

Abstract
New mapping approaches construct ordered restriction maps from fluorescence microscope images of individual, endonuclease-digested DNA molecules. In optical mapping, molecules are elongated and fixed onto derivatized glass surfaces, preserving biochemical accessibility and fragment order after enzymatic digestion. Measurements of relative fluorescence intensity and apparent length determine the sizes of restriction fragments, enabling ordered map construction without electrophoretic analysis. The optical mapping system reported here is based on our physical characterization of an effect using fluid flows developed within tiny, evaporating droplets to elongate and fix DNA molecules onto derivatized surfaces. Such evaporation-driven molecular fixation produces well elongated molecules accessible to restriction endonucleases, and notably, DNA polymerase I. We then developed the robotic means to grid DNA spots in well defined arrays that are digested and analyzed in parallel. To effectively harness this effect for high-throughput genome mapping, we developed: (i) machine vision and automatic image acquisition techniques to work with fixed, digested molecules within gridded samples, and (ii) Bayesian inference approaches that are used to analyze machine vision data, automatically producing high-resolution restriction maps from images of individual DNA molecules. The aggregate significance of this work is the development of an integrated system for mapping small insert clones allowing biochemical data obtained from engineered ensembles of individual molecules to be automatically accumulated and analyzed for map construction. These approaches are sufficiently general for varied biochemical analyses of individual molecules using statistically meaningful population sizes.
Critical binding of molecules onto surfaces forms the basis of many newly developed techniques in genomic analysis, which include optical mapping using restriction endonucleases (1–4) and hybridization-based approaches (5–12). Surface-based approaches for DNA fixation must be compatible with their respective molecular imaging techniques. Furthermore, future high-throughput systems for genomic analysis will demand high sample deposition rates, densely gridded samples, and easy access to arrayed samples. Present-day array hybridization technology already involves gridding DNA samples densely on open-faced, charged-membrane surfaces (13, 14). Massively parallel by nature, gridded sample arrays facilitate biochemical manipulations and analyses limited only by sample density and available biochemistries. Closed systems (2, 12), in contrast, where DNA is sandwiched between glass surfaces, limit access and cannot readily accommodate arrayed samples. Desirable DNA fixation attributes for single molecule analysis by optical mapping include: a usable population of elongated molecules, preservation of biochemical activity, and parallel sample processing capabilities.
Previously, we elongated and fixed DNA molecules [2–1,500 kilobases (kb)] using the flow and adhesion forces generated when a fluid sample is compressed between two glass surfaces, one derivatized with polylysine or 3-aminopropyltriethoxysilane (APTES) (2–4). Fixed molecules were digested with restriction endonucleases, fluorescently stained with YOYO-1 (oxazole yellow dimer) (15), and optically mapped (2–4). To increase the throughput and versatility of optical mapping, multiple samples need to be arrayed on a single mapping surface. Although robotic gridding techniques for DNA samples exist (6, 13, 14), such approaches were not designed to work with single molecule substrates and could not be relied on to deposit molecules retaining significant accessibility to enzymatic action. To examine molecular effects that would ensure a usable population of elongated molecules, we have investigated several approaches to molecular deposition based on placing small droplets of DNA solution onto critically derivatized glass surfaces. We have characterized a macromolecular effect that readily elongates and fixes DNA molecules—“fluid fixation.” Fluid fixation uses the flows developed within a drying droplet through evaporative means to elongate and fix DNA molecules to charged surfaces. Conveniently, application of outside forces are completely obviated, making use of electrical fields, a traveling meniscus (9), or end-tethering of molecules with beads (10) unnecessary. The passive nature of fluid fixation provides the platform needed for our efforts to automate optical mapping. In addition, biochemical versatility of fluid-fixed molecules is demonstrated by the imaging of DNA polymerase I action on these substrates.
Given the ability to grid multiple samples and assay biochemistries on the single molecule level, we have developed an integrated system to robotically deposit samples and image substrate molecules by using automated fluorescence microscopy. This system integrates with our scheme for automatic construction of restriction maps (16) to create a system that eliminates operator interaction ranging from molecular deposition through map construction. The full utility of the system presented here is derived from the synthesis of single molecule fixation effects, biochemistries, and statistical analysis approaches and should prove universally applicable to the analysis of other single molecule systems.
MATERIALS AND METHODS
Derivatized Glass Surface Preparation.
Glass coverslips were cleaned by boiling in concentrated nitric acid (6 hr) then 6 M hydrochloric acid (12 hr), followed by a thorough rinse in high-purity water. Surfaces were derivatized according to three protocols: (i) incubating in ethanol containing 10.8 μM APTES (Aldrich) (30 μl of a 2% aqueous solution of APTES, hydrolyzed for 7 hr at room temperature in 250 ml of ethanol) at 25°C for 48 hr; (ii) incubating in a 6 mM aqueous solution of APTES (pH 3.45) at 50°C for 20 hr; and (iii) incubating in a 2.5 mM aqueous solution of [3-(triethoxysilyl-propyl]trimethylammonium chloride (TESP; Aldrich) (150 μl of a 65% aqueous solution of TESP, in 150 ml of high-purity water) at 65°C for 12–16 hr.
Fixation of Arrayed DNA Samples.
DNA molecules were elongated and aligned in square arrays by spotting droplets of DNA solution onto derivatized glass surfaces, followed by air drying, using an Eppendorf micro-manipulator in combination with an x-y table (interfaced to an Apple Macintosh computer) controlled by microstepper motors. Although this instrument is not rapid, it is very precise and reproducible. A glass capillary tube (500 μm, i.d.) was used to draw DNA samples and then spot onto derivatized glass surfaces by simple contact. Each spot was typically 900 μm with a spot-to-spot variation of ±100 μm. The center-to-center spacing between spots was 1.5 mm controlled by computer program settings of the micromanipulator, and x-y table combination. Spots were deposited at the rate of one spot every 2 s. Other grids were generated by using a modified commercially available laboratory automation robot equipped with a 500-μm ID stainless steel capillary pipetting tool, and a specialized workspace deck capable of holding multiple 96-well microtiter plates and up to 12 optical mapping surfaces in a vacuum chuck. In this configuration, the robot was able to deposit one sample approximately every 10 s. Fluid droplets (5–50 pg/μl of DNA in Tris-EDTA buffer) of 10–20 nl were spotted onto open glass surfaces that had been derivatized with APTES or [3-(triethoxysilyl-propyl]trimethylammonium chloride (TESP), using several customized robots for deposition of spots.
Restriction Endonuclease Digestion of Surface-Fixed DNA Molecules.
Surface-fixed molecules were digested by adding 40 μl of 1× restriction buffer (manufacturer recommended) containing 10–20 units of the corresponding restriction endonuclease per spotted surface. Surfaces were incubated in a humidified chamber for 15 min to 2 hr, depending on the surface condition. After digestion, the overlaying buffer was removed with an aspirator, washed with high-purity water, stained with YOYO-1 fluorochrome (100 nM in 20% β-mercaptoethanol; Molecular Probes) and sealed with Cargille immersion oil to prevent drying.
Microscopy and Imaging of Surface-Fixed DNA Molecules.
Automatic imaging workstations are built around Zeiss 135 inverted microscopes equipped for epifluorescence, with 100× Zeiss plan-neofluor oil immersion objectives, numerical aperture 1.3, and fluorescein band pass filter pack (485/505/530 nm). Microscopes also are equipped with a Dage SIT68GL low light-level video camera for acquiring focus, and a Princeton Instruments cooled charge-coupled device digital camera (1,316 × 1,032 pixels, KAF 1400 chip, 12-bit digitization) for high-resolution imaging and photometry. A Ludl Electronics x-y microscope stage with 0.1-μm resolution is used for translation.
DNA molecules were imaged by using Optical Map Maker (OMM) software written “in house,” which integrates all of the workstation functions such as movement of the microscope stage, focus, and image collection. Control of light path actuators, video autofocus, and sample translation (x-y stage) is accomplished by a Ludl Electronics MAC 2000 interface bus with the following modules installed: PSSYST 200, MCMSE 500, MDMSP 503, AFCMS 801, FWSC 800, and RS232INT 400. The Ludl MAC 2000 is interfaced via RS232 serial connection to a Sun Microsystems SPARC 20 dual processor computer workstation. The Princeton Instruments charge-coupled device camera also is interfaced, via a Pentium-based microcomputer controller and distributed network, to a Sun workstation. Software for control of the above peripherals is written in the C programming language.
Digital images can be acquired by the workstation at the rate of four per min (using 10-s imaging time) and stored on hard-disk arrays for later image processing and extraction of restriction map data. The OMM system runs on a network of 15 identical dual processor Sun SPARC 20 workstations with a networked file system.
Access to all aspects of the OMM data and processing is made through one shared directory hierarchy. This file system structure and the accompanying software libraries provide uniform controlled access to all collection and processing activities and data. A distributed processing system has been developed, which allows all of the available computational resources on the network to be shared.
Nick-Translation of Surface-Fixed DNA Molecules.
The [3-(triethoxysilyl-propyl]trimethylammonium chloride (TESP)-treated surface, spotted with lambda bacteriophage DNA molecules, was washed twice with nick-translation buffer (1× Escherichia coli DNA polymerase I buffer/50 mM dNTPs/5% glycerol/100 mg/ml of BSA). Fifty microliters of nick-translation buffer containing 10 mM R110-dUTP (fluorochrome-labeled nucleotide; Perkin–Elmer), 5 μl of 10 ng/ml DNase, and 0.1 μl (0.5 units) of DNA polymerase I (Boehringer Mannheim) was pipetted onto the surface and incubated in a sealed humidified chamber (16°C overnight). The reaction solution was aspirated off the surface, which then was incubated in excess Tris-EDTA buffer for 20 min, rinsed with high-purity water, and air dried. The surface was mounted on a microscope slide with 3 μl of 20% β-mercaptoethanol in Tris-EDTA and sealed with immersion oil. R110-dUTP labeled or counterstained samples were imaged by using the fluorescein band pass and a 580/600/630 nm filter pack. DNA was counterstained with 3.5 μl of YOYO-3 fluorochrome (100 nM in 40% β-mercaptoethanol, 1% dimethyl sulfoxide). Additional surface nick-translation results and analysis will be published elsewhere.
RESULTS
Gridding of DNA Molecules onto Derivatized Surfaces.
Spots of lambda bacteriophage DNA were gridded onto derivatized surfaces using a modified commercial robot as the spotting engine and silane-derivatized surfaces (see Materials and Methods). Images of gridded DNA spots (Fig. 1A) show that they are uniform, round, and consistently packed, containing a high percentage of fully elongated DNA molecules. Spot diameters were reproducible and were varied from 500–1,000 μm by changing the width of the spotting tool (a glass capillary or cut-off stainless steel syringe needle). Because lambda bacteriophage or cosmid clones have a typical contour length of about 17 μm, we reasoned that it may be possible to create spots having diameters only two or three times as large, or approximately 50 μm across. Using small spotting tools, such spots were made. However, we found that the most satisfactory spots, in terms of facile mapping, were made with 500- to 900-μm diameter spots, with densities of 100 clones gridded onto a single 18×18 mm derivatized glass surface.
Figure 1.

Digital fluorescence micrographs of gridded spots containing fluid-fixed molecules. Droplets of lambda bacteriophage DNA dissolved in Tris-EDTA buffer containing 0.5% glycerol deposited onto APTES-treated glass surfaces, dried and stained. (A) Section of a 10×10 spot grid on a derivatized surface. Image composed by tiling a series of 16× (objective power) images. (B) Close-up of a DNA spot within the grid. Image composed by tiling a series of 16× images. (C) Elongated DNA molecules on surface before restriction digestion (16×). (D) Magnified image of elongated DNA molecules contained within the spot shown in B before restriction digestion (100×). (E) DNA molecules in B, different field, after digestion with BamHI (100×). Note appearance of gaps signaling enzyme cleavage sites. (F) DNA molecules after digestion with AvaI, from another grid spot, using the same surface and spotting conditions (100×). [Bars: 20 μm (A–C); 5 μm (D–F).]
Fluid Fixation.
When the spotted droplets dried, a significant number of fixed DNA molecules were fully elongated, aligned radially, and concentrated near the spots’ peripheries, making a “sunburst” pattern (Fig. 1 B–D). This fluid fixation effect, unlike molecular combing (9, 12), does not require deliberate end-tethering to elongate molecules. Addition of either glycerol or other polyalcohol “dopants” to the spotting solutions consistently maximized the elongation and alignment of molecules and minimized overlapping (Fig. 1), greatly facilitating image processing and analysis. No further procedures were needed to elongate the fixed molecules and, importantly, rehydration of spotted DNA samples with restriction endonuclease buffer (low, medium, or high salt) effectively restored biochemical activity because molecules could be digested with BamHI (Fig. 1E) and AvaI (Fig. 1F).
The mechanisms underlying the effect are numerous and complex. We modeled droplet drying mathematically. Given the similarity between coffee drop drying (17) and fluid fixation of DNA, our analysis is partially derived from early discussion with Todd F. Dupont at the University of Chicago. Droplet drying occurs in two phases: first, the droplet flattens until some critical contact angle is reached; second, the contact line recedes (17–19). In phase one, net flow is radially outward, with mean velocity v, which satisfies
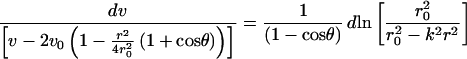 |
(R.K. and D.C.S., unpublished work) and may explain the accumulation of small molecules at the periphery of the spot. Here the contact angle θ is assumed small, k2 = (1 + cosθ) (2 − cosθ)/2 and v is flow velocity averaged over the thickness of the spot at radius r. The velocity scale v0 = l/2πr0, where, as in ref. 19, r0 is the initial radius of the spot and l is the evaporation rate per unit area.
Video Microscopy of Fluid Fixation.
Molecular fluid-fixation events were imaged by video microscopy of stained DNA molecules during droplet drying. We saw profound changes in molecular length distributions and deposition patterns correlating with variations in spot geometry. Surprisingly, we observed that molecules elongate and fix to the surface before phase two, when the receding contact line sweeps past them (Fig. 2). This suggests that high-shear fluid flows stretch molecules at least partially before they adhere to the positively charged surface. Rapid flows near the surface probably extend the molecules completely as they begin to adsorb. Thus, extreme solvent conditions near the contact line, which may alter the effective charges of the surface or of DNA, nonetheless cannot play a significant role in the fluid-fixation process (C.H., R.K., and D.C.S., unpublished work). This mechanism of elongation stands in contrast with fluid meniscus-based techniques (molecular combing) where molecules attach at one end and elongate in the fluid-air interface that sweeps past as drying occurs (12).
Figure 2.

Fluid-fixation molecular events imaged by video microscopy during droplet drying. Fluorochrome-labeled lambda bacteriophage DNA solution droplet pipetted (1 μl) onto a derivatized surface and imaged during drying. (A) Schematic detailing experimental setup: 1, droplet; 2, surface; 3, support; 4, objective. Phase one: the droplet flattens (B–E). (B) Several molecules are absorbed to the surface. A new molecule (vertical arrow) enters the field of view from the left (time = 0 s). (C) The molecule moves above the surface toward the edge of the droplet (0.10 s). (D and E) One end is adsorbed onto the surface and the molecule stretches out in the liquid flow (0.23–0.27 s). (F) The molecule elongates in the flow, sequentially attaching to the surface at several points along the backbone (0.30 s). Phase two: The contact line recedes (G–J, 2.53–3.20 s). DNA molecules are elongated and fixed before the receding liquid/air interface (horizontal arrows) sweeps by.
Evaluation of Molecular Parameters and Sizing Error.
Surface characteristics were systematically varied to balance molecular adsorption with biochemical accessibility (3, 13, 14). Excessively strong adsorption prevents molecular elongation, whereas weak adhesion does not fix a sufficient number of molecules to the surface. The distribution of molecular lengths for human adenovirus type 2 DNA molecules from 11 spots verified a high percentage of elongated molecules (Fig. 3A). No molecules appeared to be elongated longer than the full contour length of 12.3 μm even though intercalation is expected to elongate the DNA somewhat (20, 21). Longer objects all proved to be OMM-generated artifacts. The fraction of unstretched molecules varied with fixation conditions. Because optical mapping measures relative fluorescence intensity to determine restriction fragment masses, complete molecular elongation is not essential for accurate map construction. However, a narrow and reproducible distribution of elongated molecules does facilitate sizing restriction fragments by length (2). Typically, the periphery of the spot contained a higher percentage of stretched molecules than the interior (38% elongated in the outer annulus versus 30% in the core for this case). This data underrepresents the proportion of elongated molecules in the outer annulus because of difficulty in automatically scoring the densely arrayed molecules that predominate in that region.
Figure 3.

Evaluations of optical mapping molecular parameters and sizing error. (A) Histogram of lengths of spotted adenovirus type 2 DNA molecules. Lengths of 4,242 molecules from 11 spots (49 images per spot) measured by OMM were pooled and analyzed. Histogram shows the fraction (33.4%) of molecules that are sufficiently elongated for mapping (≥65% of the full contour length). The remaining fraction is primarily completely relaxed molecules or “balls,” which randomly populate the spotted areas. The average molecular length is 10 μm. (B) Sizing precision and accuracy. Restriction fragment sizing results for lambda bacteriophage DNA obtained by optical mapping plotted against sequence data. Fragment sizes range from 1,602 to 21,226 bp. Error bars represent SD of the means. Lambda DNA spotted on an APTES surface was digested with ApaLI, AvaI, BamHI, EagI, or EcoRI. Ten to 30 images were collected from one spot and analyzed by OMM.
Automation of Image Acquisition, Processing, and Map Construction.
Previous efforts constructed ordered restriction maps from images of cleaved molecules using solely manual techniques and approaches (1–4). We have developed an integrated microscope control, machine vision, and statistical analysis system, or OMM, to fully automate image collection, processing, and map construction. The computer control system advances samples for image acquisition and accumulates image files for subsequent analysis. Ordered restriction maps are derived from digital images of fully and partially digested molecules through three computational stages. First, image regions containing fragments from one molecule are identified for analysis. Second, a “backbone” of each molecule is calculated and the intensity along it used to identify enzyme cut sites and the relative mass of fragments between cut sites. OMM uses an advanced implementation of restriction fragment fluorescence intensity measurement (1) to determine the relative mass of fragments. Third, using accumulated data from all images of the same sample, a final map is computed by using Bayesian estimation (16).
To test the sizing accuracy of optical mapping, we used OMM to construct ordered restriction maps of lambda bacteriophage DNA, whose nucleotide sequence is known. Fig. 3B shows the relative fluorescence intensities of restriction fragments ranging in size from 1,602–21,226 bp, plotted against restriction fragment sizes determined from sequence. Agreement was excellent, with an average error relative to sequence-determined sizes of 217 bp. The pooled SD was 958 bp. This reflects the precision of measurements of individual molecules. Each optical map was generated from 10–40 image fields, which were collected from one digested DNA spot. These data indicate that optical size measurements are comparable in accuracy to agarose-gel electrophoresis results.
To assess the consistency of enzymatic cleavage and map construction over many gridded samples, we evaluated the distribution of BamHI cutting efficiencies over a 9×9 grid of human adenovirus type 2 DNA by tabulating the total number of scored cleavage sites per molecule. OMM accurately constructed the correct restriction map, in terms of fragment order and number, for 64 contiguous spots from the center of the 9×9 grid. Some of the spots on the periphery of the 9×9 grid failed to yield restriction maps, caused by uneven derivatization effects near the edges of the optical mapping surface. The distribution of the relative errors of the 64 restriction maps was narrow (observed fragment size vs. calculated; average 2.9%, SD 2.5%). Our map construction approach uses Bayesian inference techniques, which exhaustively evaluate models to optimally fit the map data using a set of previous assumptions such as expected number of cuts for a typical six-cutter enzyme (16). The Bayesian estimate of the precision of individual fragment sizes was 1.6 kb, and the cutting efficiency per restriction site was 73%. These cutting efficiencies are typically 30% lower than the actual number because OMM automatically discards some molecules from the analysis that otherwise would be scored manually. Molecules with scored cuts can be rejected completely (modeled as impurities) if the cuts are inconsistent with the consensus map, or individual cuts can be rejected (modeled as false) if some of the cuts are consistent and some are not. The rejection rate for molecules that already passed the morphology tests was 22% (SD 10%) and the number of false cuts per molecule was 0.32 (SD 0.12). Other runs showed similar results.
Nick-Translation Labeling of Fluid-Fixed Lambda Bacteriophage DNA Molecules.
To determine whether surface-fixed molecules might serve as substrates for other DNA modification enzymes, such as DNA ligases or polymerases, we performed a series of nick-translation reactions on surface-fixed lambda bacteriophage DNA by using E. coli DNA polymerase I and a fluorochrome-labeled nucleotide. This experiment is similar in concept to primed in situ synthesis performed on fixed intact chromosomal spreads (22). Fluorescence signals detected along molecule backbones indicated the addition of labeled nucleotides (Fig. 4 A and B). We saw labeled nucleotides added consistently over most of the DNA backbones, except for numerous small gaps. Fig. 4C shows additional molecules counterstained with YOYO-3 to confirm nick-translation results and to determine that the vast majority of gaps correspond to unlabeled regions and not double-stranded breaks.
Figure 4.

Nick-translation labeling of fluid-fixed lambda bacteriophage DNA molecules using a fluorochrome-bearing nucleotide (R110-dUTP). DNA molecules fixed onto derivatized glass surfaces before labeling by nick-translation. (A) Overview of a spot (edge) using a 16× objective. (B) The same spot portion imaged with a 100× objective. (C) Counter-staining with YOYO3 (separate experiment). The absence of heavily punctated staining patterns along molecule backbones indicates the general absence of gaps, or double-strand breaks. Staining is not robust because of fluorochrome-fluorochrome interaction. (Bars: 4 μm.)
DISCUSSION
The advent of large-scale genome sequencing necessitates the development of new approaches for the construction of physical maps. Optical mapping is an emerging application for genome analysis in which ensembles of DNA molecules are analyzed individually. Other investigators have used single molecule techniques to study DNA, manipulating molecules tethered to beads, by magnetic fields or laser trapping (10), but such approaches involve extensive manipulations and are not well suited to high-throughput analysis. Others have performed DNA biochemistries on surfaces, but used bulk deposition and analysis (5, 6, 13, 14). This report describes a highly integrated system for genomic analysis. First, we discovered a single molecule fixation effect whereby fluid flows within evaporating droplets can elongate and fix DNA molecules onto derivatized glass surfaces. Second, we discovered that such surface-mounted molecules are accessible to restriction endonucleases and DNA polymerase I. Such manipulations are amenable to high-throughput by virtue of their parallel nature, and consequently we also present results showing that many DNA spots can be applied in arrays to be manipulated biochemically and analyzed by machine vision. To complement these automation advancements, we have developed approaches for ordered restriction map construction using Bayesian inference techniques (16). These map construction algorithms work with maps automatically derived from single molecules, and are notably, practical approaches for the automated analysis of data derived from large populations of single molecules. Automated optical mapping is ideal for large-scale sequencing projects (23, 24) because optically derived maps can serve as scaffolds for aligning contigs and can provide a confident means for sequence verification.
We characterized the fluid-fixation effect, which enabled the gridding of multiple DNA spots, while also providing usable populations of well extended molecules fixed onto surfaces, competent for biochemical manipulations. Because fluid fixation relies on passive evaporative effects, large numbers of samples can be gridded onto the same surface and enzymatically treated in parallel. The mechanisms underlying fluid fixation provide insights into how fluid flows elongate populations of large coiled molecules interacting with adsorbent surfaces and furthermore provide the basis for future detailed studies of these system components. Given the results presented here, using a very simple surface geometry, it can be envisioned that microfabrication of surfaces engineered with troughs and channels will further advance the utility of this effect.
Surface-mounted DNA molecules were accessible to DNA polymerase I. We saw labeled nucleotides added consistently over most of the DNA backbones, except for numerous small gaps. These gaps did not correspond to double-stranded breaks but may be sites of particularly strong adhesion to the derivatized surface, unyielding to enzymatic action. Polymerase demands greater access to the DNA strand than do restriction endonucleases to function. Specifically, the polymerase must wrap around and travel along the DNA template, so constrictive modes of DNA adsorption would attenuate labeled nucleotide addition greatly. Surface fixation conditions would seem to be subtle enough to permit local, transient detachment of DNA, perhaps in concert with polymerase action. Both fluorescence in situ hybridization and direct visual in situ hybridization, demand less stringent strand conditions than does polymerase, and therefore could be used after optical mapping, to provide unusually well characterized and informative targets.
Our approach combines automatic analysis of restriction digestion on a surface with analysis of multiple molecules, one molecule at a time. This enables powerful statistical analyses to be performed because ensembles are created for averaging from user-designed filters to screen data derived from individual molecules. Such data, tabulated from measurements of relative fluorescence intensity of single molecules, determine the sizes of restriction fragments, enabling ordered map construction without electrophoretic analysis. The automated image acquisition and analysis presented here provides the technology required to obtain meaningful sample populations, without laborious effort. It is important to consider that manual imaging and analysis methods are often too cumbersome to permit collection of statistically meaningful sample populations. Certainly, the imaging approaches described here will be useful for the tabulation of other molecular events outside of restriction endonuclease cleavage and may be used to score hybridization results or other imagable perturbations.
Optical mapping of libraries created from entire mammalian chromosomes or genomes will require several straightforward enhancements for large-scale processing. Our recent advances in optical mapping of large insert clones including bacterial (4) and yeast artificial chromosomes (3) at high resolution point the way to optical mapping of whole genomes. Simple automation of image collection and processing currently is leading to enormous advances in throughput. We expect that refinements to OMM will soon permit us to process and create maps from 3,000–5,000 images per day per mapping workstation. Combining automatic clone analysis by optical mapping with efficient sample preparation and deposition techniques described here form the foundation of a well integrated, high-throughput restriction mapping system suitable for large-scale mapping projects. The ability to deposit tiny quantities of DNA in arrays competent for parallel biochemical manipulations and addressable by machine vision constitutes a type of “solid-state biochemistry,” in which many different samples are analyzed, in parallel, at the single molecule level. These modalities will complement genomic studies by providing platforms for sequence-function investigations.
Acknowledgments
We thank W. Cai for critical protocols and suggestions and X. Meng for helpful suggestions. We also thank E. Dimalanta and R. Rabbah for experimental assistance. Other thanks go to I. Lissanky, J.-S. Lo, and D. Geiger for early software assistance. Special thanks go to M. Waterman for early suggestions and support. Finally, special thanks go to M. Urdea, B. Warner, F. Buxton, D. Alexander, G. Kresbach, and T. F. Dupont for helpful discussions. This work was supported by grants from the National Institutes of Health (HG00225-02), the National Science Foundation, the W. M. Keck Foundation, and the Lucille P. Markey Charitable Trust.
ABBREVIATIONS
- kb
kilobase
- OMM
Optical Map Maker
- APTES
3-aminopropyltriethoxysilane
References
- 1.Schwartz D C, Li X, Hernandez L I, Ramnarain S P, Huff E J, Wang Y K. Science. 1993;262:110–114. doi: 10.1126/science.8211116. [DOI] [PubMed] [Google Scholar]
- 2.Meng X, Benson K, Chada K, Huff E J, Schwartz D C. Nat Genet. 1995;9:432–438. doi: 10.1038/ng0495-432. [DOI] [PubMed] [Google Scholar]
- 3.Cai W, Aburatani H, Stanton V P, Housman D E, Wang Y K, Schwartz D C. Proc Natl Acad Sci USA. 1995;92:5164–5168. doi: 10.1073/pnas.92.11.5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai W, Jing J, Irvin B, Ohler L, Rose E, Shizuya H, Kim U J, Simon M, Anantharaman T S, Mishra B, Schwartz D C. Proc Natl Acad Sci USA. 1998;95:3390–3395. doi: 10.1073/pnas.95.7.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schena M, Shalon D, Davis R W, Brown P O. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 6.Heller R A, Schena M, Chai A, Shalon D, Bedilion D, Gilmore J, Woolley D E, Davis R W. Proc Natl Acad Sci USA. 1997;94:2150–2155. doi: 10.1073/pnas.94.6.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erie D A, Yang G, Schultz H C, Bustamante C. Science. 1994;266:1562–1566. doi: 10.1126/science.7985026. [DOI] [PubMed] [Google Scholar]
- 8.Leuba S H, Yang G, Robert C, Samori B, van Holde K, Zlatanova J, Bustamante C. Proc Natl Acad Sci USA. 1994;91:11621–11625. doi: 10.1073/pnas.91.24.11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michalet X, Ekong R, Fougerousse F, Rousseaux S, Schurra C, Hornigold N, van Slegtenhorst M, Wolfe J, Povey S, Beckmann J S, Bensimon A. Science. 1997;277:1518–1523. doi: 10.1126/science.277.5331.1518. [DOI] [PubMed] [Google Scholar]
- 10.Strick T R, Allemand J F, Bensimon D, Bensimon A, Croquette V. Science. 1996;271:1835–1837. doi: 10.1126/science.271.5257.1835. [DOI] [PubMed] [Google Scholar]
- 11.Weier H U, Wang M, Mullikin J C, Zhu Y, Cheng J F, Greulich K M, Bensimon A, Gray J W. Hum Mol Genet. 1995;4:1903–1910. doi: 10.1093/hmg/4.10.1903. [DOI] [PubMed] [Google Scholar]
- 12.Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D. Science. 1994;265:2096–2098. doi: 10.1126/science.7522347. [DOI] [PubMed] [Google Scholar]
- 13.Craig G, Nizetic D, Hoheisel J D, Zehetner G, Lehrach H. Nucleic Acids Res. 1990;18:2653–2660. doi: 10.1093/nar/18.9.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nizetic D, Zehetner G, Monaco A P, Gellen L, Young B D, Lehrach H. Proc Natl Acad Sci USA. 1991;88:3233–3237. doi: 10.1073/pnas.88.8.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rye H S, Yue S, Wemmer D E, Quesada M A, Haugland R P, Mathies R A, Glazer A N. Nucleic Acids Res. 1992;20:2803–2812. doi: 10.1093/nar/20.11.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anantharaman T S, Mishra B, Schwartz D C. J Comp Biol. 1997;4:91–118. doi: 10.1089/cmb.1997.4.91. [DOI] [PubMed] [Google Scholar]
- 17.Deegan R D, Bakajin O, Dupont T F, Huber G, Nagel S R, Witten T A. Nature (London) 1997;389:827–829. [Google Scholar]
- 18.Chen Y L, Helm C A, Israelachvili J N. J Phys Chem. 1991;95:10736–10747. [Google Scholar]
- 19.Rowan S M, Newton M I, McHale G. J Phys Chem. 1995;99:13268–13271. [Google Scholar]
- 20.Spielmann H P, Wemmer D E, Jacobsen J P. Biochemistry. 1995;34:8542–8553. doi: 10.1021/bi00027a004. [DOI] [PubMed] [Google Scholar]
- 21.Larsson A, Carlsson C, Jonsson M, Albinsson B. J Am Chem Soc. 1994;116:8459–8465. [Google Scholar]
- 22.Koch J E, Kolvraa S, Petersen K B, Gregersen N, Bolund L. Chromosoma. 1989;98:259–265. doi: 10.1007/BF00327311. [DOI] [PubMed] [Google Scholar]
- 23.Olson M V. Science. 1995;270:394–396. doi: 10.1126/science.270.5235.394. [DOI] [PubMed] [Google Scholar]
- 24.Marshall E, Pennisi E. Science. 1996;270:188–189. doi: 10.1126/science.272.5259.188. [DOI] [PubMed] [Google Scholar]