetic modes in the trimeric G-protein system
The kinetic model of the trimeric G-protein cycle (Figure 1) is described in the Materials and methods section. Kinetic modelling of this simple trimeric G-protein cycle yields surprising diversity in the signalling responses (Figure 2) in the range of parameters, quantified from experimental data (see the Materials and methods section). This diversity is largely manifest as the Gα responses, represented as predominant production of GαGTP over GαGDP, or vice versa; furthermore, these responses can be transient (Figure 2). In contrast, βγ production is mostly sustained and uniform in response to receptor activation, and varies mostly in its level. We operationally define levels of components of the trimeric G-protein cycle (GαGDPβγ, GαGTP, GαGDP and βγ) as ‘high’ if they exceed K3 (the Michaelis–Menten constant for the GAP-accelerated GTP hydrolysis on GαGTP; set as 2 nM or <1% of the total G-protein concentration) and ‘low’ if they do not.
Figure 2. Four signalling modes predicted from kinetic modelling of the trimeric G-protein cycle.

Receptor activation starts at zero time. Mode 1 is sustained production of high levels of βγ and GαGDP, but not GαGTP, upon receptor activation. Mode 2 is production of high levels of βγ and GαGTP. Mode 3 is a sustained production of high levels of βγ, with GαGTP produced transiently and then falling to low levels, while mode 4 stabilizes GαGTP at high levels after the original overshoot. The kinetic parameters in all four modes are: starting [GαGDPβγ]=500 nM, and starting [βγ], [GαGDP] and [GαGTP]=1 nM; K2=500 nM; K3=2 nM. Other parameters are: kass=0.001 nM−1·s−1 (modes 1, 3 and 4), and 0.01 nM−1·s−1 (mode 2); kdiss=15 s−1 (mode 1), 20 s−1 (mode 2) and 25 s−1 (modes 3 and 4); khydr=15 s−1 (mode 1) and 5 s−1 (modes 2–4); [Rc*]=30 nM (modes 1 and 2), 100 nM (mode 3) and 60 nM (mode 4); [GAP]=25 nM (modes 1 and 4), 20 nM (mode 2) and 52 nM (mode 3).
The observed diversity in G-protein responses can then be categorized into four distinct signalling modes (Figure 2): sustainable production of high concentrations of βγ and GαGDP with low levels of GαGTP (mode 1); sustainable production of high concentrations of GαGTP and βγ (mode 2); transient production of high concentrations of GαGTP with sustained high βγ (mode 3); and sustainable production of high concentrations of GαGTP and βγ with an initial overshoot in GαGTP production (mode 4). This categorization has a biological rather than mathematical meaning: ‘high’ concentrations are implied to induce signal transduction, whereas ‘low’ are not. Furthermore, modes 3 and 4 are close to the mathematical description, but are vastly different biologically, as only in mode 4 can the stable activation of GαGTP-dependent responses be expected.
The conventional view of GPCR signalling implies that steady-state concentrations of GαGTP and βγ rise to high upon receptor stimulation. It is apparent from modelling presented here that this type of G-protein response is just one of multiple kinetic modes of G-protein activation (mode 2). The other three modes have not been previously characterized. The four modes of the trimeric G-protein cycle predicted here will probably produce a wider range of intracellular responses than just a sustainable production of GαGTP plus βγ. Specifically, GαGTP-dependent cellular responses cannot be expected in mode 1, and can only be transient in mode 3. Numerical analysis shows that the duration of the GαGTP transient in modes 3 and 4 is mostly under the negative control by the efficiency/concentration of the GAP, while the amplitude is largely determined by the efficiency/concentration of the activated receptor (Figure 3).
Figure 3. Transient GαGTP production in mode 3.

In conditions when GαGTP transient is produced, its amplitude is largely under GPCR control, while its period is largely under the negative GAP control. Only [βγ] (broken line) and [GαGTP] (continuous line) are shown. Curve presentation, starting concentration of the components, and the Michaelis–Menten constants are as in Figure 2. Kinetic parameters, other than those shown, are: kass=0.0001 nM−1·s−1; kdiss=20 s−1; khydr=5 s−1.
Steady-state analysis
The four kinetic modes can be placed in two separate groups: modes 2 and 4 showing high steady-state concentrations of both βγ and GαGTP; and modes 1 and 3 showing high steady-state concentration of βγ and low steady-state concentration of GαGTP. To assess whether the steady-state [GαGTP] is high or low, inequalities (eqns 12a and 12b) should be used to compare the maximal levels of the V1–V3 rates. If V2max > V3max and V1max>V3max, [GαGTP] will be high in the steady-state. In this situation, to calculate the steady-state concentrations of [GαGTP] and [βγ], eqns (9a) and (9b) should be used. For low steady-state [GαGTP], eqn (14) should be used instead.
Prediction of the kinetic modes
Three rates determine the behaviour of the trimeric G-protein cycle: the rate of dissociation of the trimeric complex (V2), the rate of GTP hydrolysis on GαGTP (V3), and the rate of re-association of the trimeric complex (V1). The choice between the four signalling modes is determined by the maximal values of these three rates. Numerical analysis reveals that:
mode 1 (low GαGTP production) is achieved if V3max>V2max;
mode 2 (sustained high GαGTP production) is achieved if V1max>V2max>V3max;
mode 3 (transient GαGTP production) is achieved if V2max>V3max>V1max;
and mode 4 (GαGTP production with overshoot) is achieved if V2max>V1max>V3max (17)
where Vmax values are as defined as in the Materials and methods section (eqn 11). Figure 4 illustrates the dependence of kinetic modes on the V1max, V2max and V3max parameters.
Figure 4. The kinetic mode can be determined based on comparison of the maximal levels of the three rates.

Top left: mode 3 occurs if V2max>V3max>V1max. Increase in V1max, so that V2max>V1max>V3max, leads to mode 4 (middle left). Further increase in V1max, so that V1max>V2max>V3max, leads to mode 2 (bottom left). Increase in V3max, so that V3max>V2max, leads to mode 1, regardless of whether V1max is high (bottom right) or low (top right).
Let us consider the situation when V3max>V2max. This situation is one of the two conditions implying low steady-state [GαGTP] (see inequalities in eqns 12a and 12b). As V3 approaches V3max if [GαGTP]>K3, the situation of V3max>V2max also means that, for all noticeable concentrations of GαGTP, the rate of its destruction through hydrolysis will be higher than the rate of its production through dissociation of the GαGDPβγ trimer. Thus [GαGTP] will always be low (mode 1), and [βγ] will depend on V1max. In essence, [βγ] can be considerably below M only in mode 1. All other modes require V2max>V3max, and have high [βγ] close to M.
Kinetic mode 3 describes the situation when [GαGTP] first rises to high but then falls back to low. Numerical and analytical calculations show that mode 3 occurs when the maximal rates relate as V2max>V3max>V1max. The condition V2max>V3max initially leads to fast dissociation of the GαGDPβγ trimers, forming high concentrations of free βγ and GαGTP. Later, the system adopts the steady-state in which the G-proteins will mostly exist in the form of βγ and GαGDP owing to low association rate V1max. Indeed, the condition V3max>V1max is the other situation when the steady-state [GαGTP] cannot be high (see inequalities in eqns 12a and 12b).
Similar analysis shows that mode 2 exists if V1max>V2max>V3max, and mode 4 exists if V2max>V1max>V3max.
Flips between modes
The multimodal behaviour of the trimeric G-protein cycle implies that the system can flip from one mode to another upon changes in concentrations of the components of the trimeric G-protein system. Such changes may arise from, e.g. increasing the concentration of the extracellular stimulus, reducing the plasma membrane levels of the receptor or changing the total receptor, GAP or G-protein levels. Figure 5 shows an example in which a system is predicted to flip from mode 1 (low GαGTP production) to mode 2 (sustained high GαGTP production) and back owing to changes in the levels of activated receptor and GAP. First, stimulation of a cell with low levels of a ligand yields relatively low concentrations of the activated receptor, which nevertheless elicits significant mode 1 responses (Figure 5). An additional increase in the ligand concentration stimulates the cell further, forcing the system to flip into mode 2. Cellular responses in these two modes would obviously be different. With time, a negative-feedback loop leading to enhanced production of the GAP protein can be activated; a 2.5-fold increase in the GAP concentration forces the system in Figure 5 back to mode 1, which would lead to termination of GαGTP-mediated responses. Importantly, changes in intracellular GAP concentration similar to those used in the modelling of Figure 5 have been observed experimentally upon prolonged stimulation of yeast cells with the mating hormone [10].
Figure 5. Flips between signalling modes.

The trimeric G-protein signalling system can flip from one mode to another and back upon changes in conditions. At zero time, the system is stimulated with 1 nM activated GPCR, and later with 5 nM; both stimulations result in increasing activation of the system in mode 1. Further increase in the GPCR activation (20 nM and then 90 nM) results in a switch to mode 2 of system activation. Concentration of the GAP is originally modelled at 10 nM; stepwise increases in GAP concentration lead to reduction and then loss of the GαGTP levels, switching the system back to mode 1; importantly, the system is still activated, and levels of βγ remain high. Kinetic parameters, other than those shown, are as in Figure 3, except for kass which is 0.0005 nM−1·s−1.
The steady-state flux
At the steady state, the three rates governing the G-protein system equalize. The resultant steady-state flux rate determines the velocity of turnover of the trimeric G-protein through the trimeric, active and monomeric inactive conformations. We have found that the steady-state flux rate is determined by the smallest of the Vmax values in eqn (17). For example, mode 2 occurs if V1max>V2max>V3max and the steady-state flux rate is determined by V3max, but is practically independent of V1max or V2max. Figure 6 illustrates this feature. The steady-state flux rate is approximately linearly proportional to the lowest Vmax value, while steady-state concentrations of βγ and GαGTP are affected to a considerably lower degree. For example, a 4-fold increase in GAP concentration (and thus V3max) on Figure 6 leads to a 4-fold increase in the steady-state flux rate, but only a 2-fold decrease in the steady-state [GαGTP], and an approx. 50% decrease in the steady-state [βγ]. It is possible that certain intracellular responses to the trimeric G-protein activation might be determined not only by the absolute concentrations of βγ and GαGTP, but also, or rather, by the steady-state flux rate.
Figure 6. The steady-state flux rate is under the control of the smallest of the three Vmax values.

The steady-state flux rate is achieved when the rates V1–V3 (thin lines) equalize. In mode 2, which occurs if V1max>V2max>V3max, the steady-state flux rate is independent of the concentration of the activated receptor and thus V2max (upper row), but is increased proportionally to the increase in the concentration of GAP and thus V3max (bottom row). The steady-state concentrations of βγ and GαGTP are affected only moderately by changes in [GAP], as compared with the effect on the flux rate. Parameters are as in Figure 3, except for those shown, and kass=0.01 nM·s−1.
Oscillations
It is well established that GPCR-elicited signalling over time stimulates uncoupling of the GPCR from G-proteins, either through the action of protein kinases A or C, or through the action of GRKs (GPCR kinases) and β-arrestins [24]. The GRK/β-arrestin signalling can lead further to receptor internalization. This regulation serves as a negative feedback terminating the response, with uncoupling being a faster acting, and internalization being a slower-acting negative feedback. Kinetic modelling confirms response termination by the negative feedback (Figures 7A and 7B). Similar modelling results are obtained for βγ- or GαGTP-initiated negative feedback on GPCRs, and both can operate in cells [25].
Existence of the negative-feedback loop in the trimeric G-protein cycle makes the system highly sensitive to incorporation of additional feedback regulation. For example, experiments and modelling show that coexistence of a positive- and a negative-feedback loop can result in oscillations in concentrations of system components [22,26,27]. In agreement with this, we show here that addition of a positive feedback to the trimeric G-protein cycle with signal-mediated receptor uncoupling or internalization leads not to termination of the response, but to stable oscillations in the trimeric G-protein cycle activation in a broad range of conditions (Figures 7D and 7E). The nature of the positive-feedback loop shown in Figure 7 is GαGTP-mediated activation of the GPCR-catalysed dissociation of the trimeric GαGDPβγ complex. Similar results are obtained by the βγ-mediated positive-feedback loop. Such stimulation of the GPCR by GαGTP or βγ can be either direct or indirect. The frequency and shape of the predicted oscillations in the trimeric G-protein cycle depend on the initial conditions (Figures 7D and 7E), such as the enzymatic potency of the receptor, or the nature of the GAP protein utilized.
Possible implications of the described variability in trimeric G-protein responses are elaborated in the Discussion.
DISCUSSION
The main result of the present study is a prediction of an unappreciated diversity in kinetic and steady-state responses of the trimeric G-protein system upon activation. Specifically, four distinct kinetic modes of the system behaviour are predicted: sustained production of βγ and GαGDP with only low levels of GαGTP (mode 1); sustained production of both βγ and GαGTP (mode 2); transient production of GαGTP with sustained βγ (mode 3); and sustained production of both βγ and GαGTP with an initial overshoot in GαGTP production (mode 4) (Figure 2). We performed extensive analysis of the experimentally measured parameters, such as kinetic constants and concentrations of components (see the Materials and methods section). The four kinetic modes exist in the identified range of parameters, and the choice between the four modes depends on the exact combination of parameter values. Once activated, the system can flip from one mode to another if parameters change (Figure 5). The prediction of choice of the kinetic mode by the G-protein system can be made by comparing the maximal values the three rates governing the trimeric G-protein cycle (eqn 17), these rates being the rate of formation of the trimeric G-protein complex (V1), the rate of GPCR-catalysed dissociation of the complex (V2) and the rate of GAP-accelerated hydrolysis of GTP on Gα-subunits (V3).
The typically assumed way of trimeric G-protein activation is production of high steady-state concentrations of both βγ and GαGTP in response to cell stimulation – mode 2 in our classification. Although several models for GPCR–trimeric G-protein activation have been proposed previously [7–11,28], the three other kinetic modes that we describe here have not been predicted before. Our kinetic modelling shows that mode 2 can only be achieved if among the three rates governing the G-protein cycle, V1max is the highest, and V3max is the lowest (eqn 17). Previous models strongly overestimate V1 and/or strongly underestimate V3, thus being biased towards mode 2 and omitting the other modes. Several models are built implying V1 to be not rate-limiting and the reaction of re-association of the trimeric G complex from βγ and GαGDP to be essentially instantaneous [8,10,11,28]. In other cases [7,9], the kass rate constant describing formation of the trimeric complex is set at approx. 30 nM−1·s−1, which is several orders of magnitude higher than that measured experimentally or used in our modelling (see the Materials and methods section). The kinetic modes 3 and 4, which require that V1max is not the highest of the three rates, thus could not be predicted by the previous models.
Some earlier models were built without any consideration of the role of GAP proteins, thus underestimating V3max by orders of magnitude [11,28]. In other models [7,9], the khydr constant for the Sst2p (yeast GAP)-catalysed GTP hydrolysis on Gpa1p (yeast Gα) was set at 0.11 s−1 which rather corresponds to the basic GAP-independent GTP hydrolysis rate for mammalian and also yeast Gα subunits (0.05 s−1 [3] and 0.21 s−1 [10] respectively). Such underestimations of V3max make the previous models unable to predict the kinetic mode 1, which requires that V3max>V2max (eqn 17).
The form of equations used to model the GPCR-catalysed dissociation of the trimeric complex (V2) and the GAP-catalysed hydrolysis of GTP (V3) are usually not of the Michaelis–Menten form in previous models. For example, both reactions are modelled as linear in [7,9]. The Michaelis–Menten reactions V2 and V3 can only be reduced to the linear form if the concentration of the substrate is much lower than the respective Michaelis–Menten constant. As shown in the Materials and methods section, V3 can be reduced to the linear form only for the kinetic modes 1 and 3. In contrast, in kinetic modes 2 and 4 with high steady-state [GαGTP], V3 is not linear but constant.
Thus the main difference between our kinetic model and those published previously is a more careful assignment and description of the rates governing the trimeric G-protein cycle, which allowed us observe the unexpected diversity in kinetic responses of this system.
Current experimental techniques used to monitor trimeric G-protein activation in vitro or in cell populations can hardly detect the kinetic diversity in G-protein responses predicted here. First, these techniques analyse activation of the G-protein system normalized for a population of cells. As a result, the kinetic aspects of individual cell behaviour, such as transient GαGTP production or oscillations in the G-protein cycle, become overlooked. Secondly, current methods do not measure the concentrations of the components of the trimeric G-protein cycle, but rather the steady-state flux rates of the system. For example, activation of GTP hydrolysis on trimeric G-proteins upon cell stimulation is often taken as a sign of formation of high concentrations of free GαGTP and thus activation of GαGTP-dependent responses. However, such accelerated GTP hydrolysis shows only that the G-protein system is active and constantly goes through the trimeric G-protein cycle, while the concentration of GαGTP can in fact be minuscule (as in kinetic modes 1 and 3). Other techniques, focusing on concentrations of individual components, should be developed to monitor kinetic behaviour of the trimeric G-protein system in individual cells.
As current techniques measure the steady-state flux rates, rather than concentrations of the components of the G-protein system, some unexpected experimental observations become apparent. For example, under some conditions, increase in the concentration of the GAP proteins enhances not only the rate of GTP hydrolysis in GPCR-stimulated cell membranes (V3), but also the rate of GPCR-induced incorporation of GTP into trimeric G-proteins (V2) [8]. As our modelling shows (see the Results section and Figure 6), in kinetic modes 2 and 4, the steady-state flux rate is directly proportional to V3max. Thus an increase in the concentration of GAP, increasing V3max, will increase the steady-state flux rate and, as a result, all three rates governing the cycle, including the rate of GTP incorporation (V2).
Analysis of the steady-state flux rates offers explanations for the paradoxical experimental observations that certain GPCR-triggered cellular responses are enhanced by addition of a GAP [29,30]. We propose that certain cellular responses are under the influence of the flux rate of G-protein turnover through the trimeric G-protein cycle, rather than solely on the absolute concentrations of active components of the trimeric G-protein system. A similar hypothesis was put forward for the small GTPase Rab5-controlled vesicular trafficking, where it was not the concentrations of the GTP- compared with GDP-bound forms of Rab5 that activated trafficking, but rather the speed of Rab5 cycling between the GTP- and GDP-bound forms [31]. In the case of trimeric G-proteins, when kinetic modes 2 or 4 are in place, increasing [GAP] will increase the flux rate and thus stimulation of some intracellular read-out mechanisms.
The four modes of the trimeric G-protein cycle predicted here will produce a wider range of intracellular responses than a sustainable production of GαGTP plus βγ. For example, mode 1 (production of high βγ with low GαGTP) will elicit βγ-dependent, but not GαGTP-dependent, responses, and might be used in βγ-activated signalling in yeast mating [32] or leucocyte chemotaxis [33]. Mode 1 might also be involved in responses controlled by free GαGDP (e.g. in the GPCR-mediated control of asymmetric cell divisions [34]). Modes 3 and 4 (transient production of GαGTP) are attractive for explaining transient cellular responses to stimuli. So far, negative-feedback loops have been implicated in explaining such transient responses. In the present study, we show that such transience may be a result of intrinsic properties of the trimeric G-protein cycle.
Kinetic modelling shows that the duration of the GαGTP transient in modes 3 and 4 is determined largely by the efficiency/concentration of the GAP, while the amplitude is mostly determined by the efficiency/concentration of the activated receptor (Figure 3). As different signalling complexes comprising the activated receptor, trimeric G-protein complex and the GAP protein can emerge [5,35,36], they may result in production of different GαGTP transients decoded differently by the downstream signalling components.
The four kinetic modes arise in the experimentally measured kinetic and concentration range of parameters. For example, kass values for different Gα subunits vary by one or two orders of magnitude [17], which leads to a prediction that trimeric G-proteins with higher kass (such as Gi) can be found with more likelihood in mode 2 than trimeric G-proteins with lower kass (such as Gs or Go) for which modes 3 and 4 are more likely.
GRK- and β-arrestin-dependent desensitization/internalization of GPCRs after prolonged stimulation is a well-known feedback mechanism of adaptation and signal termination [24]. Interestingly, a body of evidence shows that the GPCR desensitization pathway is also required for obtaining certain cellular responses and not just termination of cell activation. For example, leucocyte chemotaxis is defective in mice lacking GRK6 or β-arrestin 2 [37]. Chemotactic leucocytes exhibit short- and long-scale oscillatory behaviour (with periods of approx. 10 and 45 s) necessary for chemotaxis [38]. One might speculate that repetitive termination of the GPCR response, mediated by the GRK6/β-arrestin 2, directs this oscillatory chemotactic behaviour. Similarly, some GPCR-stimulated oscillations in intracellular Ca2+ are dependent on protein kinase C-mediated GPCR uncoupling [39,40], supporting the hypothesis that cyclic GPCR activation translates into Ca2+ oscillations [41].
However, kinetic modelling of the G-protein cycle with the negative feedback does not produce oscillations, but a single transient response (Figure 7B). [Under some marginal conditions, damped oscillations can be obtained (results not shown).] However, addition of a positive feedback to such modified G-protein system produces oscillations under a broad range of conditions. The nature of the positive-feedback loop in our modelling is GαGTP- or βγ-mediated activation of the GPCR-catalysed dissociation of the trimeric GαGDPβγ complex. Such stimulation of the GPCR by GαGTP or βγ can be either direct or indirect, and may include enhancement of the enzymatic activity of the receptor or increase in the affinity of ligand–receptor interaction. Potentially, the positive feedback could be achieved by substrate inhibition of the GAP-catalysed GTPase reaction [22]. Free βγ has been found to inhibit the GAP-catalysed GTPase reaction [5,42]. However, the mechanism of this inhibition appears to be competitive, with βγ increasing the apparent Michaelis–Menten constant of the reaction, without changing Vmax [43]. Incorporation of such inhibition into the kinetic modelling did not significantly change the behaviour of the system and was not sufficient to induce oscillations (results not shown).
The frequency and shape of the predicted oscillations in the trimeric G-protein cycle depend on the initial conditions (Figures 7D and 7E), such as the properties of the receptor and the GAP. They also depend on other receptor properties, such as dephosphorylation/trafficking rates. Thus a plethora of kinetic responses can be elicited within a cell by activation with different stimuli, even if the same or similar trimeric G-protein complexes are used to transduce them. The downstream signalling components are then predicted to read different oscillations of the G-protein cycle differently, which would then elicit different cellular responses.
Several experimental observations are explained by the kinetic modelling of oscillations presented here. For example, it has been shown that a large increase in GAP concentration turns GPCR-induced Ca2+ oscillations into a sustained Ca2+ increase [44]. This is in a good agreement with the cessation of trimeric G-protein oscillations predicted to result from a large increase in GAP concentration (Figure 7C).
Experimental demonstration of the positive feedback in the trimeric G-protein system is lacking so far, but is a frequent phenomenon in a variety of chemical, metabolic, genetic or signalling cascades (see, e.g., [27,45,46]). Such a positive feedback is known to exist in the endocytic traffic controlled by the small G-protein Rab5. There, membrane-bound GTP-loaded Rab5 molecules recruit a complex of the Rab5 effector Rabaptin-5 and the Rab5-specific GEF Rabex-5, thus amplifying the activation of membrane-bound Rab5 molecules [47]. We predict the existence of a similar positive-feedback loop in the trimeric G-protein cycle. Oscillation in the G-protein cycle would be able to convey a wealth of information (encoded in the form of oscillation frequency, shape and amplitude) to the downstream signalling components. Such G-protein oscillations can translate into oscillations in the activation of downstream signalling intermediates, demonstrated for cAMP [48], inositol 1,4,5-trisphosphate [40] and Ca2+ [39]. They may also be decoded by non-oscillating read-outs, perhaps similarly to the way different calcium oscillations translate differently into transcriptional responses [49].
The simple kinetic modelling shown in the present paper predicts an unappreciated diversity in G-protein responses, highlighting that behaviour of even simple systems can be far more complicated than intuitively expected. The diversity of G-protein responses comes in the format of four different kinetic modes and flips between them, as well as two different steady-state conditions (with high or low [GαGTP]). Furthermore, steady states with different flux rates can be produced. The model further serves as a framework to incorporate positive- and negative-feedback loops, predicting oscillations in the trimeric G-protein cycle. The following features (and their various combinations) of the trimeric G-protein cycle are predicted to determine specific activation of the downstream signalling: (i) the concentration of free βγ; (ii) the steady-state concentration of GαGTP; (iii) the amplitude and duration of the GαGTP transient; (iv) the steady-state flux rate; and (v) in the oscillatory mode, the frequency and the amplitude of the βγ and GαGTP peaks.
This wealth of kinetic and steady-state responses produced by the simple trimeric G-protein system may serve to encode signal specificity in GPCR signalling.
Online data
Acknowledgments
We thank Oleg Sarbey, Viktor Kalyta, Andrew Tomlinson, Mikhail Titov and Salavat Aglyamov for useful discussions. This work was supported by the Deutsche Forschungsgemeinschaft DFG (SFB-TR11) to V. L. K.
References
- 1.Fredriksson R., Lagerstrom M. C., Lundin L. G., Schioth H. B. The G-protein-coupled receptors in the human genome form five main families: phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 2.Nambi P., Aiyar N. G protein-coupled receptors in drug discovery. Assay Drug Dev. Technol. 2003;1:305–310. doi: 10.1089/15406580360545116. [DOI] [PubMed] [Google Scholar]
- 3.Gilman A. G. G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 4.Clapham D. E., Neer E. J. G protein βγ subunits. Annu. Rev. Pharmacol. Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- 5.Ross E. M., Wilkie T. M. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 6.Janetopoulos C., Jin T., Devreotes P. Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science. 2001;291:2408–2411. doi: 10.1126/science.1055835. [DOI] [PubMed] [Google Scholar]
- 7.Yi T. M., Kitano H., Simon M. I. A quantitative characterization of the yeast heterotrimeric G protein cycle. Proc. Natl. Acad. Sci. U.S.A. 2003;100:10764–10769. doi: 10.1073/pnas.1834247100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong H., Wade S. M., Woolf P. J., Linderman J. J., Traynor J. R., Neubig R. R. A spatial focusing model for G protein signals: regulator of G protein signaling (RGS) protein-mediated kinetic scaffolding. J. Biol. Chem. 2003;278:7278–7284. doi: 10.1074/jbc.M208819200. [DOI] [PubMed] [Google Scholar]
- 9.Kofahl B., Klipp E. Modelling the dynamics of the yeast pheromone pathway. Yeast. 2004;21:831–850. doi: 10.1002/yea.1122. [DOI] [PubMed] [Google Scholar]
- 10.Hao N., Yildirim N., Wang Y., Elston T. C., Dohlman H. G. Regulators of G protein signaling and transient activation of signaling: experimental and computational analysis reveals negative and positive feedback controls on G protein activity. J. Biol. Chem. 2003;278:46506–46515. doi: 10.1074/jbc.M308432200. [DOI] [PubMed] [Google Scholar]
- 11.Adams J. A., Omann G. M., Linderman J. J. A mathematical model for ligand/receptor/G-protein dynamics and actin polymerization in human neutrophils. J. Theor. Biol. 1998;193:543–560. doi: 10.1006/jtbi.1998.0721. [DOI] [PubMed] [Google Scholar]
- 12.Wettschureck N., Moers A., Offermanns S. Mouse models to study G-protein-mediated signaling. Pharmacol. Ther. 2004;101:75–89. doi: 10.1016/j.pharmthera.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Katanaev V. L., Ponzielli R., Semeriva M., Tomlinson A. Trimeric G protein-dependent frizzled signaling in Drosophila. Cell. 2005;120:111–122. doi: 10.1016/j.cell.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Hermans E. Biochemical and pharmacological control of the multiplicity of coupling at G-protein-coupled receptors. Pharmacol. Ther. 2003;99:25–44. doi: 10.1016/s0163-7258(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 15.Figler R. A., Lindorfer M. A., Graber S. G., Garrison J. C., Linden J. Reconstitution of bovine A1 adenosine receptors and G proteins in phospholipid vesicles: βγ-subunit composition influences guanine nucleotide exchange and agonist binding. Biochemistry. 1997;36:16288–16299. doi: 10.1021/bi972000q. [DOI] [PubMed] [Google Scholar]
- 16.Sarvazyan N. A., Remmers A. E., Neubig R. R. Determinants of Gi1α and βγ binding: measuring high affinity interactions in a lipid environment using flow cytometry. J. Biol. Chem. 1998;273:7934–7940. doi: 10.1074/jbc.273.14.7934. [DOI] [PubMed] [Google Scholar]
- 17.Sarvazyan N. A., Lim W. K., Neubig R. R. Fluorescence analysis of receptor–G protein interactions in cell membranes. Biochemistry. 2002;41:12858–12867. doi: 10.1021/bi026212l. [DOI] [PubMed] [Google Scholar]
- 18.Leskov I. B., Klenchin V. A., Handy J. W., Whitlock G. G., Govardovskii V. I., Bownds M. D., Lamb T. D., Pugh E. N., Jr, Arshavsky V. Y. The gain of rod phototransduction: reconciliation of biochemical and electrophysiological measurements. Neuron. 2000;27:525–537. doi: 10.1016/s0896-6273(00)00063-5. [DOI] [PubMed] [Google Scholar]
- 19.Voit E. O. A Practical Guide for Biochemists and Molecular Biologists. Cambridge: Cambridge University Press; 2000. Computational Analysis of Biochemical Systems. [Google Scholar]
- 20.Heck M., Hofmann K. P. Maximal rate and nucleotide dependence of rhodopsin-catalyzed transducin activation: initial rate analysis based on a double displacement mechanism. J. Biol. Chem. 2001;276:10000–10009. doi: 10.1074/jbc.M009475200. [DOI] [PubMed] [Google Scholar]
- 21.Wang J., Ducret A., Tu Y., Kozasa T., Aebersold R., Ross E. M. RGSZ1, a Gz-selective RGS protein in brain: structure, membrane association, regulation by Gαz phosphorylation, and relationship to a Gz GTPase-activating protein subfamily. J. Biol. Chem. 1998;273:26014–26025. doi: 10.1074/jbc.273.40.26014. [DOI] [PubMed] [Google Scholar]
- 22.Kholodenko B. N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006;7:165–176. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reference deleted.
- 24.Ferguson S. S. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol. Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 25.Kohout T. A., Lefkowitz R. J. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 2003;63:9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- 26.Zhabotinsky A. M. Concentrational Self-Oscillations. Moscow: Nauka Publishers; 1974. [Google Scholar]
- 27.Bhalla U. S., Iyengar R. Robustness of the bistable behavior of a biological signaling feedback loop. Chaos. 2001;11:221–226. doi: 10.1063/1.1350440. [DOI] [PubMed] [Google Scholar]
- 28.Shea L., Linderman J. J. Mechanistic model of G-protein signal transduction: determinants of efficacy and effect of precoupled receptors. Biochem. Pharmacol. 1997;53:519–530. doi: 10.1016/s0006-2952(96)00768-x. [DOI] [PubMed] [Google Scholar]
- 29.Doupnik C. A., Davidson N., Lester H. A., Kofuji P. RGS proteins reconstitute the rapid gating kinetics of Gβγ-activated inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. U.S.A. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saitoh O., Kubo Y., Miyatani Y., Asano T., Nakata H. RGS8 accelerates G-protein-mediated modulation of K+ currents. Nature. 1997;390:525–529. doi: 10.1038/37385. [DOI] [PubMed] [Google Scholar]
- 31.Rybin V., Ullrich O., Rubino M., Alexandrov K., Simon I., Seabra M. C., Goody R., Zerial M. GTPase activity of Rab5 acts as a timer for endocytic membrane fusion. Nature. 1996;383:266–269. doi: 10.1038/383266a0. [DOI] [PubMed] [Google Scholar]
- 32.Dohlman H. G., Thorner J. W. Regulation of G protein-initiated signal transduction in yeast: paradigms and principles. Annu. Rev. Biochem. 2001;70:703–754. doi: 10.1146/annurev.biochem.70.1.703. [DOI] [PubMed] [Google Scholar]
- 33.Katanaev V. L. Signal transduction in neutrophil chemotaxis. Biochemistry (Moscow) 2001;66:351–368. doi: 10.1023/a:1010293809553. [DOI] [PubMed] [Google Scholar]
- 34.Katanaev V. L., Tomlinson A. Dual roles for the trimeric G protein Go in asymmetric cell division in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 2006;103:6524–6529. doi: 10.1073/pnas.0601853103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeng W., Xu X., Popov S., Mukhopadhyay S., Chidiac P., Swistok J., Danho W., Yagaloff K. A., Fisher S. L., Ross E. M., et al. The N-terminal domain of RGS4 confers receptor-selective inhibition of G protein signaling. J. Biol. Chem. 1998;273:34687–34690. doi: 10.1074/jbc.273.52.34687. [DOI] [PubMed] [Google Scholar]
- 36.Benians A., Nobles M., Hosny S., Tinker A. Regulators of G-protein signaling form a quaternary complex with the agonist, receptor, and G-protein: a novel explanation for the acceleration of signaling activation kinetics. J. Biol. Chem. 2005;280:13383–13394. doi: 10.1074/jbc.M410163200. [DOI] [PubMed] [Google Scholar]
- 37.Fong A. M., Premont R. T., Richardson R. M., Yu Y. R., Lefkowitz R. J., Patel D. D. Defective lymphocyte chemotaxis in β-arrestin2- and GRK6-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 2002;99:7478–7483. doi: 10.1073/pnas.112198299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coates T. D. Behavioral aspects of neutrophil motility. Curr. Opin. Hematol. 1996;3:41–47. doi: 10.1097/00062752-199603010-00007. [DOI] [PubMed] [Google Scholar]
- 39.Kawabata S., Tsutsumi R., Kohara A., Yamaguchi T., Nakanishi S., Okada M. Control of calcium oscillations by phosphorylation of metabotropic glutamate receptors. Nature. 1996;383:89–92. doi: 10.1038/383089a0. [DOI] [PubMed] [Google Scholar]
- 40.Nash M. S., Young K. W., Challiss R. A., Nahorski S. R. Intracellular signalling: receptor-specific messenger oscillations. Nature. 2001;413:381–382. doi: 10.1038/35096643. [DOI] [PubMed] [Google Scholar]
- 41.Cobbold P., Cuthbertson R., Woods N. The generation of repetitive free calcium transients in a hormone-stimulated hepatocyte. In: Nunez J., Dumont J. E., Carafoli J. E., editors. Proceedings of the 12th Symposium on Hormones and Cell Regulation. Paris: John Libbey Eurotext; 1988. pp. 134–146. [Google Scholar]
- 42.Chidiac P., Ross E. M. Phospholipase C-β1 directly accelerates GTP hydrolysis by Gαq and acceleration is inhibited by Gβγ subunits. J. Biol. Chem. 1999;274:19639–19643. doi: 10.1074/jbc.274.28.19639. [DOI] [PubMed] [Google Scholar]
- 43.Tang W., Tu Y., Nayak S. K., Woodson J., Jehl M., Ross E. M. Gβγ inhibits Gα GTPase-activating proteins by inhibition of Gα–GTP binding during stimulation by receptor. J. Biol. Chem. 2006;281:4746–4753. doi: 10.1074/jbc.M510573200. [DOI] [PubMed] [Google Scholar]
- 44.Luo X., Popov S., Bera A. K., Wilkie T. M., Muallem S. RGS proteins provide biochemical control of agonist-evoked [Ca2+]i oscillations. Mol. Cell. 2001;7:651–660. doi: 10.1016/s1097-2765(01)00211-8. [DOI] [PubMed] [Google Scholar]
- 45.Angeli D., Ferrell J. E., Jr, Sontag E. D. Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feedback systems. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1822–1827. doi: 10.1073/pnas.0308265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stelling J., Sauer U., Szallasi Z., Doyle F. J., 3rd, Doyle J. Robustness of cellular functions. Cell. 2004;118:675–685. doi: 10.1016/j.cell.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 47.Horiuchi H., Lippe R., McBride H. M., Rubino M., Woodman P., Stenmark H., Rybin V., Wilm M., Ashman K., Mann M., Zerial M. A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell. 1997;90:1149–1159. doi: 10.1016/s0092-8674(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 48.Dyachok O., Isakov Y., Sagetorp J., Tengholm A. Oscillations of cyclic AMP in hormone-stimulated insulin-secreting β-cells. Nature. 2006;439:349–352. doi: 10.1038/nature04410. [DOI] [PubMed] [Google Scholar]
- 49.Lewis R. S. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.