

Abstract
We isolated a membrane-bound metallopeptidase, DINE (damage-induced neuronal endopeptidase), by differential display PCR using rat normal and axotomized hypoglossal nuclei. The most marked properties of DINE were neuron-specific expression and a striking response to axonal injury in both the central nervous system and peripheral nervous system. For instance, cranial and spinal nerve transection, ischemia, corpus callosum transection, and colchicine treatment increased DINE mRNA expression in the injured neurons, whereas kainate-induced hyperexcitation, immobilization, and osmotic stress failed to up-regulate DINE mRNA. Expression of DINE in COS cells partially inhibited C2-ceramide-induced apoptosis, probably because of the activation of antioxidant enzymes such as Cu/Zn-superoxide dismutase, Mn-superoxide dismutase, and glutathione peroxidase through the proteolytic activity of DINE. These data provide insight into the mechanism of how injured neurons protect themselves against neuronal death.
Peripheral nerve regeneration entails sequential changes in the expression of thousands of genes, which are necessary to protect damaged neurons from death, activate surrounding glial cells, and accelerate neurite elongation. For the last few years, we have attempted to identify molecules involved in this process by using a technique known as differential display PCR (DD-PCR) and random cloning with a specific cDNA library derived from nerve-injured hypoglossal nuclei (1, 2).
Among the molecules we have identified as being markedly up-regulated in response to nerve injury (3, 4), growth factors, cytokines, and neuropeptides are well established as survival factors for injured neurons (5). These molecules might participate in the protective process as intercellular signaling molecules via secretion in an autocrine or paracrine manner. Generally, secreted proteins such as neuropeptides and growth factors are biosynthesized as large precursor proteins, and processing occurs in the trans-Golgi network by endoproteolytic serine proteases, which are members of the proprotein convertase (PC) family (6). An increasing number of other secreted proteins now are recognized as being derived from integral plasma membrane proteins by hydrolysis (shedding) on the cell surface (7). Proteins secreted in this fashion include some membrane receptors, receptor ligands, ectoenzymes, and cell adhesion molecules. These ectodomain shedding events have been shown to be associated with metalloprotease inhibitors (8). Since identification of the ADAM (a disintegrin and metalloproteinase) family (9) and MMP (matrix metalloprotease) family, our understanding of the shedding events on the cell surface has greatly improved in recent years.
As for nerve regeneration, the repertoire of proteases involved in the process is limited. Among regeneration processes, the roles of proteases in a process of axon elongation are relatively well studied both in vitro and in vivo. It has been assumed that this axonal behavior is, for instance, a consequence of the balance between (i) thrombin (serine protease) and its inhibitor nexin-I (10), (ii) Ca2+-dependent calpain and its inhibitor, calpastatin (11), and (iii) extracellular matrix degradation enzymes, matrix metalloproteases, and their inhibitors, TIMPs, tissue inhibitor of metalloproteinases (12, 13). These studies imply that nerve regeneration requires a wide range of active proteases, and the existence of additional proteases is very likely.
In the present study, we identified one clone, which encodes a membrane-bound metallopeptidase, whose expression is markedly increased in response to nerve injury. We termed this molecule DINE (damage-induced neuronal endopeptidase) because of its characteristics (which will be described below). DINE shares homology with endothelin converting enzyme (ECE) (14), which catalyzes the conversion of big-endothelin to biologically active endothelin. However, DINE does not have any activity like ECE. Instead, we here demonstrate the characteristics of DINE such as a higher association with neuronal damage in the central nervous system (CNS) and peripheral nervous system, and activity in promoting the expression and activation of antioxidant enzymes.
Methods
Animal Operation.
As described previously, hypoglossal nerve transection (1), facial nerve transection (15), sciatic nerve transection (16), optic nerve crush (17), intrahippocampal kainic acids injection (18), colchicine injection, osmotic stress, and immmobilization stress (19), middle cerebral artery (MCA) occlusion (20), and cortical and corpus callosum injury (21) were performed. All of the experiments using male Wistar rats weighing about 150 g were carried out in accordance with the guidelines specified in the National Institutes of Health guide for the care and use of laboratory animals.
DD-PCR.
DD-PCR was carried out by using hypoglossal nuclei of the control and operated side obtained from 70 Wistar male rats as described (1). Subsequently, each pool of cDNA was amplified by PCR in the presence of [35S] dATP (New England Nuclear-DuPont) by using a single primer, 5′-ACTGGCCGAGGG-3′, which was randomly selected from among 87 arbitrary primers.
Histology.
For in situ hybridization, animals were decapitated, and the brain was removed quickly and frozen in powdered dry ice. Then, 20-μm-thick sections were cut on a cryostat, thaw-mounted onto 3-aminopropyltriethoxysilane-coated slides, and stored at −80°C until use. In situ hybridization was performed as described (1).
Relative Quantification of mRNA.
The relative area occupied by autoradiographic grains in the hypoglossal nuclei was measured bilaterally on the x-ray film by using a computerized image analysis system (MCID, Imaging Research, Ontario, Canada). In the same sections, we calculated the difference in optical density between the right (ipsilateral side) and left hypoglossal nuclei (contralateral side). For statistical analysis, at least eight sections from three rats each were studied.
Molecular Cloning of DINE.
5′ rapid amplification of cDNA ends (RACE).
Four micrograms total RNA from the operated rat hypoglossal nucleus was reverse-transcribed to cDNA by using DD-PCR fragment-specific primer. An anchor oligomer (5′-PO4-GTAGGAATTCGGGTTGTAGGGAGGTCGACATTGCC-NH2-3′) was added to the 3′ end by T4 RNA ligase. The RNA then was digested with 4 units of RNaseH (Toyobo, Osaka) at 37°C for 30 min. The reverse transcription reaction product was purified by phenol/chloroform extraction. The first-round PCR was performed with DD-PCR fragment-specific 3′ primer and anchor B primer (5′-GGCAATGTCGACCTCCCTACAAC-3′) using LA Taq polymerase (Takara, Kyto, Japan). The PCR conditions were as follows: 30 cycles with denaturation at 98°C for 20 sec, annealing and extension at 68°C for 4 min. A nested PCR was performed by using another specific primer and primer C (5′-CTCCCTACAACCCGAATTCCTAC-3′). The 5′ RACE product was subcloned with pGEM-T vector (Promega) and sequenced by an Applied Biosystems model 373A DNA sequencer. Both strands of cDNA were covered at least twice. Further 5′ RACE reactions were performed based on the obtained sequence of 5′ RACE product until full-length cDNA encoding DINE was isolated.
3′ RACE.
The first-strand cDNA was synthesized with Superscript reverse transcriptase (GIBCO/BRL) using 5′-GAGTCGACTCGAGAATTC-(T)7-3′ oligo primer. The initial 3′ RACE reaction was performed with DD-PCR-specific 5′ primer and 5′-GAGTCGACTCGAGAATTC-3′ primer. A one-side nested PCR was performed by using another specific primer and 5′-GAGTCGACTCGAGAATTC-3′ primer.
Plasmid Construction.
For Northern blot and protection assay, partial cDNAs of DINE [corresponding to amino acids 396–477 (probe a) and DD-PCR fragment (probe b)] were subcloned in pBluescript KS. Mn-superoxide dismutase (SOD), Cu/Zn-SOD, catalase, and glutathione peroxidase (GPX) were amplified by PCR using cDNA derived from COS-7 cells and inserted into pGEM-T Easy vector (Promega). For expression experiments, cDNAs for full-length DINE and DINE lacking the protease domain (amino acids 612–720) were subcloned into pME18Sf expression vectors (donated by T. Masaki, Kyoto University).
Northern Hybridization.
Five micrograms of poly(A)+ RNA or 20 μg total RNA derived from hypoglossal nuclei was separated by formaldehyde/0.8% agarose gel electrophoresis and transferred to a Hybond N+ membrane (Amersham Pharmacia). Two 32P-labeled RNA probes were prepared by in vitro transcription of DINE cDNA by using T7 polymerase and [32P] UTP (New England Nuclear-DuPont). Hybridization was performed at 55°C in hybridization buffer [5 × 105 cpm/ml labeled probe/50% formamide, 10% dextransulfate/1.5× standard saline phosphate/EDTA (0.18 M NaCl/10 mM phosphate, pH 7.4/1 mM EDTA)/1% SDS/0.5% Blotto/0.2 mg/ml yeast RNA/0.5 mg/ml salmon sperm DNA]. The membranes were washed in 2× SSC/0.1% SDS, 0.5× SSC/0.1% SDS, and then 0.1× SSC/0.1% SDS, rinsed with PBS, incubated in RNase buffer containing RNaseA (10 μg/ml), rinsed with PBS, and autoradiographed for 1 day at −80°C.
RNase Protection Assay.
RNase protection assays were performed with a RPAII Ribonuclease Protection Assay Kit (Ambion). To generate cRNA probes, plasmids were linearized and transcribed in vitro with T7 RNA polymerase and [32P] UTP. Then, 10 μg total RNA was hybridized with labeled probe (2.5 × 105 cpm/reaction) at 45°C overnight. The reaction products were digested with RNaseA/T1, electrophoresed on 4% sequencing gel, and visualized by autoradiography.
Cell Culture.
COS-7 cells were grown in DMEM containing 10% FBS, penicillin, and streptomycin. Cells were transfected with Lipofectamine reagents according to the manufacturer's protocol (GIBCO/BRL).
Polyclonal Antibody.
Antibody against DINE was produced by immunizing rabbits with synthetic peptide, MTAHYDEFQEVKYESRC, corresponding to amino acids 7–13 of rat DINE. Rabbits were immunized with keyhole limpet hemocyanin-coupled peptide in complete adjuvant four times at 2-week intervals, and then the antiserum was affinity-purified (Tana Laboratories, Houston, Texas).
Western Blotting.
Cells were collected, washed with PBS, and homogenized in 20 mM Tris⋅HCl (pH 7.5), 5 mM MgCl2, 0.1 mM PMSF, 20 μM pepstatin, and 20 μM leupeptin. The homogenates were centrifuged at 15,000 rpm for 45 min. The membrane fractions obtained were solubilized with the homogenizing buffer containing 0.5% (wt/vol) Triton X-100. The samples were stirred for 30 min and then centrifuged at 15,000 rpm for 60 min. The supernatant was used for further analysis. Immunoblot analysis was performed by using DINE antibody and horseradish peroxidase-conjugated anti-rabbit IgG antibodies, and the procedure was followed as recommended by the manufacturer (Amersham Pharmacia).
Peptidase Assay.
Recombinant DINE was derived from a baculovirus expression system (GIBCO/BRL). DINE cDNA lacking the cytoplasmic region and transmembrane domain was subcloned into pFASTBAC HTb expression vector. Purified proteins were incubated with fluorogenic substrate, Z-Gly-Gly-Leu-pNA (Peptide Institute, Osaka) for 30 min, and released pNA was measured with a 405-nm absorption filter. For the inhibition studies, EDTA (10 μM), phophoramidon (100 μM), 1,10-phenanthroline (100 μM), and thiorphan (50 μM) were preincubated for 15 min, and then substrate was added to the reaction mixture.
Cell Viability.
Transfected cells were maintained in serum-free DMEM for 24 h before treatment with C2-ceramide (Calbiochem). C2-ceramide was diluted with serum-free DMEM at 50 μM. Viability of cells was determined by trypan blue dye exclusion. In brief, a cell suspension was mixed with trypan blue and examined by low-power microscopy. For nuclear staining, cells were fixed with 1% glutaraldehyde and stained with Hoecst 33258 dye (Sigma).
Measurement of SOD Activity.
Total SOD activity was determined by monitoring spectrophotometrically at 560 nm the rate of reduction of nitroblue tetrazolium by O2-, using a hypoxanthine-xanthine oxidase system as the source of O2- to reduce nitroblue tetrazolium (22).
Results
Isolation of DINE.
Seven days after rat hemi-hypoglossal nerve transection, the hypoglossal nuclei of the injured side and contralateral side (as control) were collected and processed for DD-PCR (Fig. 1A). Histological survey using DD-PCR fragment as a probe (corresponding to probe b in Fig. 1D) also demonstrated a significant increase in mRNA expression in the injured hypoglossal motoneurons (Fig. 1B). An increase in the hybridization signal was first observed in the ipsilateral hypoglossal nucleus 6 h after nerve injury. The intensity of the hybridization signal markedly increased to a peak at 3 days, and thereafter gradually decreased to the control level over the next 5 weeks (Fig. 1C).
Figure 1.

Isolation of DINE by DD-PCR and its structure. (A) DD-PCR demonstrates 35S-labeled PCR products in hypoglossal nuclei of normal (cont) and operated (ope) sides (7 days after injury). The arrow indicates the differentially expressed band, which we detected in the present study. (B) Expression of the isolated cDNA fragment was further examined by in situ display. A section was obtained from a unilateral hypoglossal nerve transected-animal (7 days after surgery). The arrow indicates that the expression of the candidate gene is observed only in the hypoglossal nucleus of the injured side. (Bar = 3 mm.) (C) mRNA expression profile after hypoglossal nerve transection. The relative mRNA signal intensity in control and operated sides was measured and presented as mean +/− SD; *, P < 0.01 (ANOVA test). (D) Isolated cDNA structure encoding DINE. Closed box, predicted transmembrane domain; hatched box, zinc-binding motif; dotted box, conserved amino acids; circle, conserved cystein residues. Probe b corresponds to the initially derived DD-PCR fragment. Northern blotting was carried out by using probes a and b. In probe a, 20 μg total RNA extracted from control (cont) and axotomized (ope) hypoglossal nuclei was loaded and hybridized with probe a. In probe b, 5 μg poly(A)+ RNA from injured nuclei was loaded and hybridized with probe b.
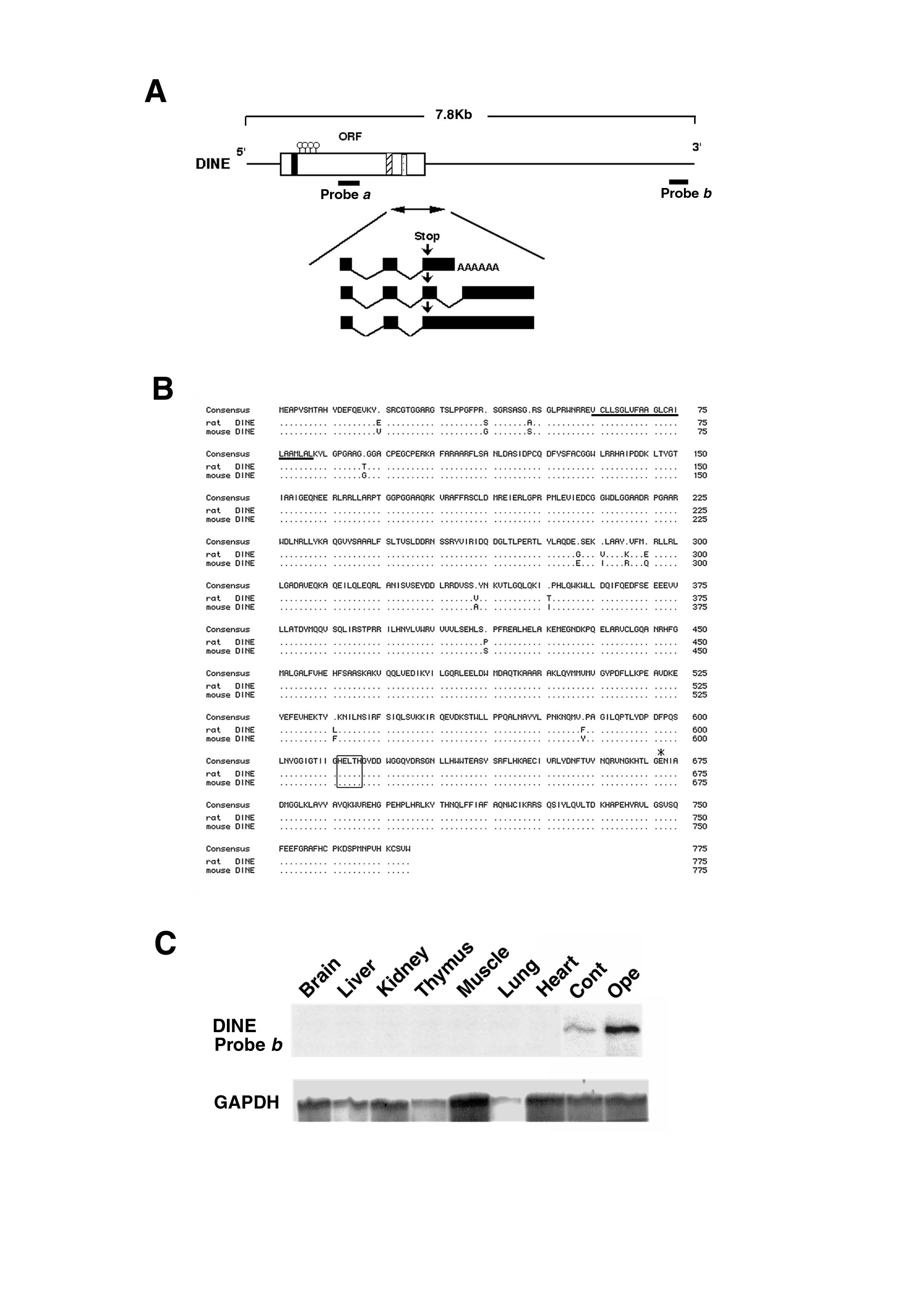
To isolate the full length of cDNA encoding DINE, we first prepared a 3′ and 5′ RACE library using rat axotomized hypoglossal nuclei and designed specific primers to the DD-PCR fragment, because the expression was restricted only to injured hypoglossal nuclei (Fig. 1B and Fig. 5C, which is published as supplementary material on the PNAS web site, www.pnas.org). The 7.8-kb full-length nucleotide sequence had an ORF that was preceded by an in-frame stop codon and a long 3′ noncoding region containing AU- and AC-rich sequences. The initial DD-PCR fragment was detected in the 3′ noncoding region (corresponding to probe b in Fig. 1D). Northern blot analysis detected a weak single 7.8-kb transcript with a cRNA probe generated from the DINE DD-PCR fragment, probe b (Fig. 1D). However, additional Northern blots hybridized with a probe corresponding to the ORF (probe a in Fig. 1D) demonstrated an additional 2.9-kb transcript that was much more abundant than the 7.8-kb band. Analysis of the genomic DNA sequence around the 3′ end region of the ORF suggested that these transcripts are caused by alternative splicing of the 3′ noncoding region and/or differential polyadenylation (see Fig. 5A).
The rat and mouse full-length cDNA sequence encoded 775 aa, which predict a type II integral membrane protein and highly conserved consensus sequence of a zinc-binding motif, HEXXH, that is shared by many known metalloproteases (see Fig. 5B). Protein homology revealed 36% sequence identity with bovine ECE-1 and 32% identity with neutral endopeptidase (NEP). Thus, we concluded that this clone is a novel membrane-bound metalloprotease and termed it DINE because of its functional characteristics (see below). Valdenaire et al. (23) recently have reported a CNS-specific XCE, which may be a human homologue to DINE, although no functional data were presented.
DINE Specifically Expressed in Neuronal Tissue.
RNase protection assay using probe a in Fig. 1D confirmed the existence of DINE mRNA in brain (Fig. 2A), although such a signal was not detected by using probe b (Fig. 5C). This discrepancy may be caused by the quite low amount of 7.8-kb transcript. To examine the detailed distribution of DINE mRNA in the brain, we carried out in situ hybridization using probe a. As shown in Fig. 2B, a strong hybridization signal was observed in neurons in the caudate putamen, diagonal band, paraventricular nucleus of the thalamus, a wide area of the hypothalamus, cranial motor nuclei, inferior olive, and substantia gelatinosa of the spinal tract trigeminal nucleus. In contrast, no hybridization signal was observed in cerebral cortex, hippocampus, and cerebellum. These signals all were restricted to neurons and were not detected in glia (Fig. 2Bg).
Figure 2.

Localization of DINE mRNA. (A) Tissue localization of DINE mRNA is demonstrated by RNase protection assay using probe a (Fig. 1D) and a probe for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. (B) DINE mRNA expression in rat brain by in situ hybridization. Sagittal section (a) and coronal sections (b–f) represent DINE mRNA hybridization signal under dark-field illumination. Coronal sections at various levels: caudate putamen (b), hypothalamus and thalamus (c), pons (d), rostral part of medulla oblongata (e), and caudal part of medulla oblongata (f). Cpu, caudate putamen; LC, locus coeruleus; PVP, paraventricular thalamic nucleus; 7, facial nucleus; Amb, ambiguus nucleus; IO, inferior olive; Sp5C, spinal trigeminal tract nucleus caudalis. (Scale bar: a = 2 mm; b–e = 3 mm; f = 1 mm.) (g) Bright-field micrograph stained with thionin shows localization of mRNA on neurons (arrows). (Scale bar = 20 μm.)
Neuronal Damage Induces DINE Expression.
Because DINE mRNA was markedly increased in axotomized hypoglossal motor neurons (Fig. 3A), we further examined whether this up-regulation is specific to hypoglossal nerve injury. A variety of CNS lesions, including mechanical, ischemic, and excitotoxic were induced. As shown in Fig. 3 and Table 1 (which is published as supplementary material on the PNAS web site, www.pnas.org), cranial and spinal nerve lesions such as those produced by hypoglossal, facial, and sciatic nerve transection dramatically up-regulated DINE mRNA expression in injured motor neurons and sensory neurons in the dorsal root ganglia. In the case of CNS axonal injury, similar marked up-regulation was observed in retinal ganglion cells (after optic nerve crush) and neurons in the cerebral cortex after corpus callosum transection. Injection of colchicine, which inhibits axonal flow, also resulted in increase of DINE mRNA. In the MCA occlusion model of ischemia, marked expression of DINE mRNA was found in neurons near the area of infarction, in particular in the cerebral cortex and thalamus. These DINE mRNA-positive neurons probably had some connection with the infarction area and were damaged secondary to the focal cell death. By contrast, no increase of DINE mRNA was observed after psychological stress (immobilization stress), osmotic stress (hypertonic saline injection), or kainate-induced hyperexcitation (Table 1).
Figure 3.

DINE mRNA expression up-regulated by a variety of neuronal damage. (A) Hypoglossal nerve transection (arrow indicates hypoglossal nucleus of injured side). (B) Cortical and corpus callosum transection (the right cerebral cortex was cut by a knife sagittally; broken line). DINE mRNA is expressed in the cortical neurons located near the injured region (arrow in B) and in the thalamus of the injured side (arrow head in B). (C and D) L4 dorsal root ganglia (DRG) after sciatic nerve transection (D). (C) Sham-operated DRG. (E and F) Optic nerve transection: sham-operated (E) and optic nerve-crushed (F) retina. Arrow indicates that the axon-injured retinal ganglion cells express DINE mRNA. (G and H) MCA occlusion 7 days after unilateral MCA occlusion (right side), DINE mRNA is expressed on the injured side in the cerebral cortex (arrow in H) and thalamus (arrowhead in H), but not in that of sham operated (G). [Scale bars: 500 μm (A), 1 mm (B, G, and H), 40 μm (C and D), and 450 μm (E and F).]
Characterization of DINE Protein.
We generated an antibody against the intracelluar N terminus of DINE. We performed Western blotting using the cytosol and membrane fractions generated from full-length DINE and mock-transfected COS-7 cells. Fig. 4A shows that the antibody detected a single band located at approximately 90 kDa in the membrane fraction and not in the cytosol fraction of DINE-transfected or mock-transfected cells. DINE tagged with C-terminus myc also was recognized as a same band by both myc antibody and N-terminus antibody against DINE (data not shown).
Figure 4.

Characterization of DINE protein. (A) Western blotting using cytosol (Cyto.) and membrane (Mem.) fractions collected from COS-7 cells, which were transfected with either full-length DINE or mock. (B) Proteolytic activity of recombinant DINE. For inhibition experiments, typical metalloprotease inhibitors were added before incubation with substrate. (C) C2-ceramide-induced apoptosis in COS cells was significantly inhibited by the expression of DINE. Cell survival assay using trypan blue was carried out. Hatched bar, vehicle-plasmid expressing cells; closed bars, DINE-expressing cells. Bars represent mean +/− SD; *, P < 0.05 (ANOVA test). (D) Apoptotic nuclei (arrows) were identified by staining with Hoecst 33528 dye 5 h after exposure to C2-ceramide. (Bar = 20 μm.) (E) RNase protection assay showed that the proteolytic activity of DINE promotes Cu/Zn-SOD, Mn-SOD, and GPX expression, but not catalase at 1 h after C2-ceramide exposure. Cont, vehicle-plasmid expressing cells; Full, full-length DINE-expressing cells; ΔHEXXH, mutated DINE, lacking zinc-binding domain, expressing cells. (F) SOD activity induced by proteolytic activity of DINE. Bars represent means +/− SD; *, P < 0.05 (ANOVA test).
We initially examined whether big-endothelins were the substrates of DINE because of the sequence homology between DINE and ECE. However, DINE did not have any activity like ECE. Although the endogenous substrate for DINE is unknown, the sequence similarity between DINE and ECE suggests that DINE probably functions as a membrane-bound metallopeptidase. Therefore, to assess whether DINE cleaves a synthetic fluorescent substrate by proteolysis, a biochemical approach was taken. A recombinant protein lacking cytoplasmic and transmembrane domains was produced by Sf9 cells using the baculovirus expression system. To demonstrate endopeptidase activity, purified DINE was incubated with fluorogenic peptide, Z-Gly-Gly-Leu-pNA, which is a known NEP substrate. Metalloprotease inhibitors such as phosphoramidon, EDTA, 1,10-phenanthroline, and thiorphan blocked its hydrolysis (Fig. 4B). Therefore, DINE does have the pharmacological characteristics of a metalloprotease.
DINE Inhibits C2-Ceramide-Induced Apoptosis in COS-7 Cells.
Although the endogenous substrate for DINE is still unknown, the data described above imply that cellular homeostasis in damaged neurons may be altered by DINE proteolysis. We therefore examined the effect of DINE expression on the C2-ceramide-induced model of cell death. About 9 h after exposure to 50 μM C2-ceramide, 90% of COS-7 cells were killed. We thus examined whether overexpression of DINE could inhibit C2-ceramide-induced apoptosis. DINE-transfected COS-7 cells were exposed to C2-ceramide and their viability was measured. The number of dying cells was markedly decreased in cultures transfected with DINE compared with mock-transfected cultures (Fig. 4 C and D). However, DINE did not succeed in preventing ceramide-induced cell death completely. Although the mechanism responsible for the apoptotic effects of ceramide is poorly understood, it is evident that initially a rapid decline of mitochondrial oxidative phosphorylation is followed by an increased generation of oxygen radicals (24). Thus, we focused on the regulation of antioxidant enzyme genes after exposure to C2-ceramide. To determine whether the protease activity of DINE can influence intracellular events, we generated three types of expression vector constructs, full-length DINE, DINE lacking the zinc-binding domain, and the mock. Strikingly, we found that expression of Cu/Zn-SOD, Mn-SOD, and GPX mRNAs was up-regulated only in the full-length DINE-expressing cells (Fig. 4E). Furthermore, the activity of SOD also was induced by DINE (Fig. 4F).
Discussion
In the present study, a 200-bp cDNA fragment of DINE initially was isolated as a novel hypoglossal nerve injury response gene by DD-PCR. Subsequent cloning of the rat and mouse full-length cDNA revealed that DINE is a membrane-bound metallopeptidase and has high similarity to ECE. A search of the database revealed a human homologue of DINE, XCE, which was isolated in an attempt to identify a third member of the ECE family; however, XCE did not have ECE activity (23). Deficiency of XCE is lethal just after birth because of respiratory malfunction (25). These studies did not address the function of XCE, which has been left unidentified. The present study demonstrated striking characteristics of DINE. Expression of DINE is markedly up-regulated in response to a wide spectrum of nerve injury in the CNS and peripheral nervous system. Although the endogenous substrates of this enzyme are not yet clear, its proteolytic activity seems to activate, at least in part, free radical scavenging in damaged neurons.
Structure of DINE.
The deduced amino acid sequence of DINE showed that it was a membrane-bound metalloprotease with a conserved typical HEXXH zinc-binding motif and an ENXADX consensus sequence, consistent with gluzintin (26). Sequence similarity studies indicated that DINE is structurally a member of the ECE (14, 27) and NEP (28) superfamily that metabolizes/catabolizes neuropeptides. It generally is considered that ECE can hydrolyze big-endothelin to the final active peptide, whereas NEP inactivates a wide range of peptides such as substance P, enkephalins, cholecystokinin, neurotensin, and somatostatin by degradation. However, recently ECE-1 has been shown to possess a broad substrate specificity (29) and can hydrolyze neurotensin, substance P, and bradykinin as efficiently as big-endothelin-1. This lower substrate selectivity may be caused by the structural similarity in the C-terminus extracellular domain. As for the N-terminus cytoplasmic region, DINE shares only 12% similarity with ECE. Emoto et al. (30) have provided evidence that ECE-1 includes lysosomal targeting signal in its cytoplasmic tail. Thus, it is likely that DINE and related family members also may possess targeting sequences in their cytoplasmic domain. Although the natural substrate and function of DINE remain unknown, it may be possible that these family members function with quite low substrate selectivity whereas their diversity may be determined by the recruited and assembled proteins at the functional site.
Localization of DINE in Neuronal Tissue.
RNase protection analysis demonstrated that expression of DINE is restricted to brain tissue, and subsequent in situ hybridization studies localizing the mRNA to restricted brain regions confirmed this observation. The strongest expression of DINE mRNA was observed in the hypothalamus, large cholinergic cells in the striatum, and some cranial motor nerve nuclei. This characteristic localization of DINE mRNA reminds us that neuropeptides are primarily abundant in the hypothalamus, that the large neurons in the striatum also contain neuropeptides such as somatostatin, that cranial motor neurons express galanin and calcitonin gene-related peptide as well, and that several neuropeptides also are induced by peripheral nerve axotomy (5). Although no neuropeptides demonstrate exactly the same localization pattern as DINE, we cannot rule out the possibility that DINE may be a peptidase for a group of neuropeptides. DINE thus may be a potential regulator to elicit the function of neuropeptides, which would modulate synaptic transmission and/or neuronal survival as neurotrophic factors.
Response to Nerve Damage.
One of the striking characteristics of DINE is its robust response to widespread types of nerve damage. We initially isolated DINE from a hypoglossal motor nerve injury model by DD-PCR and subsequently demonstrated that DINE can respond to spinal sensory, optic, cortical, and thalamic nerve injury. The expression of DINE is always restricted to injured neurons and is not seen in glial cells such as injured nerve Schwann cells, oligodendrocytes, astrocytes, or microglia. In ischemic injury, MCA occlusion induced obvious expression of DINE in neurons that projected or passed through the focal region. Colchicine injection causes a chemical disorder in axons, where tubulin polymerization is disturbed and thereby the axonal flow is blocked. This chemical is used to block first axonal flow causing accumulation of an antigen within the soma for immunohistochemical detection, but also causes severe pathological change of axons. Therefore, in the case of colchicine injection, axonal damage is evident. However, stimuli such as immobilization and osmotic stress and kainate-induced hyperexcitation did not up-regulate DINE mRNA. The latter group of paradigms does not affect the axon directly. This may explain why these nonaxonal injuries or stresses to neuronal cells fail to elicit DINE expression. Thus, we conclude that mechanical and chemical injury to the neuronal axon may be required to increase DINE mRNA.
It is well known that after peripheral nerve injury, neuropeptides, growth factors, cytokines, and their receptors, which all are secreted and require processing to be activated or mature, are markedly induced in injured neurons, and these molecules probably give rise to a protective effect. However, to our knowledge, general processing enzymes such as PC family members were not up-regulated in damaged hypoglossal nuclei (unpublished data). ECE showed a slight increase in mRNA (unpublished data), whereas NEP, also termed CD10, was down-regulated in axotomized hypoglossal motor neurons (31). No other protease except DINE showed such a striking response in axotomized motor neurons. Although DINE expression alone is insufficient to rescue all injured neurons, the unique response to nerve injury might play a crucial modulatory function such as an activator of free radical scavengers.
C2-Ceramide-Induced Apoptosis Is Partially Inhibited by DINE.
As DINE mRNA is markedly increased in response to damage, the possibility arises that the control of cell death may be mediated by its proteolytic activity. A variety of apoptotic inducers such as tumor necrosis factor α, Fas ligand, deprivation of growth factors, and UV and x-ray irradiation are assumed to increase ceramide production, and a strong correlation between the production of ceramide and subsequent cell death is evident (32). Application of a cell-permeable ceramide analogue (C2-ceramide) mimics induction of intracellular ceramide level, and thereby induces apoptosis in many different cell systems (33, 34). We examined a function of DINE by using this apoptosis model. C2-ceramide induces apoptosis in COS cells, and the expression of DINE partially inhibited cell death. The rescue activity of DINE is not as great as that of antiapoptotic genes such as Bcl-2 (35) and Akt (36); however, expression of DINE appears to postpone the onset of apoptosis. Similar prolongation of apoptosis rather than complete rescue also is seen in nerve growth factor-deprived sympathetic neurons after injecting Cu/Zn-SOD (37). Therefore, we examined the expression level of Cu/Zn-SOD together with that of related antioxidant enzymes in DINE-expressing COS cells. Strikingly, Cu/Zn-SOD, Mn-SOD, and GPX mRNA were up-regulated in DINE-transfected cells. However, such induction was not seen in COS cells expressing mutant DINE, which lacked the proteolytic activity. Similarly, COS cells expressing the full length of DINE showed up-regulated activity of SOD. These results suggest that the catalytic activity of DINE evoked some intracellular signal to promote transcription and activity of the enzymes, which is crucial for reducing oxidative stress caused by superoxide production. Mn-SOD is a stress responsive molecule, and hypoglossal nerve injury and MCA occlusion induce Mn-SOD expression in injured neurons. Cerebral ischemic injury is exacerbated both in Mn-SOD-deficient (38) and Cu/Zn-SOD-deficient mice (39). Cu/Zn-SOD-deficient mice exhibit enhanced cell death after facial nerve injury (40). These reports suggest that the expression of SOD is pivotal for the survival of injured neurons. However, the molecular mechanism underlying the induction of SOD mRNA is poorly understood. The present study demonstrated that DINE significantly enhanced the expression of antioxidant enzymes such as Cu/Zn-SOD, Mn-SOD, and GPX under oxidative stress. Although enhanced expression alone is insufficient to rescue injured cells, it can modulate the apoptotic profile.
We conclude that expression of DINE is correlated with enhanced expression and activity of antioxidant enzymes, and thereby diminishes the impact of oxidative stress induced by nerve injury. The protease-mediated mechanism might contribute to reduced susceptibility after nerve injury and prevent neuronal death.
Supplementary Material
Acknowledgments
We are grateful to Dr. N. Mori for useful advice, Dr. S. J. Augood for critical reading and English correction, and Mr. K. Hazawa and Mr. T. Sasaki for technical assistance. This work was partly supported by Sumitomo Electric Inc. and grants from the Ministry of Education, Science, Sports and Culture, Ministry of Health and Welfare, and Ministry of Science and Technology, Japan.
Abbreviations
- DINE
damage-induced neuronal endopeptidase
- DD-PCR
differential display PCR
- PC
proprotein convertase
- ECE
endothelin converting enzyme
- CNS
central nervous system
- MCA
middle cerebral artery
- RACE
rapid amplification of cDNA ends
- SOD
superoxide dismutase
- GPX
glutathione peroxidase
- NEP
neutral endopeptidase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AB023896, AB026293, and AB026294).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.070509897.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.070509897
References
- 1.Kiryu S, Yao G L, Morita N, Kato H, Kiyama H. J Neurosci. 1995;15:7872–7878. doi: 10.1523/JNEUROSCI.15-12-07872.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanabe K, Nakagomi S, Kiryu-Seo S, Namikawa K, Imai Y, Ochi T, Tohyama M, Kiyama H. Mol Brain Res. 1999;64:34–40. doi: 10.1016/s0169-328x(98)00302-7. [DOI] [PubMed] [Google Scholar]
- 3.Toki H, Namikawa K, Su Q, Kiryu-Seo S, Sato K, Kiyama H. J Neurochem. 1998;71:913–919. doi: 10.1046/j.1471-4159.1998.71030913.x. [DOI] [PubMed] [Google Scholar]
- 4.Morita N, Kiryu S, Kiyama H. J Neurosci. 1996;16:5961–5966. doi: 10.1523/JNEUROSCI.16-19-05961.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hokfelt T. Neuron. 1991;7:867–879. doi: 10.1016/0896-6273(91)90333-u. [DOI] [PubMed] [Google Scholar]
- 6.Steiner D F. Curr Opin Chem Biol. 1998;2:31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- 7.Hooper N M, Karran E H, Turner A J. Biochem J. 1997;321:265–279. doi: 10.1042/bj3210265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong J, Opresko L K, Dempsey P, Lauffenburger D A, Coffey R J, Wiley H S. Proc Natl Acad Sci USA. 1999;96:6235–6240. doi: 10.1073/pnas.96.11.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Werb Z, Yan Y. Science. 1998;282:1279–1280. doi: 10.1126/science.282.5392.1279. [DOI] [PubMed] [Google Scholar]
- 10.Smirnova I V, Ma J Y, Citron B A, Ratzlaff K T, Gregory E J, Akaaboune M, Festoff B W. J Neurochem. 1996;67:2188–2199. doi: 10.1046/j.1471-4159.1996.67052188.x. [DOI] [PubMed] [Google Scholar]
- 11.Gitler D, Spira M E. Neuron. 1998;20:1123–1135. doi: 10.1016/s0896-6273(00)80494-8. [DOI] [PubMed] [Google Scholar]
- 12.Zuo J, Ferguson T A, Hernandez Y J, Stetler-Stevenson W G, Muir D. J Neurosci. 1998;18:5203–5211. doi: 10.1523/JNEUROSCI.18-14-05203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.La Fleur M, Underwood J L, Rappolee D A, Werb Z. J Exp Med. 1996;184:2311–2326. doi: 10.1084/jem.184.6.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, Yanagisawa M. Cell. 1994;78:473–485. doi: 10.1016/0092-8674(94)90425-1. [DOI] [PubMed] [Google Scholar]
- 15.Kitahara T, Kiryu S, Ohno K, Morita N, Kubo T, Kiyama H. Neurosci Res. 1994;20:275–280. doi: 10.1016/0168-0102(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 16.Noguchi K, Senba E, Morita Y, Sato M, Tohyama M. Mol Brain Res. 1989;6:327–330. doi: 10.1016/0169-328x(89)90077-6. [DOI] [PubMed] [Google Scholar]
- 17.Isenmann S, Bahr M. Exp Neurol. 1997;147:28–36. doi: 10.1006/exnr.1997.6585. [DOI] [PubMed] [Google Scholar]
- 18.Lurton D, Coussemacq M, Barrow P, Sundstrom L E, Rougier A. Neurosci Lett. 1996;213:181–184. doi: 10.1016/0304-3940(96)12854-8. [DOI] [PubMed] [Google Scholar]
- 19.Hannibal J, Mikkelsen J D, Fahrenkrug J, Larsen P J. Endocrinology. 1995;136:4116–4124. doi: 10.1210/endo.136.9.7649120. [DOI] [PubMed] [Google Scholar]
- 20.Inuzuka T, Tamura A, Sato S, Kirino T, Toyoshima I, Miyatake T. Brain Res. 1990;526:177–179. doi: 10.1016/0006-8993(90)90269-h. [DOI] [PubMed] [Google Scholar]
- 21.Kiryu-Seo S, Matsuo N, Wanaka A, Ogawa S, Tohyama M, Kiyama H. Mol Brain Res. 1998;62:220–223. doi: 10.1016/s0169-328x(98)00255-1. [DOI] [PubMed] [Google Scholar]
- 22.Spitz D R, Oberley L W. Anal Biochem. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- 23.Valdenaire O, Richards J G, Faull R L M, Schweizer A. Mol Brain Res. 1999;64:211–221. doi: 10.1016/s0169-328x(98)00321-0. [DOI] [PubMed] [Google Scholar]
- 24.France-Lanord V, Brugg B, Michel P P, Agid Y, Ruberg M. J Neurochem. 1997;69:1612–1621. doi: 10.1046/j.1471-4159.1997.69041612.x. [DOI] [PubMed] [Google Scholar]
- 25.Schweizer A, Valdenaire O, Koster A, Lang Y, Schmitt G, Lenz B, Bluethmann H, Rohrer J. J Biol Chem. 1999;274:20450–20456. doi: 10.1074/jbc.274.29.20450. [DOI] [PubMed] [Google Scholar]
- 26.Hooper N M. FEBS Lett. 1994;354:1–6. doi: 10.1016/0014-5793(94)01079-x. [DOI] [PubMed] [Google Scholar]
- 27.Shimada K, Takahashi M, Tanzawa K. J Biol Chem. 1994;269:18275–18278. [PubMed] [Google Scholar]
- 28.Malfroy B, Schofield P R, Kuang W J, Seeburg P H, Mason A J, Henzel W J. Biochem Biophys Res Commun. 1987;144:59–66. doi: 10.1016/s0006-291x(87)80475-8. [DOI] [PubMed] [Google Scholar]
- 29.Johnson G D, Stevenson T, Ahn K. J Biol Chem. 1999;274:4053–4058. doi: 10.1074/jbc.274.7.4053. [DOI] [PubMed] [Google Scholar]
- 30.Emoto N, Nurhantari Y, Alimsardjono H, Xie J, Yamada T, Yanagisawa M, Matsuo M. J Biol Chem. 1999;274:1509–1518. doi: 10.1074/jbc.274.3.1509. [DOI] [PubMed] [Google Scholar]
- 31.Back S A, Gorenstein C. J Comp Neurol. 1994;340:149–160. doi: 10.1002/cne.903400202. [DOI] [PubMed] [Google Scholar]
- 32.Hannun Y A. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 33.Hannun Y A, Obeid L M. Trends Biochem Sci. 1995;20:73–77. doi: 10.1016/s0968-0004(00)88961-6. [DOI] [PubMed] [Google Scholar]
- 34.Obeid L M, Linardic C M, Karolak L A, Hannun Y A. Science. 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 35.Kane D J, Sarafian T A, Anton R, Hahn H, Gralla E B, Valentine J S, Ord T, Bredesen D E. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 36.Zhou H, Summers S A, Birnbaum M J, Pittman R N. J Biol Chem. 1998;273:16568–16575. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
- 37.Greenlund L J S, Deckwerth T L, Johnson E M., Jr Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 38.Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen S F, Chan P H. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kondo T, Reaume A G, Huang T-T, Carlson E, Murakami K, Chen S F, Hoffman E K, Scott R W, Epstein C J, Chan P H. J. Neurosci. 1997. 4180–4189. [Google Scholar]
- 40.Reaume A G, Elliott J L, Hoffman E K, Kowall N W, Ferrante R J, Siwek D F, Wilcox H M, Flood D G, Beal M F, Brown R H, Jr, et al. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}