

Abstract
In the genome era, there is great hope that genetic approaches such as linkage equilibrium mapping can be used to study common human disorders using a case-control population association study design. Ideally, the parental chromosomes are marked so that chromosomal regions in the form of haplotypes are compared in these studies to increase the power of association. Determining the haplotypes in a diploid individual is a major technical challenge in genetic studies of complex traits. A molecular approach to haplotyping is therefore highly desirable. Recent advances in DNA preparation, separation, labeling, and image analysis provide hope that a strategy of using a three-dye system coupled with DNA distance measurements between alleles will yield haplotype information of sufficiently high quality for genetic studies. In this work, we present the outline of the major challenges one must meet in developing a robust strategy for SNP detection and molecular haplotyping using single molecule analysis. Hum Mutat 23:442–446, 2004.
Keywords: haplotyping, SNP, mutation detection
INTRODUCTION
With the completion of the sequencing of the human genome and technological advances in molecular genetics, there is great hope that genetic approaches can be used to study common human disorders [Collins et al., 2003]. One of the approaches favored by proponents of genetic studies of common diseases is linkage disequilibrium mapping using a case-control population association study design [Botstein and Risch, 2003]. Unlike simple Mendelian disorders, however, the inheritance pattern of common disorders is complex, with several, if not many, genetic factors involved. In addition, the expression of the disease phenotype is greatly affected by environmental factors and lifestyle, leading to incomplete penetrance. The chromosomal segments shared by those suffering from the disorder in the population are predicted to be small, since the genetic factors associated with the common adult-onset disorders are presumably not under natural selection [Lohmueller et al., 2003]. In order to compare the genetic constitution of patients and controls in a comprehensive way, one needs to obtain the complete DNA sequences of the two sets of parental chromosomes of each individual in a study. Because this is not practical, genetic markers are used to track the chromosomal segments that segregate between the patients and controls. Ideally, the parental chromosomes are marked so that chromosomal regions (rather than single nucleotides) are compared in population-based genetic association studies. In other words, one analyzes haplotypes rather than individual genetic markers to increase the power of association [Fan and Knapp, 2003].
Although genotypes for thousands of single nucleotide polymorphisms (SNPs) can be obtained quite readily, determining which alleles of neighboring SNPs are found on the same chromosome (the haplotype) in an individual is still difficult. This is due to the fact that humans are diploid and current genotyping methods use genomic DNA as targets without separating the parental chromosomes. Haplotypes are therefore inferred by analyzing the genotypes of family members or predicted by computer algorithms [Long et al., 1995]. The family approach is very costly, since several additional individuals have to be typed to obtain the information for one individual in the study. The computational approach is useful but has several limitations, including uncertainties in the predicted haplotypes, especially when some of the SNPs have low heterozygosities [Fallin and Schork, 2000]. One of the remaining technical hurdles of genetic studies of complex traits is therefore a molecular approach to haplotyping.
CURRENT APPROACHES
Several published approaches of molecular haplotyping are found in the literature; as presented in the next paragraph. These approaches can be divided into two general categories. One is based on physically separating the parental chromosomal segments, followed by genotyping the isolated (and amplified) chromosomal segments. The second category is based on analyzing individual DNA molecules.
A number of methods have been used to separate the parental chromosomal segments, including cloning [Patil et al., 2001; Douglas et al., 2001], single-molecule PCR [Ruano et al., 1990; Tost et al., 2002; Mitra et al., 2003; Ding and Cantor, 2003], and allele-specific PCR [Michalatos-Beloin et al., 1996]. One extreme example of the cloning approach is to create somatic hybrid cell lines in which entire human chromosomes are separated into different cell lines [Patil et al, 2001]. A more conventional approach is to clone PCR products into plasmid or cosmid libraries in which individual clones represent DNA segments from one of the parents.
A promising cloning approach for whole genome analysis uses the polymerase colony (POLONY) strategy, in which individual DNA with ligated universal adapters is amplified within a thin acrylamide gel [Mitra et al., 2003]. At limiting dilution, the PCR products are derived from individual DNA molecules and are far removed from each other in the gel. These PCR product ‘‘colonies’’ can then serve as templates for genotyping and haplotyping.
The allele-specific PCR approach is straightforward. By designing PCR primers that end with the allelic bases of a SNP in the target region and pairing them with a reverse PCR primer far enough away to encompass other SNPs in the same region, only one parental chromosome is amplified in heterozygous individuals [Michalatos-Beloin et al., 1996]. Needless to say, this approach is limited by the exacting demands of allele-specific PCR and the fact that the approach will not work for individuals homozygous for the SNP that anchors the region.
With technological advances in microscopy, one group has shown that single DNA molecules and labels hybridized to the DNA molecule can be visualized by atomic force microscopy (AFM) [Woolley et al., 2000]. Single-molecule analysis as an approach to molecular haplotyping is therefore within our grasp.
ISSUES FOR CONSIDERATION
In molecular haplotyping, one has to determine which allele of each SNP is found on the piece of DNA being analyzed. Therefore, six critical issues must be considered: allele-specific tagging of SNPs, detection of individual labels, determination of the relationship between labels, preparation of DNA fragments, physical method for DNA separation, and image analysis for haplotypes. These issues are interrelated, with choice of one method in each area influencing and limiting the approach one can take in the other areas.
Allele-Specific Tagging of SNPs
There are many ways to distinguish between the two alleles of a SNP and specifically label the individual alleles [Syvanen, 2001]. Labeling strategies in SNP genotyping include hybridization of labeled probes, allele-specific primer extension, and allele-specific ligation of labeled probes. Not all labeling strategies will work in molecular haplotyping by single-molecule analysis, however, because an absolute requirement for labeling alleles of a SNP in single-molecule analysis is that the label has to stay on the DNA molecule at the time the label is detected. For detection methods with immobilized DNA targets, hybridization of labeled oligonucleotide probes works well [Cutler et al., 2001]. For detection systems where the DNA molecules flow by the detector, the labeled probes have to stay on the DNA molecule as they move in solution. Closed circular padlock probes are more likely to stay in place while the DAN target is in motion [Nilsson et al., 1994].
Another important consideration is the efficiency and specificity of the labeling of alleles. Because several alleles on the same DNA molecule have to be labeled at the same time in order to yield a complete haplotype for the locus, inefficient labeling will result in missing data that may lead to ambiguities in haplotype assignment. For example, if the labeling efficiency is 90% for each probe, only 59% of the DNA molecules will have all alleles labeled if there are five SNPs present in that region. When the labeling efficiency drops to 80%, only 33% of the DNA molecules are fully labeled at five sites. If enough DNA molecules are analyzed to piece together the two haplotypes of that person, incomplete labeling is not a serious issue when determining the haplotypes found in one individual.
Detection of Individual Labels
Single-molecule detection in genetics is not new. Fluorescent in situ hybridization (FISH) has been used to detect unique loci on individual chromosomes [Pinkel et al., 1986]. Large probes (several kilobase pairs in size) containing multiple fluorescent molecules are needed to visualize the hybridization event because of the lack of sensitivity in fluorescence microscopy. In haplotype analysis, the markers are usually so close together that there will not be room to place a large probe on a marker without interfering with the neighboring markers. Moreover, much smaller probes are used to increase the power of discrimination of the assay. The small probes (tens of bases in size) can only accommodate at most a handful of fluorescent dyes. As fluorescent dyes have variable intensities and absorption/emission characteristics, not all dyes can be detected above the background noise when only one dye molecule is present. With improved optics, brighter dyes, and signal analysis algorithms, it is now feasible to detect single fluorescent labels [Yildiz et al., 2003].
Determination of the Relationship Between Labels
If there were enough dyes available for labeling the DNA molecules, all one had to do was to use one dye for each allele of the SNPs making up the haplotype and look for coincidence of fluorescence to infer the haplotype. This is not feasible for haplotypes consisting of multiple markers, however, because it will take 2n dyes to cover n SNPs with two alleles each. In other words, 10 dyes are needed for a haplotype consisting of five SNPs. With the limited visible spectrum and with the high cost involved in building sophisticated detection systems, it is impractical to use more than three or four dyes.
The only way to move forward, then, is to create a haplotype barcode using spacing information and a two-dye label system for each SNP (Fig. 1). For example, one can use dye-1 for the common alleles for all the SNPs in the haplotype and dye-2 for the minor alleles. If one can determine the distance between each SNP by labeling the DNA target with dye-3, one can read out the haplotype by stringing together the labels and distances. As shown in Figure 1, some of the haplotypes are:
FIGURE 1.
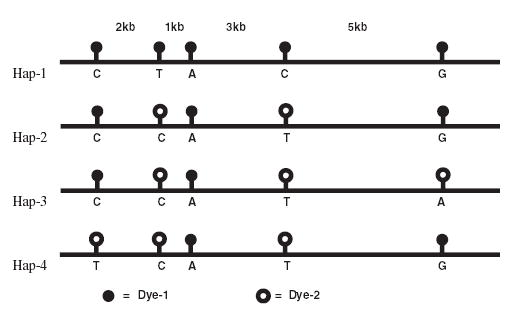
Haplotypes represented by barcodes consisting of two sets of labels with positional information.
Dye-1 : 2 kb : Dye-1 : 1 kb : Dye-1 : 3 kb : Dye-1 : 5 kb :
Dye-1 = CTACG [Hap-1]
Dye-1 : 2 kb : Dye-2 : 1 kb : Dye-1 : 3 kb : Dye-2 : 5 kb :
Dye-1 = CCATG [Hap-2]
Dye-1 : 2 kb : Dye-2 : 1 kb : Dye-1 : 3 kb : Dye-2 : 5 kb :
Dye-2 = CCATA [Hap-3]
Dye-2 : 2 kb : Dye-2 : 1 kb : Dye-1 : 3 kb : Dye-2 : 5 kb :
Dye-1 = TCATG [Hap-4]
The bar-code is uniquely suitable for haplotype determination because one can infer the haplotypes from either orientation.
The challenge is to resolve the fluorescent signals from the dyes and measure the distance between the signals. The assay format has great implications for how this can be achieved. In a flow system, DNA fragments are linearized as they are being pushed through microchannels. Fluorescent labels found on the DNA fragments can be detected by the detectors just above the microchannels. The spacing information between different SNPs can be deduced by the arrival time of the fluorescent labels as they sequentially pass by the detector. In a static system where DNA molecules are immobilized on a flat surface, tracing the molecule along the DNA target is not easy when hundreds or thousands of molecules are to be analyzed [Jing et al., 1998].
Preparation of DNA Fragments
In a perfect world, a chromosome is stretched out end-to-end and a scanner is used to run along the chromosome and read the alleles one by one. Since this is not achievable any time soon, one has to work with DNA fragments of more realistic sizes. Genomic DNA can be sheared into random fragments of a certain size range or cut with restriction enzymes into fragments of a large range of sizes. The problem is that these approaches are more or less random and only two copies of the locus of interest are found in each cell. An enormous amount of DNA will be needed to get the number of DNA fragments required to achieve statistical confidence in calling the haplotypes for a particular locus. In addition, the noise from the remaining 3 billion base pairs of DNA are likely to overwhelm the system as one tries to focus on the locus in question.
It is therefore necessary to amplify the locus one wishes to study to both increase the number of DNA molecules for the study and to reduce the background noise. Long-range PCR is the obvious choice at present and DNA in the range of 10–20 kilobase pairs (kb) can be amplified quite routinely [Barnes, 1994; Cheng et al., 1994]. Amplifying DNA fragments larger than 20 kb is more difficult to achieve and the larger DNA fragments break easily if not handled with great care [Cheng et al., 1994]. Fortunately, the mean size of haplotype blocks in the human genome is around 20 kb, so being able to prepare DNA samples in the 20 kb size range is quite adequate for most studies [Gabriel et al., 2002]. Overlapping DNA fragments can be prepared if longer range haplotypes must be determined.
Physical Method for DNA Separation
Unlike approaches where genomic DNA is separated into individual pieces and then amplified for detection, single-molecule detection approaches require the separation of amplified DNA into individual molecules for detection. Currently, two strategies are feasible. In a flow system, a dilute solution of DNA is made to sequentially stream pass the detector [Goodwin et al., 1993]. The DNA dilution is such that most DNA molecules do not travel with any other DNA molecule and the time interval between two DNA molecules as they arrive at the detector is not too long. Since many molecules must be analyzed in an experiment, it will take much too long if the DNA solution is too dilute. In the stationary system, DNA fragments are immobilized on a support surface and imaged and the pattern of labels is analyzed. The labeling can be done prior to or after DNA immobilization. Once again, DNA dilution must be such that the molecules are spread out across the support surface individually but not so far apart so that many molecules cannot be analyzed per unit area.
In both the flow and stationary systems, the DNA molecules have to be linearized so that both the order and spacing between the alleles can be determined unambiguously. For the flow system, the DNA passes through a stricture either just before or at the point at which detection occurs. In one configuration, DNA passes through a maze of posts to disentangle the DNA molecules before they enter a microchannel where the detectors are found [Huang et al., 2002]. For the stationary system, DNA in solution is drawn across a treated surface so that the DNA molecules are kept separate from each other as they are stretched out across the surface [Michalet et al., 1997; Skiadas et al., 1999]. The more consistent the stretching, the more precise the distance between markers can be measured.
Image Analysis for Haplotypes
Because of the inefficiencies in allele-specific labeling, to achieve statistical confidence in haplotype calls, many molecules must be analyzed for each individual. Each DNA molecule must be imaged and analyzed. In the flow system, one simply plots the time course of fluorescence detected for the DNA molecule and superimposes on it the points at which the allele-specific labels are seen. Thousands of such images (plots) are then analyzed to deduce the two haplotypes that are consistent with the pattern observed. In the stationary system, the surface with immobilized DNA is imaged and each DNA molecule is traced from end to end and the allele-specific labels are placed along the DNA molecule. Once again, thousands of DNA images are combined to produce the two haplotypes that are consistent with the images.
Although robust protocols for DNA labeling and stretching may take a lot of work to perfect, image analysis may turn out to be the hardest problem to solve both in terms of algorithm development and in practice during the course of the experiment. In this regard, the flow system may pose a much easier problem in data analysis than the stationary system, in which the DNA molecules have to be located, traced, and digitally linearized before analysis can take place. Efficient and robust algorithms for image analysis must be developed for single-molecule detection to be a reality in molecular haplotyping.
CONCLUSIONS
Molecular haplotyping is the only way to obtain unambiguous haplotypes for individuals in a case-control association study. It is therefore highly desirable to develop methods that can meet the demands of large-scale studies involving thousands of samples. The methods must be robust yet easy to perform. They must also be applicable to targeted regions of the genome but with universal designs.
Recent advances in DNA preparation, separation, labeling, and image analysis provide hope that a strategy of using a three-dye system (two to label the alleles of each SNP and one to label the DNA backbone), coupled with DNA distance measurements between alleles, will yield haplotype information of sufficiently high quality for genetic studies. The challenges ahead are in integrating the various aspects of this strategy in a way that an average laboratory can afford to implement it in terms of cost and experimental expertise. With physicists, biochemists, geneticists, and engineers working together, molecular haplotyping by single-molecule analysis will soon become a reality.
References
- Barnes WM. PCR amplification of up to 35 kb DNA with high fidelity and high yield from l bacteriophage templates. Proc Natl Acad Sci USA. 1994;91:2216–2220. doi: 10.1073/pnas.91.6.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet. 2003;33:228–237. doi: 10.1038/ng1090. Suppl. [DOI] [PubMed] [Google Scholar]
- Cheng S, Fockler C, Barnes WM, Higuchi R. Effective amplification of long targets from cloned inserts and human genomic DNA. Proc Natl Acad Sci USA. 1994;91:5695–5699. doi: 10.1073/pnas.91.12.5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Green ED, Guttmacher AE, Guyer MS US National Human Genome Research Institute. A vision for the future of genomics research. Nature. 2003;422:835–847. doi: 10.1038/nature01626. [DOI] [PubMed] [Google Scholar]
- Cutler DJ, Zwick ME, Carrasquillo MM, Yohn CT, Tobin KP, Kashuk C, Mathews DJ, Shah NA, Eichler EE, Warrington JA, Chakravarti A. High-throughput variation detection and genotyping using microarrays. Genome Res. 2001;11:1913–1925. doi: 10.1101/gr.197201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C, Cantor CR. Direct molecular haplotyping of long-range genomic DNA with M1-PCR. Proc Natl Acad Sci USA. 2003;100:7449–7453. doi: 10.1073/pnas.1232475100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas JA, Boehnke M, Gillanders E, Trent JM, Gruber SB. Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies. Nat Genet. 2001;28:361–364. doi: 10.1038/ng582. [DOI] [PubMed] [Google Scholar]
- Fallin D, Schork NJ. Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet. 2000;67:947–959. doi: 10.1086/303069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R, Knapp M. Genome association studies of complex diseases by case-control designs. Am J Hum Genet. 2003;72:850–868. doi: 10.1086/373966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Goodwin PM, Johnson ME, Martin JC, Ambrose WP, Marrone BL, Jett JH, Keller RA. Rapid sizing of individual fluorescently stained DNA fragments by flow cytometry. Nucleic Acids Res. 1993;21:80380–80386. doi: 10.1093/nar/21.4.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LR, Tegenfeldt JO, Kraeft JJ, Sturm JC, Austin RH, Cox EC. A DNA prism for high-speed continuous fractionation of large DNA molecules. Nat Biotechnol. 2002;20:1048–1051. doi: 10.1038/nbt733. [DOI] [PubMed] [Google Scholar]
- Jing J, Reed J, Huang J, Hu X, Clarke V, Edington J, Housman D, Anantharaman TS, Huff EJ, Mishra B, Porter B, Shenker A, Wolfson E, Hiort C, Kantor R, Aston C, Schwartz DC. Automated high resolution optical mapping using arrayed, fluid-fixed DNA molecules. Proc Natl Acad Sci USA. 1998;95:8046–8051. doi: 10.1073/pnas.95.14.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- Long JC, Williams RC, Urbanek M. An E-M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet. 1995;56:799–810. [PMC free article] [PubMed] [Google Scholar]
- Michalatos-Beloin S, Tishkoff SA, Bentley KL, Kidd KK, Ruano G. Molecular haplotyping of genetic markers 10 kb apart by allele-specific long-range PCR. Nucleic Acids Res. 1996;24:4841–4843. doi: 10.1093/nar/24.23.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalet X, Ekong R, Fougerousse F, Rousseaux S, Schurra C, Hornigold N, van Slegtenhorst M, Wolfe J, Povey S, Beckmann JS, Bensimon A. Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science. 1997;277:1518–1523. doi: 10.1126/science.277.5331.1518. [DOI] [PubMed] [Google Scholar]
- Mitra RD, Butty VL, Shendure J, Williams BR, Housman DE, Church GM. Digital genotyping and haplotyping with polymerase colonies. Proc Natl Acad Sci USA. 2003;100:5926–5931. doi: 10.1073/pnas.0936399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary BP, Landegren U. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science. 1994;265:2085–2988. doi: 10.1126/science.7522346. [DOI] [PubMed] [Google Scholar]
- Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, Kautzer CR, Lee DH, Marjoribanks C, McDonough DP, Nguyen BT, Norris MC, Sheehan JB, Shen N, Stern D, Stokowski RP, Thomas DJ, Trulson MO, Vyas KR, Frazer KA, Fodor SP, Cox DR. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science. 2001;294:1719–1723. doi: 10.1126/science.1065573. [DOI] [PubMed] [Google Scholar]
- Pinkel D, Straume T, Gray JW. Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci USA. 1986;83:2934–2938. doi: 10.1073/pnas.83.9.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano G, Kidd KK, Stephens JC. Haplotype of multiple polymorphisms resolved by enzymatic amplification of single DNA molecules. Proc Natl Acad Sci USA. 1990;87:6296–6300. doi: 10.1073/pnas.87.16.6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skiadas J, Aston C, Samad A, Anantharaman TS, Mishra B, Schwartz DC. Optical PCR: genomic analysis by long-range PCR and optical mapping. Mamm Genome. 1999;10:1005–1009. doi: 10.1007/s003359901148. [DOI] [PubMed] [Google Scholar]
- Syvanen AC. Accessing genetic variation: genotyping single nucleotide polymorphisms. Nat Rev Genet. 2001;2:930–942. doi: 10.1038/35103535. [DOI] [PubMed] [Google Scholar]
- Tost J, Brandt O, Boussicault F, Derbala D, Caloustian C, Lechner D, Gut IG. Molecular haplotyping at high throughput. Nucleic Acids Res. 2002;30:e96. doi: 10.1093/nar/gnf095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley AT, Guillemette C, Li Cheung C, Housman DE, Lieber CM. Direct haplotyping of kilobase-size DNA using carbon nanotube probes. Nat Biotechnol. 2000;18:760–763. doi: 10.1038/77760. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]