

Abstract
The nuclear organization of specific endogenous chromatin regions can be investigated only by fluorescence in situ hybridization (FISH). One of the two fixation procedures is typically applied: (1) buffered formaldehyde or (2) hypotonic shock with methanol acetic acid fixation followed by dropping of nuclei on glass slides and air drying. In this study, we compared the effects of these two procedures and some variations on nuclear morphology and on FISH signals. We analyzed mouse erythroleukemia and mouse embryonic stem cells because their clusters of subcentromeric heterochromatin provide an easy means to assess preservation of chromatin. Qualitative and quantitative analyses revealed that formaldehyde fixation provided good preservation of large-scale chromatin structures, while classical methanol acetic acid fixation after hypotonic treatment severely impaired nuclear shape and led to disruption of chromosome territories, heterochromatin structures, and large transgene arrays. Our data show that such preparations do not faithfully reflect in vivo nuclear architecture.
Electronic supplementary material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s00412-006-0084-2 and is accessible for authorized users.
Introduction
The nuclear architecture and the organization of DNA in the interphase nucleus have attracted considerable interest (for reviews, see Spector 2003; Cremer et al. 2004; Foster and Bridger 2005; Sproul et al. 2005). The only method to label specific endogenous DNA sequences is fluorescence in situ hybridization (FISH) which requires access to the target DNA and DNA denaturation, both conflicting with preservation of nuclear morphology. Thus, preparation methods must seek a compromise. Published studies using FISH mostly relied on one of the two methods: either fixation with buffered formaldehyde (BF) with subsequent permeabilization or hypotonic treatment with methanol:acetic acid (75%:25%, MAA) fixation, dropping of nuclei on slides, and air drying (Hypo-MAA). The two methods are also known as 3D and 2D FISH, respectively (Croft et al. 1999). Few studies used glutaraldehyde, a fixative that is widely used for electron microscopy studies, in the presence of detergent (e.g., Brown et al. 1997; Osborne et al. 2004).
Preservation of large-scale chromatin structure during the fixation with BF was demonstrated by in vivo imaging of GFP fusions to DNA binding proteins such as centromere binding proteins CENP-B (Shelby et al. 1996), CENP-A (Mahy et al. 2002b), or histone H2B (Kanda et al. 1998; Solovei et al. 2002). Preservation during subsequent FISH was demonstrated by comparison of centromere distribution before and after FISH in the same nuclei (Cremer et al. 1993; Kurz et al. 1996) and by detection of PML bodies before and after FISH (Verschure et al. 1999). In vivo replication labeling with fluorescent nucleotides introduces a label that is visible from the living cell to after FISH. With such a label, it was shown that while DNA denaturation causes significant damage on the electron microscopic level, with the limited resolution of conventional and confocal light microscopy, chromatin structure appears preserved (Solovei et al. 2002). For light microscopy, BF-fixed nuclei thus provide a gold standard for interphase FISH against which other preparation methods can be compared.
Hypo-MAA fixation protocols have been originally developed for the preparation of metaphase chromosome spreads. Hypotonic shock before fixation in combination with dropping of nuclei to glass slides and subsequent air drying lead to well-spread metaphase chromosomes sticking firmly to the slide. Interphase nuclei in such preparations are flattened and have an increased diameter (Kozubek et al. 2000 and present study). Hypotonic treatment (Kobliakova et al. 2005) and other changes in ion concentration (Belmont et al. 1989) have been shown to disturb chromosome morphology. Distributions of core histones and of the splicing factor SC-35 were observed to change substantially under these conditions (Hendzel and Bazett-Jones 1997), suggesting a redistribution of chromatin components. Extraction of proteins (Sumner et al. 1973; Fraschini et al. 1981), RNA (Lawrence and Singer 1985), and DNA (Raap et al. 1986) has been described, the latter being amplified by denaturation and hybridization. In spite of these issues, MAA fixation is still often used in studies on nuclear organization (e.g., Nikiforova et al. 2000; Roix et al. 2003; Chambeyron and Bickmore 2004) for three apparent reasons: (1) it is easier to produce bright FISH signals on Hypo-MAA-fixed nuclei; (2) image acquisition of flat structures is faster (Boyle et al. 2001); and (3) microscopic equipment is less expensive if single images are made instead of 3D-image stacks, the latter requiring motorized z-drive, motorized filter wheel, and automated camera or a confocal microscope (Kozubek et al. 2000).
Published differences between BF- and Hypo-MAA-fixed nuclei and the widespread use of Hypo-MAA fixation for examination of large-scale chromatin structure raise concerns to which extend data from such preparations reflect the in vivo situation. To allow correct interpretation, qualitative and quantitative comparisons of BF- and Hypo-MAA-fixed cells to a level not available so far are required. In this study, we investigated chromatin morphology after both fixation methods qualitatively and quantitatively. To be able to separate effects from hypotonic treatment and MAA fixation per se, we included MAA fixation procedures without hypotonic treatment and dropping of cells (Table 1). We investigated nuclear shape and preservation of DNA counterstain patterns directly after fixation and after FISH. We selected mouse cells for our study because their prominent centromeric heterochromatin clusters (chromocenters) allow an easy assessment of preservation of this fraction of chromatin in interphase nuclei. As targets for FISH hybridization, we used large and small transgene arrays in mouse erythroleukemia (MEL) cells as well as chromosome territories and loci detected by BAC clones in mouse embryonic stem cells. We measured nuclear shape, chromocenter preservation, compactness of detected regions, distances from BAC signals to the surface of their harboring chromosome territory, and distances from BACs and territories to the nuclear surface. In addition, we compared large-scale chromatin structure in living MEL cells and after BF fixation. Our results confirm that nuclear shape and 3D distribution of chromatin are preserved in BF-fixed nuclei at the light microscopic level. We show that hypotonic treatment and air drying in typical MAA fixation protocols cause severe distortions in nuclear shape and chromatin structure.
Table 1.
Overview over prefixation, fixation, and postfixation steps applied to MEL cells which were subjected to FISH
| BF 3.7 | BF 1.8 | Hypo-MAA I | Hypo-MAA II | Attach-MAA-dried | Attach-MAA-SSC | Attach-MAA-FA | |
|---|---|---|---|---|---|---|---|
| Attachment (MEL cells only) | With poly-L-lysine | With poly-L-lysine | With poly-L-lysine | With poly-L-lysine | With poly-L-lysine | ||
| Pretreatment | Centrifugation, hypotonic shock with 0.5% sodium citrate, 0.25% KCl for 10 min at room temperature, centrifugation | Centrifugation, hypotonic shock with 0.56% KCl for 15 min at 37°C, centrifugation | |||||
| Fixation | 3.7% PBS-buffered formaldehyde for 10 min | 1.8% PBS-buffered formaldehyde for 15 min | Addition of MAA (room temperature) to resuspended pellet, 10 min at room temperature, over night 4°C | Addition of MAA (−20°C) to resuspended pellet, 30 min −20°C | Replacement of medium with MAA (−20°C), 30 min −20°C | Replacement of medium with MAA (−20°C), 30 min −20°C | Replacement of medium with MAA (−20°C), 30 min −20°C |
| Intermediate steps | Permeabilization (see text) | Permeabilization (see text) | Dropping on glass slides, air drying | Dropping on glass slides, air drying, aging 1 h at 60–65°C | Air drying | 2 × 3 min 2×SSC | |
| Storage | 50% formamide/50% 2×SSC at 4°C | 50% formamide/50% 2×SSC at 4°C | Dry at −20°C | Dry at −20°C | Dry at −20°C | 50% formamide/50% 2×SSC at 4°C | 50% formamide/50% 2×SSC at 4°C |
| FISH pretreatment | RNase, wash in 2×SSC, dehydration in ethanol series (70, 90, and 100%), air-dried | ||||||
| Denaturation | 75°C for 2 min in 50% formamide/50% 2×SSC | 75°C for 2 min in 50% formamide/50% 2×SSC | 5 min, at 70°C, 2–3 min, in 70% formamide/30% 2×SSC preheated to 72°C | 75°C for 2 min. in 50% formamide/ 50% 2×SSC | 75°C for 2 min in 50% formamide/50% 2×SSC | 75°C for 2 min in 50% formamide/50% 2×SSC | 75°C for 2 min in 50% formamide/50% 2×SSC |
Materials and methods
Cells
PALZ39E and PALZ39M cells are MEL cells having multiple integrations of a 15-kbp plasmid (Dietzel et al. 2004). They were grown in suspension in DMEM with 10% fetal calf serum at 37°C with 5% CO2. To perform all tested fixation methods on cells from the same culture, a respectively large amount of cells was accumulated. For fixation procedures that require adherent cells, autoclaved microscopic coverslips (170 μm thick) were preincubated with poly-L-lysine (1 mg/ml; MW 150000, Sigma, Deisenhofen, Germany, P1399) for 30 min, washed, and dried before adding the cell culture. After 30–60 min, the cells were sufficiently attached to remove the medium and add the fixative.
Mouse embryonic stem cells of the CCE line were kindly provided by C. Bonifer, Leeds, UK. They were cultured on STO fibroblasts in a DMEM-based medium with 15% fetal calf serum and LIF as described elsewhere (Faust et al. 1997).
Fixation methods
An overview over performed procedures is given in Table 1. Formaldehyde solution was freshly made from paraformaldehyde as described (Dernburg and Sedat 1998). Bottled formaldehyde was not used because formaldehyde can polymerize or oxidize, particularly if older (Kiernan 2000). BF was applied to cells as 3.7% (MEL and ES cells) or 1.8% (MEL cells only) in PBS for 10–15 min and washed 3 × 3 min in PBS. Nuclei not subjected to FISH were immediately counterstained with TO-PRO-3 (1 μM in buffer; Molecular Probes Europe, Leiden, The Netherlands), which has AT-preference like DAPI and is excitable with 633 or 648 nm laser lines, and mounted in Vectashield (Vector, Burlingame, CA, USA) for microscopic observation. Nuclei for FISH were permeabilized for 15 min in 0.5% Triton-X-100 in PBS, immersed in 20% glycerol/80% PBS for at least 1 h, and subjected to 5 freeze/thaw cycles with liquid nitrogen. After three washes in PBS (3 min each) and a short equilibration in 0.1 N HCL, the cells were incubated 10 min in fresh 0.1 N HCL, washed 3 × 3 min in 2×SSC, and stored (1 h—weeks) in 50% formamide/50% 2×SSC. Hybridization mixture was added to wet slides, and air drying was carefully avoided.
For MAA fixation with hypotonic treatment and dropping of cells, two variations were used. Hypo-MAA I was previously used in studies on nuclear architecture (Croft et al. 1999; Boyle et al. 2001; Mahy et al. 2002a,b; Chambeyron and Bickmore 2004), and a detailed protocol was kindly provided by W. Bickmore, Edinburgh, UK. The cells were centrifuged and resuspended in 14 ml hypotonic solution (0.5% sodium citrate, 0.25% KCl, 10 min at room temperature). After centrifugation, leaving 1 ml of supernatant, a 1-ml MAA (methanol 75%, acetic acid 25%) at room temperature was added and the pellet was resuspended. After dropwise addition of 10–12 ml MAA, 10 min incubation at room temperature, and centrifugation, the pellet was resuspended in 1 ml supernatant, diluted with 12 ml MAA, and incubated overnight at 4°C. After concentration by centrifugation, the nuclei were dropped on glass slides, air-dried overnight at room temperature, and stored at −20°C under desiccating conditions. Nuclei not subjected to FISH were counterstained with TO-PRO-3 and embedded in Vectashield for microscope observation. Before FISH, the slides were incubated 1 h with RNase at 37°C, washed in 2×SSC dehydrated in increasing ethanol concentrations (70, 90, and 100%), and air-dried. Hypo-MAA I fixation was applied to MEL cells and, after trypsinization, to ES cells.
Hypo-MAA II fixation is used in our lab to prepare metaphase spreads. It is similar to the above procedure (Table 1), but hypotonic treatment is performed with 14 ml 0.56% KCl for 15 min at 37°C. A 1-ml MAA is added before centrifugation, then the pellet is carefully resuspended under dropwise addition of 10–12 ml MAA (−20°C) and stored for 30 min at −20°C. Nuclei are centrifuged again and resuspended in an amount of MAA that is suitable for dropping the cells on microscopic slides according to Deng et al. (2003). Artificial aging of slides was performed for 1 h at 60–65°C.
In MAA fixation protocols without hypotonic treatment and dropping, the cells were attached to poly-L-lysinated coverslips (see above). The medium was replaced by cold MAA, the cells were transferred to −20°C for at least 30 min, and one wash with MAA was performed. For air drying, MAA was poured off (attach-MAA-dried). In one protocol, MAA was replaced by 2×SSC for two washes (3 min each) before transfer to 50% formamide/50% 2×SSC (attach-MAA-SSC), while in another protocol, the 2×SSC washing step was omitted (attach-MAA-FA).
FISH
The plasmid pPALZ8.8 for which the investigated MEL cells are transgenic (Dietzel et al. 2004) was also used as a FISH probe and labeled with digoxigenin-dUTP by nicktranslation. Probe concentration in the hybridization mix (50% formamide, pH = 7, 10% dextran sulfate in 1×SSC) was 10 ng/μl. Salmon sperm DNA was used as carrier DNA (2 μg/μl). For hybridization on ES cells, BAC RP23-6I17 with 199.7 kbp (http://www.ensembl.org) was obtained from the BACPAC resource center (http://bacpac.chori.org) and labeled with digoxigenin-dUTP by nicktranslation. It delineates a region on mouse chromosome 7 including Phlda2 (= Tssc3) and a part of Osbpl5 (= Obph1; http://www.ensembl.org). The region detected in a previous study by the BAC 245N5 (Mahy et al. 2002a) appears to include some additional sequences toward the centromeric end. Mouse chromosome 7 paint probe, produced and labeled with DNP-dUTP by DOP-PCR (Telenius et al. 1992) from sorted chromosomes, was kindly provided by N. Carter, Cambridge, UK (Rabbitts et al. 1995).
Hybridization mix was sealed with rubber cement air-free between coverslip and glass slide. Probe and target DNA were denatured simultaneously on a metal plate at 75°C for 2 min except for nuclei subjected to Hypo-MAA I fixation. Here, the slides were incubated 5 min at 70°C, denatured 2–3 min in 70% formamide (pH = 7)/30% 2×SSC preheated to 72°C, immediately dehydrated in an ice-cold ethanol series (70, 90, and 100%), and air-dried in a vacuum. The hybridization mix with the probe DNA was denatured separately for 5 min at 70°C before application on the dry slides.
Hybridization for all specimens was for 2–3 days at 37°C, followed by two washes in 2×SSC (37°C) and three stringent washes in 0.1×SSC (60°C). After blocking in 4% bovine serum albumin (ICN Biochemicals, Eschwege, Germany, #160069) in 4×SSC with 0.2% Tween20, haptens were detected with antibodies specified below in blocking solution at 37°C, 30–45 min for each layer. For MEL cells, sheep-anti-dig-FITC (1:100, Dianova, Hamburg, Germany) was incubated together with 0.1 μg/μl RNase except for Hypo-MAA I-treated cells where RNase treatment had already occurred (see above). For ES cells, haptens were detected by rabbit-anti-DNP (1:200, Sigma), goat-Alexa488-anti rabbit (1:200, Molecular Probes Europe, Leiden, The Netherlands), mouse-anti-dig-Cy3 (1:100, Dianova), and sheep-anti-mouse-Cy3 (1:500, Dianova). DNA was counterstained with TO-PRO-3 (1 μM in 4×SSC/Tween) followed by a short rinsing step in 4×SSC/Tween. Preparations that were previously air-dried were washed with demineralized water, dried again, and embedded in Vectashield. Others were transferred directly from washing buffer to Vectashield.
Confocal microscopy
Confocal image stacks were generated on a Leica TCS4D (for MEL cells, 488, 568, and 648 nm excitation, PlanApo 100× NA 1.4) or on a Zeiss LSM 410 (for ES cells, 488, 543, and 633 nm, PlanApo 63× NA 1.4) with voxel sizes of 0.08 × 0.08 × 0.24 or 0.09 × 0.09 × 0.25 μm3, respectively. Micrographs shown in the figures were not computationally “enhanced” except for linear adjustment of gray levels of whole images with the levels command in Adobe Photoshop.
Image analysis
Measurements of nuclear width and height were performed with the open source software ImageJ (http://rsb.info.nih.gov/ij/). The number of objects to which a structure disintegrates at increasing thresholds (“object counting”) was measured with a program described elsewhere (Stadler et al. 2004) or a newly developed version thereof. While the same results are obtained with both programs (data not shown), the new version is much faster. Briefly, each image stack is first Gauss-filtered and normalized to 256 gray values before increasing thresholds are applied at intervals of 5. At each level, the number of independent objects with at least 10 voxels is determined. For significance calculations, to represent each nucleus, we used the maximal number of objects at any threshold above 90 (to safely exclude background influence) and applied the Mann–Whitney rank-sum test in SigmaStat 3.0 (SPSS, Chicago, IL, USA).
To determine the distance of BAC signals to the surface of their harboring chromosome territory, the program “Enhanced Distance Measurement Tool”, kindly provided by T. Thormeyer from our institute, was used (Albiez et al. 2006). It is modeled after the program ADS developed by one of the coauthors (von Hase et al., in preparation) but is written in Mathematica under Windows instead of using the Khoros environment in Linux. The reference structure (cell nucleus or chromosome territory) is segmented by setting the threshold interactively. Layers of equal thickness (0.25 μm) are then computed around the surface, inside, and outside. The program then assigns each voxel of the investigated signal (BACs, territories, and, in the case of nuclei as reference, nuclear counterstain) to the respective layer. Intensities of all voxels in a given layer are summed up to calculate the percentage of total signal intensity in each layer. If territories were the reference structure, for each nucleus, two stacks with only one territory in each were generated in ImageJ before processing. For a series of nuclei, values were averaged to produce curves as shown. p-values were calculated with the signed rank test (Wilcoxon) in SigmaStat 3.0 using for each signal the intensity weighted mean value of the distance to the surface of the reference structure. p-values for relative radial distributions (Cremer et al. 2001) were calculated with the two-sided Kolmogorov–Smirnov test (Young 1977).
Live cell microscopy
Live cell imaging was performed in an open POC cell chamber on a VisiScope Cell Explorer (Visitron Systems, Puchheim, Germany) based on a Zeiss Axiovert 200 and a Spot TR-SE6 CCD Camera with Sony ICX285 chip. The cells were incubated on poly-L-lysinated coverslips overnight to ensure attachment. The medium was supplemented with 15 mM HEPES to avoid pH changes under atmospheric conditions (without CO2 addition). Addition of the live cell nuclear counterstain Hoechst 33342 (0.1 μ/ml; Sigma, B-2261) was 1–2 h before recording. Excitation was with a 100-W Hg arc lamp with a neutral density filter (10% transmission), and exposure time was 1 s for GFP (filter: 470/40, 497LP, 522/40) and 0.1 s for Hoechst (360/40, 400LP, 470/40). 3D stacks were recorded with a 100× N.A. 1.4 PlanApo oil objective with 0.5 μm between sections. Without antifade reagents, the GFP signal was prone to bleaching. Therefore, GFP signals (with 17 sections) and whole nuclei (with 40 sections) were recorded from different preparations. After recording of the live cells, the medium was pipetted off and replaced with 3.7% buffered formaldehyde (see above) at 37°C. After 10 min, the fixation solution was removed and replaced with PBS at 37°. The strong refractive index mismatch due to the use of aqueous medium or buffer in combination with oil immersion causes a notable deviation of the nominal from the actual focal position, the focal shift (Hell and Stelzer 1995). This was corrected by multiplying measured z-distances with a factor of 0.82 (Hell and Stelzer 1995).
Results
Experimental setup of MEL cell fixation
Cultures of MEL cells were split in several fractions to allow different fixation procedures on cells from the same culture flask. Of two identical preparations from each fixation procedure, one was only DNA-stained, while the other was subjected to FISH before confocal microscopy. We carried out three independent experimental series. Two series were performed with the MEL cell line PALZ39E which carries a transgene array of ∼50 Mbp interspersed with host DNA, and one series was performed with PALZ39M which carries a much smaller transgene array (Dietzel et al. 2004). Within a series, FISH was performed with aliquots from the same hybridization mix, the same antibody detection, and the same confocal microscope settings.
Preservation of nuclear shape in MEL cells
The most obvious difference between nuclei from different fixation procedures is a distortion of nuclear shape by some methods. We measured the diameter of nuclei in the xy plane (width) and the height along the optical axis in 3D confocal image stacks. Because the nuclei of MEL cells are almost spherical, a good preservation of nuclear shape leads to a width/height ratio close to 1 while flattened nuclei have higher values. In preparations fixed with BF (BF 3.7 and BF 1.8, Fig. 1a), the nuclei had an average ratio between 1 and 1.5 (blue dots in Fig. 2, see supplemental online material S1 for mean values). While we observed some intra- and interexperimental variability, this variability was limited to a rather narrow range for BF-fixed nuclei.
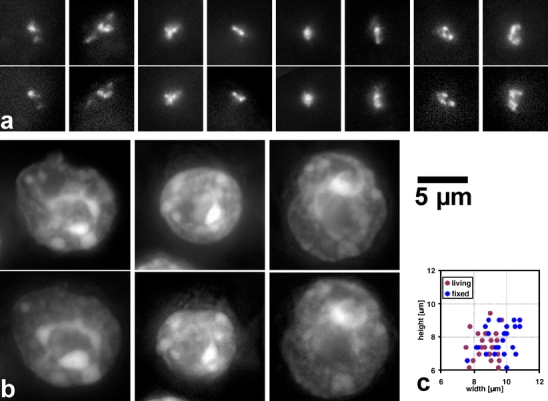
Fig. 1.
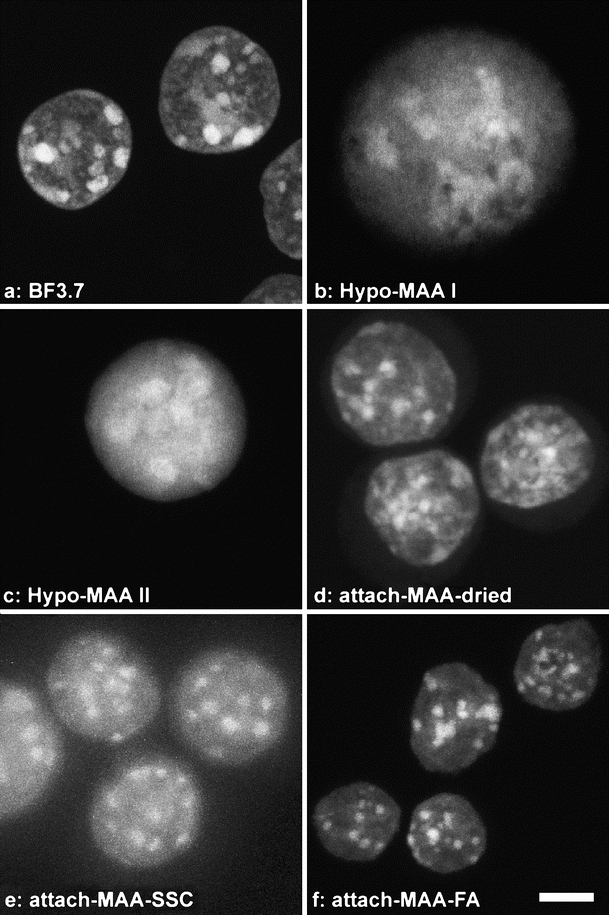
Nuclei of MEL cells after different fixation methods at the same scale. Nuclei were fixed and immediately counterstained, embedded in mounting medium, and imaged. All images are at the same scale. Bar 5 μm. Fixation methods: a 3.7% buffered formaldehyde on adherent cells (BF 3.7). b, c Suspension cells treated with hypotonic shock, MAA fixation, dropping, and air drying (Hypo-MAA I in (b) and II in (c)). d–f Adherent cells were immersed in MAA and air-dried (attach-MAA-dried, (d)) or washed in aqueous buffer (attach-MAA-SSC, (e)), or directly transferred to formamide/SSC (attach-MAA-FA, (f)). Projections of confocal image stacks are shown. Note the differences in preservation of chromocenters and nuclear size
Fig. 2.
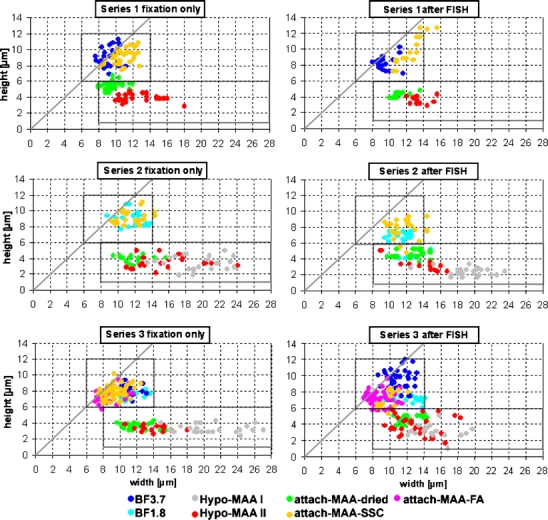
Nuclear shape of MEL cells after different fixation procedures. Each dot represents one nucleus, indicating its diameter in the central confocal section (width) and its height in the confocal image stack. Nuclei with identical height and width fall on the gray bisecting diagonal. Results are shown for nuclei directly after fixation (graphs on the left) and for those after FISH (graphs on the right). Each of the three experimental series was performed on nuclei from one cell culture flask. While values for BF-fixed nuclei (dark and light blue dots) cluster in a relative narrow window around the bisecting diagonal (upper box), the results for Hypo-MAA-fixed nuclei (red and gray dots) show flattening of nuclei and a high variability in width (lower box). See Table 1 for fixations
The two protocols that involved hypotonic swelling of nuclei before fixation with MAA, dropping of swollen, fixed nuclei to glass slides, and subsequent air drying (Hypo-MAA I and II, Fig. 1b,c) resulted in the largest distortions (red and gray dots, respectively, in Fig. 2) with average width/height ratios between 3.2 and 6.3. Nuclei were flattened to less than half the height of formaldehyde-fixed nuclei. Their width varied considerably, with diameters from normal to up to twice as large. Increased diameters were a consequence of hypotonic swelling and/or dropping but not of fixation with MAA per se: If living cells were attached to coverslips and fixed without hypotonic shock by immersion in MAA, after evaporation of MAA (attach-MAA-dried, Fig. 1d), nuclear width was maintained (green dots in Fig. 2a,c, and e). Due to air drying, their height was about halved. When MAA was replaced directly with another liquid and evaporation was avoided (attach-MAA-SSC and attach-MAA-FA, Fig. 1e,f), nuclear shape was similar to formaldehyde-fixed preparations (yellow and pink dots in Fig. 2). For all fixation methods, nuclear shape was similar after FISH (Figs. 2, 3, and 4, supplemental online material S1).
Fig. 3.
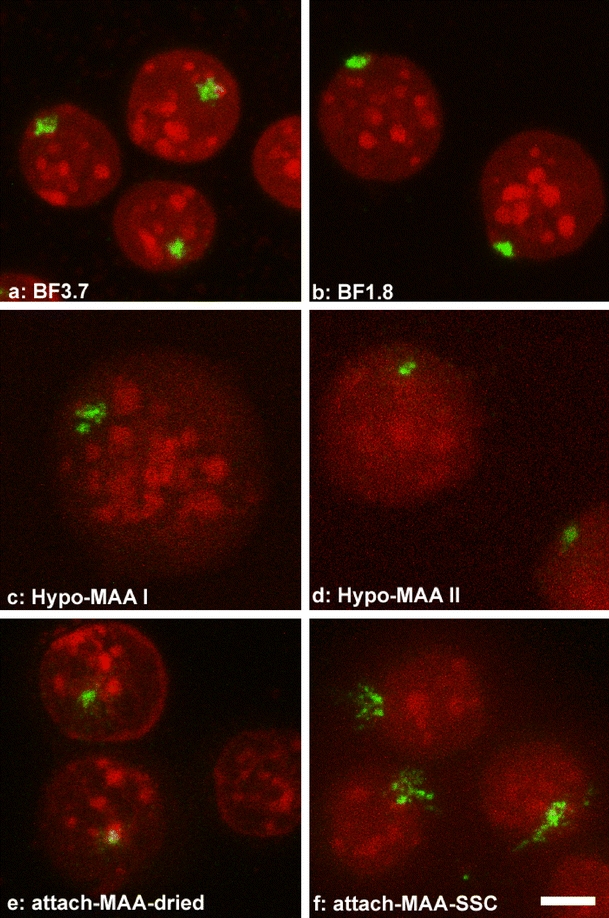
Nuclei of the MEL cell line PALZ39E subjected to different fixation methods after FISH. All images are shown at the same scale (bar 5 μm). The transgene array (green FISH signal) in this cell line is about 50 Mbp in size. While the FISH signals in BF-fixed nuclei show relatively little variation with a compact core (a, b), signals in Hypo-MAA-fixed cells range from divided (c) to compact (d). Signals in attach-MAA-dried cells (e) resembled those from BF-fixed cells, while in cells transferred from MAA to aqueous buffer (attach-MAA-SSC, (f)) signals had an exploded appearance. Nuclear shape and chromocenter appearance are similar to those observed directly after fixation (Fig. 1). DNA counterstain in red. Projections of confocal image stacks are shown
Fig. 4.
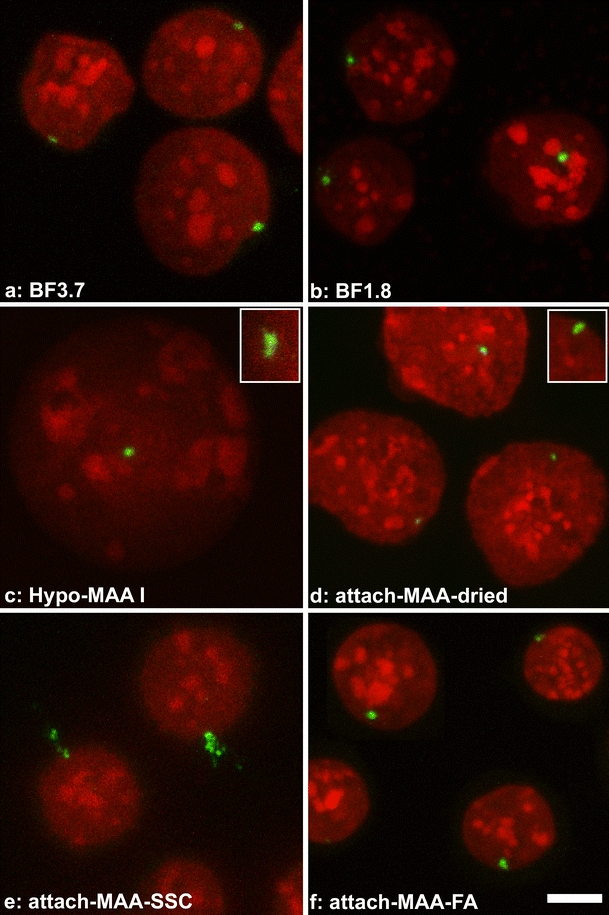
Nuclei of the MEL cell line PALZ39M after different fixation methods and FISH. All images are shown at the same scale (bar 5 μm). The small transgene array in this cell line (green FISH signal) appears dot-like in all preparations (a–d, f) except after transfer of cells from MAA to aqueous solution (attach-MAA-SSC, (e)). Insets show additional examples from other nuclei. See Table 1 for details on fixation methods. DNA counterstain in red. Projections of confocal image stacks are shown
Preservation of chromocenters
A characteristic of mouse cell nuclei is the clustering of their pericentromeric regions to so-called chromocenters (Hsu et al. 1971; Mayer et al. 2005 and references therein). In structurally preserved mouse nuclei, chromocenters are easily recognized when AT-specific DNA counterstains such as DAPI or TO-PRO-3 are applied. Accordingly, in BF-fixed nuclei, chromocenters appeared distinct with well-defined borders and an intensity high above other nuclear staining (Fig. 1a). When attached cells were fixed by immersion in MAA with or without air drying (attach-MAA-dried, attach-MAA-SSC, and attach-MAA-FA; see Table 1), the visual appearance of chromocenters in projections of confocal stacks was similar to formaldehyde-fixed nuclei (Fig. 1d–f), although attach-MAA-SSC nuclei appeared somewhat hazy (Fig. 1e). In nuclei subjected to hypotonic swelling followed by MAA fixation and dropping (Hypo-MAA I and II), however, the structural preservation of heterochromatin varied largely. While in a fraction chromocenters appeared normal, most nuclei appeared homogeneous or had only diffuse areas of brighter intensity with borders blurring into the nuclear background where chromocenters had been. Such examples are shown in Fig. 1b,c. Changes in chromocenter appearance were also found in ES cells (data not shown).
Described differences in preservation of chromocenters were confirmed by quantitative, automated computational image analyses of 3D confocal stacks (object counting, supplementary online material S2).
Comparison of FISH signals in MEL cells
Cells fixed according to procedures described above were subjected to FISH. The cell line PALZ39E (Fig. 3) contains a region of about 50 Mbp comprised of transgenes intermingled with host DNA (Dietzel et al. 2004). We labeled DNA of the plasmid used to make the transgenes and applied it as a FISH probe. Nuclei with FISH signals were randomly selected for microscopic recording, independent of signal quality, to minimize bias. Visual inspection of projections of confocal image stacks revealed differences in the structure of FISH signals, depending on the fixation procedure (Fig. 3). To quantify these differences, we categorized the signals as “compact” and “spread” (Table 2, series 1 and 2). Compact signals had a core homogeneous in intensity (Fig. 3a,b, and d). Spread signals had two or more areas of high signal intensity, separated by low-intensity or unstained regions (Fig. 3c,f).
Table 2.
Appearance of FISH signals of transgene arrays in MEL cells
| Series 1 | Series 2 | Series 3 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fixation procedure | BF 3.7 | Hypo-MAA II | Attach-MAA-dried | Attach-MAA-SSC | BF 1.8 | Hypo-MAA I | Hypo-MAA II | Attach-MAA-dried | Attach-MAA-SSC | BF 3.7 | BF 1.8 | Hypo-MAA I | Hypo-MAA II | Attach-MAA-dried | Attach-MAA-SSC | Attach-MAA-FA |
| Compact | 88 | 50 | 44 | 7 | 88 | 55 | 46 | 35 | 0 | 68 | 55 | 69 | 86 | 84 | 6 | 71 |
| Spread | 12 | 50 | 35 | 64 | 12 | 45 | 54 | 10 | 78 | 5 | 10 | 13 | 14 | 4 | 88 | 7 |
| Weak | – | – | 21 | 29 | – | – | – | 55 | 22 | 27 | 35 | 19 | – | 12 | 6 | 21 |
Series 1 and 2 were performed with the line PALZ39E (large transgene array), series 3 with PALZ39M (small transgene array). If FISH signals were bright enough, their appearance was classified as either compact or spread, otherwise as weak. All numbers are percentages. See Table 1 for explanation of fixation procedures.
In formaldehyde-fixed nuclei (BF 3.7 and BF 1.8), the majority of signals (88%) was compact (Table 2, series 1 and 2). Only 12% of signals were spread, leading to a ratio of about 7:1. In Hypo-MAA I and II nuclei, the percentage of spread signals about quadrupled, so that both types of appearances occurred approximately equally often (Table 2, series 1 and 2). In agreement with the differences in nuclear shape (see above), we found that FISH signals in hypotonically treated nuclei were flatter than those in formaldehyde-fixed nuclei (data not shown). In BF- as well as Hypo-MAA-fixed nuclei, all recorded FISH signals were intense enough to allow assignment to one of the two categories. This was not the case in preparations where cells attached to the coverslip had been fixed with MAA. In this study, the recorded FISH signals were sometimes too weak, preventing their categorization. In the remaining nuclei from attach-MAA-dried cells, the ratios of compact to spread signals were 1:1 and 3.5:1, respectively, in the two series of experiments (Table 2, series 1 and 2). In attach-MAA-SSC cells, signals were distributed over a large area, often with parts outside the nucleus, resulting in an “exploded” appearance (Fig. 3f). Differences in signal appearances were confirmed by automated quantitative 3D-image analysis using object counting (supplementary online material S2c,d).
In PALZ39M cells (Fig. 4), the region containing transgenes is much smaller than in PALZ39E cells (Dietzel et al. 2004). All fixation procedures led mostly to compact FISH signals (Fig. 4a–d, Table 2, series 3), with the exception of attach-MAA-SSC cells. In this study, signals again appeared exploded, often with parts outside the nucleus (Fig. 4e). We suspected this to be a consequence of the direct transfer from MAA to an aqueous solution due to the hygroscopic properties of MAA. We therefore included in this experimental series a protocol where the washing step in SSC was avoided and cells were transferred directly from MAA to formamide/SSC (attach-MAA-FA). Indeed, FISH signals had the typical dot-like shape in this preparation (Fig. 4f). Automated 3D-image analysis with object counting confirmed the visual impression of FISH signals in PALZ39M cells with on average between 1 and 1.1 objects for thresholds from 80 to 170 for all fixation procedures, with the exception of attach-MAA-SSC cells (data not shown).
Comparison of living cells with formaldehyde fixed cells
Preservation of morphology during fixation with BF has been demonstrated previously for other systems (see “Introduction”). In this study, we tested whether fixation with BF would also preserve the morphology of counterstained DNA and GFP-labeled transgene arrays in PALZ39E cell nuclei. Living cells were recorded three-dimensionally and then fixed on the microscope stage. After removal of the fixative and addition of buffer, previously recorded cells were relocated and recorded again. GFP signals of transgene arrays as well as counterstained nuclei were generally very similar before and after fixation (Figs. 5 and 1). Because a time lag between recording of optical sections and fixation is unavoidable, completely identical structures before and after fixation cannot be expected. Indeed, differences increased when the time interval between live cell imaging and fixation was prolonged. Images of GFP signals often had to be rotated to obtain the best possible match, suggesting a certain local movement of chromatin structures. No rotation was required to match images of nuclei. Measurements of height and width of nuclei (Fig. 5c) revealed a slight swelling upon formaldehyde fixation, in agreement with earlier findings (Solovei et al. 2002). Average width increased from 8.8 to 9.6 μm, and average height from 7.5 to 7.9 μm (n = 27). The lesser height of BF-fixed nuclei in this experiment compared to others (Fig. 2 and supplemental online material S1) is probably due to the prolonged attachment on poly-L-lysinated slides overnight. We conclude that fixation with BF preserves chromatin structure for nuclear subregions such as the GFP-labeled transgene array and for whole nuclei. Small changes in the positioning of chromatin regions may be due to intranuclear movement in the time period between recording and immobilization by fixation or due to a perturbation by the fixation itself.
Fig. 5.
Comparison of nuclei from living cells and after fixation with BF 3.7. Living PALZ39E cells were recorded with a widefield microscope, fixed on the microscope stage, and recorded again. Projections of consecutive optical sections are shown. Scale bar is valid for all micrographs. a GFP signals from the transgene array in living cells (top row) and fixed cells (bottom row). Projections from fixed GFP signals were rotated to match those from living cells as closely as possible. The time interval between recording of the GFP signal in living cells and the start of fixation was between approximately 1 min for the two leftmost signals and 5 min for the rightmost signal. Some differences in the distribution of substructures can be observed, in particular for the rightmost signal. However, in general, the morphology is very well maintained. b Nuclei stained with Hoechst 33342 in living cells (top) and after fixation (bottom). While overall shape and chromatin distribution is well maintained, some intensely stained regions have changed their position relative to each other, although only approximately 5 min have passed between recording and start of fixation. This is most obvious in the leftmost nucleus. c Shape of nuclei. As in Fig. 2, each dot represents one nucleus, either before or after fixation. The same 26 nuclei were measured. A slight shift to larger values can be observed for fixed nuclei
Chromosome territories in mouse embryonic stem cells
We next asked whether differences in morphology between formaldehyde and Hypo-MAA-fixed nuclei also occur in endogenous chromatin regions. Because large transgene arrays showed a stronger disruption in Hypo-MAA-fixed nuclei than small transgene arrays, we suspected that FISH-labeled chromosome territories might be more affected than FISH signals from small regions delineated by a BAC. We thus hybridized a mouse (Mus musculus) chromosome 7 (MMU7) paint probe together with a BAC for a region near the MMU7 q-arm telomere on mouse embryonic stem (ES) cells (Fig. 6). This gene-rich region on MMU7F5 was previously found at the edge of the MMU7 territory in Hypo-MAA I-fixed mouse ES cells (Mahy et al. 2002a).
Fig. 6.
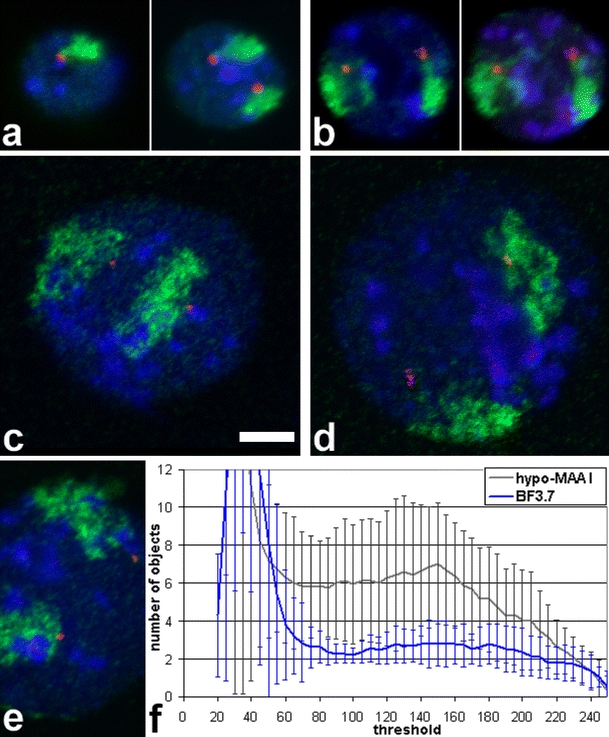
Dual color FISH on mouse ES cells with a paint probe to MMU7 (green), a BAC (red) and DNA counterstain (blue). All micrographs are at the same scale (bar 5 μm). While territories in BF-fixed nuclei (a, b) appear compact, territories after Hypo-MAA fixation (c–e) mostly have a disrupted, torn appearance which is in line with the increased diameter of their nuclei. In BF-fixed nuclei, BAC signals are connected to chromosome territory signals. For nuclei in (a) and (b), a single confocal section (left) and a projection of the stack (right) are shown. (c)–(e) are single confocal sections. (f) Object counting reveals a much higher disintegration of chromosome territories in Hypo-MAA-fixed nuclei (n = 33) than in BF-fixed nuclei (n = 39). While values below a threshold of 80 are influenced by nuclear background, above 200 chromosome territories start to fall below threshold. From 80 to 200, territories in BF-fixed nuclei show separation in about two objects, one for each chromosome territory, while in Hypo-MAA-fixed nuclei, the values are around 6. Also, the variation is much larger, indicated by the larger standard deviation (error bars)
When we compared confocal images from BF 3.7 and Hypo-MAA I-fixed nuclei, we observed a clear difference not only in nuclear shape but also in the appearance of painted chromosome territories (Fig. 6). While in formaldehyde-fixed nuclei territories were compact, in MAA-fixed nuclei, they appeared spread out, often disrupted, with borders difficult to define. FISH signals of the BAC probe revealed no noticeable differences in accordance with their smaller size and the results for transgene arrays described above. We measured the disruption of chromosome territories in MAA-fixed nuclei quantitatively by counting the number of independent objects in which the territories disintegrate when increasing thresholds were applied (Fig. 6f). While the two homologous territories in BF 3.7-fixed nuclei (n = 39) formed on average between two and three objects at a meaningful threshold range (see figure legend), in Hypo-MAA I-fixed nuclei (n = 33), six or more objects were detected (p < 0.001). As expected, no difference was found for BAC signals (data not shown).
We next measured the absolute distances of BAC signals to the surface of their chromosome territories (Fig. 7a,b). After both fixation methods, BAC signals were on average closer to the surface of the chromosome territory than the bulk signal of the territory (p < 0.001). However, while in BF 3.7 nuclei, 16% of BAC signal intensity was detected outside of the territories; in Hypo-MAA I-fixed nuclei, this fraction was 31% (p = 0.01 for the two distributions). This difference in distribution suggests an artificial looping out of peripheral regions to external areas during hypotonic shock, MAA fixation, dropping and/or air drying. Intensity weighted mean distances of territory signals to the territory surface were significantly smaller in MAA-fixed cells compared to formaldehyde-fixed cells (p < 0.001).
Fig. 7.
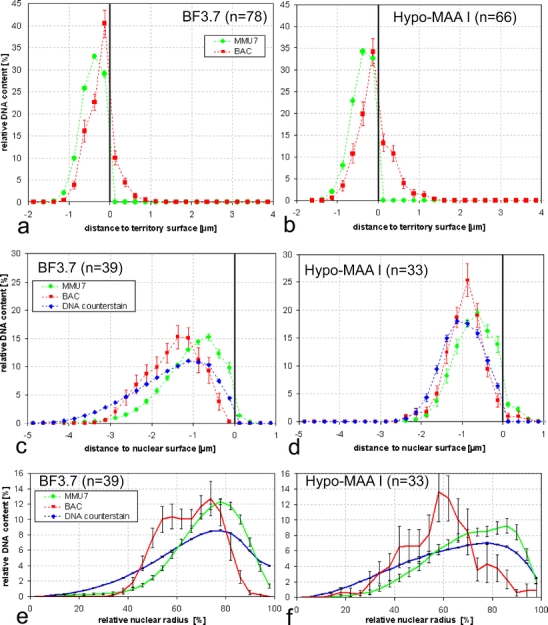
Quantitative distribution of FISH signals in mouse ES cells. a–d Absolute distances to the surface of segmented chromosome territories (a, b) or the cell nucleus (c, d) in BF 3.7 (a, c) and Hypo-MAA I-fixed (b, d) preparations. Voxels of each signal were assigned to layers of equal thickness (0.25 μm). Each dot represents the average relative signal content of one such layer. Negative distances are inside the reference structure, positive distances outside. See “Materials and methods” for details. e, f Relative radial distribution of FISH signals and DNA counterstain in BF 3.7 (e) and Hypo-MAA I-fixed nuclei (f). The method applied (Cremer et al. 2001) assigns each voxel of the nuclear volume to one of the 25 shells, each with the same thickness along a ray from the nuclear center to the edge. The outermost shell is fitted to the nuclear edge, and inner shells are adapted accordingly. The distribution of the DNA counterstain in BF 3.7-fixed ES cell nuclei (e) was very similar to the one previously reported (Mayer et al. 2005), confirming reproducibility. Hypo-MAA I-fixed cells (f) differed significantly (p < 0.001) and resembled more the distribution in structurally preserved, formaldehyde-fixed fibroblasts with their flatter nuclei (Mayer et al. 2005) with less DNA in outer shells and more in inner shells. BAC signals were also relatively more internal than their harboring territories after both fixation methods (p < 0.001 for BF 3.7, p < 0.005 for Hypo-MAA I). Nuclei included in this study were selected for separate MMU7 territories. This excludes nuclei where both MMU7 territories are in the center, thus causing a bias toward more external position
Measurements of absolute distances from FISH signals and nuclear counterstain to the surface of the nucleus (Fig. 7c,d) confirmed that Hypo-MAA I-fixed nuclei were flattened. While in the latter counterstained DNA was within 2.5 μm of the nuclear surface, in BF 3.7-fixed nuclei, distances farther than 4 μm away from the surface were reached (p < 0.001). Notably, in formaldehyde-fixed nuclei, no BAC signals and only 1.6% of territory signal intensity were found outside the segmented nucleus, while the respective percentages for MAA-fixed nuclei were 2.2% for BAC signal intensity and 5.9% for territory signal intensity, indicating a dislocation of these structures. After both fixation methods, BAC signals were found on average further away from the nuclear edge, toward the interior, than territory signals (p < 0.001), indicating a certain robustness of radial distributions in the nucleus against distortions. Measurements of relative radial distributions (Fig. 7e,f) confirmed differences found with absolute distances.
Discussion
In this study, we investigated preservation of chromatin structure after different fixation procedures. We quantitatively measured and compared nuclear shape and chromocenter morphology directly after fixation and after FISH as well as morphology of FISH signals. Previous studies demonstrated a well-maintained nuclear morphology after fixation with BF in the absence of detergent (Cremer et al. 1993; Kurz et al. 1996; Shelby et al. 1996; Kanda et al. 1998; Verschure et al. 1999; Mahy et al. 2002b; Solovei et al. 2002). Our current comparison of live and BF-fixed cells confirms these results. This fixation method therefore provides a gold standard against which other fixation methods must measure up. In the present study, preparations made with hypotonically swollen, MAA-fixed, dropped and air-dried nuclei (Hypo-MAA I and II) showed the following artifacts: (1) nuclear width was increased while height decreased. This has been documented earlier (Kozubek et al. 2000), and the flattening is also reflected in the term “2D-FISH” used for this procedure (Croft et al. 1999); (2) the organization of chromocenters, which in mouse contain the subcentromeric repetitive sequences, was severely disturbed; (3) in contrast to FISH signals from a BAC or a small transgene array which showed similar appearance in formaldehyde and Hypo-MAA-fixed cells, larger structures such as a ∼50-Mbp transgene array or a chromosome territory disintegrated and got a spread appearance after hypotonic shock and MAA fixation; (4) BAC signals in hypotonically treated nuclei were found more often outside their chromosome territory than in formaldehyde-fixed cells; and (5) chromosome territories and BAC signals had higher percentages of signal outside the nucleus. Our results strongly discourage the assumption that hypotonically treated, MAA-fixed nuclei faithfully represent in vivo large-scale chromatin organization.
Several studies investigating nuclear architecture in Hypo-MAA-fixed preparations also have included smaller samples of formaldehyde-fixed nuclei. Despite the artifacts induced by hypotonic MAA fixation, the radial nuclear distributions of chromosome territories were remarkably stable (Croft et al. 1999; Bridger et al. 2000; Boyle et al. 2001 and the current study). The spatial positioning of nearby structures relative to each other, however, was affected. The gene-rich MHC locus looped out of its chromosome territory more often in MAA-fixed nuclei than in formaldehyde-fixed nuclei (Volpi et al. 2000). The same was found for the EDC locus (Williams et al. 2002). In contrast, several probes in a region of moderate gene density on human chromosome 11p13 showed a rather interior position in Hypo-MAA-fixed lymphoblast nuclei, while in BF-fixed nuclei, they were closer to the territory edge (Mahy et al. 2002b). This difference decreased when distances were normalized by territory size. The authors assumed that territories in hypotonically treated, MAA-fixed cells were swollen. However, in a follow-up study on the gene-rich region 11p15, five of nine sites investigated with both fixation methods were more external in Hypo-MAA-fixed nuclei, while the others had a similar distance to the territory edge after both fixation methods (Mahy et al. 2002a). The interpretation was that Hypo-MAA fixation may preferentially loosen or decondense chromatin located at the surface of chromosome territories or that formaldehyde fixation condenses these regions. The dislocation was not linear and thus unpredictable: two sites that were outside in 30 and 28% of BF-fixed nuclei were found outside in 43 and 72% of Hypo-MAA-fixed nuclei, respectively (Mahy et al. 2002a). In mouse embryonic stem cells, the Hoxb1 gene was found more often outside of its harboring chromosome 11 territory in hypotonically treated, MAA-fixed cells than in cells fixed with formaldehyde after permeabilization (Chambeyron and Bickmore 2004).
In our study, we found a change of the relative positioning of BAC-delineated sequences relative to their chromosome territory, a dispersion of chromocenters into the surrounding nuclear volume in a majority of Hypo-MAA-fixed cells, and a disaggregation of chromosome territories. In combination, the present data suggest that in Hypo-MAA-fixed cells, chromatin is unpredictably dispersed but dispersion is restricted to an area around the original position. Because this effect is not spatially directed, whole chromosome territories maintain their relative nuclear position except for adaptations to the distorted nuclear shape.
The observed artifacts in large-scale chromatin structure in hypotonically treated, MAA-fixed cells may be more or less pronounced for different genomic regions or in different cell types. It is possible, that “open” chromatin structures are more easily dislocated than silent, compact chromatin structures. MMU7 is the second most gene-rich chromosome in mouse, and the region delineated by the BAC is particularly gene rich (http://www.ensembl.org). Thus, one could assume that less gene-rich regions or territories may show less pronounced artifacts. However, such an assumption would have to be tested for every sequence investigated, voiding advantages of potentially easier protocols.
It has been argued that hypotonically treated, MAA-fixed nuclei are projections of the in vivo situation, and true distances between sites can be obtained from resulting data by mathematical correction (Yokota et al. 1995). It appears, however, that artifacts induced by Hypo-MAA fixation are more complex than a flattening of nuclei. Still, results from such nuclei may be useful when direct comparisons are made, for example, between genes before and after induction. One should be aware, however, that both of these distributions may differ significantly from the in vivo situation. With this inherent limitation, it may be more promising to perform such experiments on structurally preserved cells in the first place.
Fixation by formaldehyde has been shown to depend on concentration and incubation time (Linden et al. 1997; Guillot et al. 2004). In the present study, we tested fixation for 10 min in 3.7% as well as 15 min in 1.8% BF. While beta-galactosidase activity is largely destroyed in the first case, it is well detectable in the latter (Cheng et al. 1999), arguing for a weaker fixation. However, we did not detect differences in structural preservation after the two procedures. Apparently, differences in fixation strength are not large enough to induce significant differences in resistance against permeabilization or denaturation of the nuclei during FISH. The study originally introducing the GFP-lac repressor–lac operator system compared chromatin structure of transgene arrays in live cells and after BF fixation and FISH, describing “noticeable blurring of the fine structures” (Robinett et al. 1996). In this study, fixation was for 3 h in 2.5% BF and denaturation was in 70% formamide at 80°C for 10 min. While fixation thus was much stronger than in our study, denaturation also was much harsher, most likely contributing to the observed changes. In our protocol, we could observe reduced preservation of nuclear morphology when denaturation at 75°C in 50% formamide was increased from 2 to 4 min (data not shown). Some studies have used formaldehyde fixation after permeabilization of the cells. Then, however, chromatin preservation is not ensured because permeabilization of unfixed cells can severely impair chromatin structure (Belmont et al. 1989; Mongelard et al. 1999).
Increased diameter parallel to the slide and flattening orthogonal to it in Hypo-MAA-fixed nuclei can be uncoupled as shown by our data on attached nuclei which were immersed in MAA with subsequent air drying without hypotonic treatment. Here, the nuclei were flattened but had normal diameters in xy. In projections of confocal image stacks, chromocenters looked very similar to those in formaldehyde-fixed cells. This suggests that disruption of chromatin structure is a consequence of the hypotonic shock rather than of MAA fixation per se. However, FISH signals in attached cells fixed with MAA (without hypotonic shock) were often weak, even for the large transgene array, despite its repetitive character. We therefore assume that the hypotonic shock facilitates access of the DNA probe to the target sequence. If the hypotonic shock is omitted, additional permeabilization steps, as they are used in formaldehyde fixations, might help to enhance signal strength but would also reduce the ease of handling currently offered by these protocols when compared to formaldehyde fixation. In addition, preservation of morphology of the large transgene array did not reach the quality of formaldehyde-fixed preparations, as indicated by a larger ratio of distributed to compact transgene FISH signals (Table 2).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Parameters of nuclear shape. Nuclei were evaluated in 3 independent series of experiments directly after fixation or after FISH. Given are mean values (mv), standard deviation (sd), medians of maximal nuclear width (w) and height (h), the ratio (r) of width/height and the number of examined nuclei (n) in each experiment (DOC 44 kb)
Below is the image is a link to a high resolution version.
{kind=link}
(JPEG 147 kb)
High resolution image file (TIFF 658 kb)
Acknowledgments
We thank C. Cremer for supporting JvH from his funding. We thank C. Bonifer for providing mouse ES cells, W. Bickmore for providing a detailed protocol for “procedure I”, and T. Thormeyer for providing the EDMT software. We thank T. Cremer for continuous support and helpful discussions. This work was financially supported by the Deutsche Forschungsgemeinschaft.
Footnotes
Electronic supplementary material
Supplementary material is available in the online version of this article at http://dx.doi.org/10.1007/s00412-006-0084-2 and is accessible for authorized users.
References
- Albiez H, Cremer M, Tiberi C, Vecchio L, Schermelleh L, Dittrich S, Küpper K, Joffe B, Thormeyer T, von Hase J, Yang S, Rohr K, Leonhardt H, Solovei I, Cremer C, Fakan S, Cremer T (2006) Chromatin domains and the interchromatin compartment form structurally defined and functionally interacting nuclear networks. Chromosome Res 14 (in press) [DOI] [PubMed]
- Belmont AS, Braunfeld MB, Sedat JW, Agard DA (1989) Large-scale chromatin structural domains within mitotic and interphase chromosomes in vivo and in vitro. Chromosoma 98:129–143 [DOI] [PubMed]
- Boyle S, Gilchrist S, Bridger JM, Mahy NL, Ellis JA, Bickmore WA (2001) The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum Mol Genet 10:211–219 [DOI] [PubMed]
- Bridger JM, Boyle S, Kill IR, Bickmore WA (2000) Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr Biol 10:149–152 [DOI] [PubMed]
- Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG (1997) Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 91:845–854 [DOI] [PubMed]
- Chambeyron S, Bickmore WA (2004) Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev 18:1119–1130 [DOI] [PMC free article] [PubMed]
- Cheng G, Thompson RP, Gourdie RG (1999) Improved detection reliability of beta-galactosidase in histological preparations. Biotechniques 27:438–440 [DOI] [PubMed]
- Cremer T, Kurz A, Zirbel R, Dietzel S, Rinke B, Schröck E, Speicher MR, Mathieu U, Jauch A, Emmerich P, Scherthan H, Ried T, Cremer C, Lichter P (1993) Role of chromosome territories in the functional compartmentalization of the cell nucleus. Cold Spring Harb Symp Quant Biol 58:777–792 [DOI] [PubMed]
- Cremer M, von Hase J, Volm T, Brero A, Kreth G, Walter J, Fischer C, Solovei I, Cremer C, Cremer T (2001) Non-random radial higher-order chromatin arrangements in nuclei of diploid human cells. Chromosome Res 9:541–567 [DOI] [PubMed]
- Cremer T, Küpper K, Dietzel S, Fakan S (2004) Higher order chromatin architecture in the cell nucleus: on the way from structure to function. Biol Cell 96:555–567 [DOI] [PubMed]
- Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA (1999) Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol 145:1119–1131 [DOI] [PMC free article] [PubMed]
- Deng W, Tsao SW, Lucas JN, Leung CS, Cheung AL (2003) A new method for improving metaphase chromosome spreading. Cytometry A 51:46–51 [DOI] [PubMed]
- Dernburg AF, Sedat JW (1998) Mapping three-dimensional chromosome architecture in situ. In: Berrios M (ed) Methods in cell biology: nuclear structure and function. Academic, New York, pp 187–233 [DOI] [PubMed]
- Dietzel S, Zolghadr K, Hepperger C, Belmont AS (2004) Differential large-scale chromatin compaction and intranuclear positioning of transcribed versus non-transcribed transgene arrays containing beta-globin regulatory sequences. J Cell Sci 117:4603–4614 [DOI] [PubMed]
- Faust N, König S, Bonifer C, Sippel AE (1997) Transgene analysis in mouse embryonic stem cells differentiating in vitro. In: Adolph KW (ed) Human genome methods. CRC Press, Boca Raton, FL, pp 185–205
- Foster HA, Bridger JM (2005) The genome and the nucleus: a marriage made by evolution. Genome organisation and nuclear architecture. Chromosoma 114:212–229 [DOI] [PubMed]
- Fraschini A, Pellicciari C, Biggiogera M, Manfredi Romanini MG (1981) The effect of different fixatives on chromatin: cytochemical and ultrastructural approaches. Histochem J 13:763–769 [DOI] [PubMed]
- Guillot PV, Xie SQ, Hollinshead M, Pombo A (2004) Fixation-induced redistribution of hyperphosphorylated RNA polymerase II in the nucleus of human cells. Exp Cell Res 295:460–468 [DOI] [PubMed]
- Hell SW, Stelzer EHK (1995) Lens aberations in confocal fluorescence microscopy. In: Pawley JB (ed) Handbook of biological confocal microscopy. Plenum, New York, pp 347–354
- Hendzel MJ, Bazett-Jones DP (1997) Fixation-dependent organization of core histones following DNA fluorescent in situ hybridization. Chromosoma 106:114–123 [DOI] [PubMed]
- Hsu TC, Cooper JE, Mace ML Jr, Brinkley BR (1971) Arrangement of centromeres in mouse cells. Chromosoma 34:73–87 [DOI] [PubMed]
- Kanda T, Sullivan KF, Wahl GM (1998) Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 8:377–385 [DOI] [PubMed]
- Kiernan JA (2000) Formaldehyde, formalin, paraformaldehyde and glutaraldehyde: what they are and what they do. Microscopy Today 00-1:8–12
- Kobliakova I, Zatsepina O, Stefanova V, Polyakov V, Kireev I (2005) The topology of early- and late-replicating chromatin in differentially decondensed chromosomes. Chromosome Res 13:169–181 [DOI] [PubMed]
- Kozubek S, Lukasova E, Amrichova J, Kozubek M, Liskova A, Slotova J (2000) Influence of cell fixation on chromatin topography. Anal Biochem 282:29–38 [DOI] [PubMed]
- Kurz A, Lampel S, Nickolenko JE, Bradl J, Benner A, Zirbel RM, Cremer T, Lichter P (1996) Active and inactive genes localize preferentially in the periphery of chromosome territories. J Cell Biol 135:1195–1205 [DOI] [PMC free article] [PubMed]
- Lawrence JB, Singer RH (1985) Quantitative analysis of in situ hybridization methods for the detection of actin gene expression. Nucleic Acids Res 13:1777–1799 [DOI] [PMC free article] [PubMed]
- Linden E, Skoglund P, Rundquist I (1997) Accessibility of 7-aminoactinomycin D to lymphocyte nuclei after paraformaldehyde fixation. Cytometry 27:92–95 [DOI] [PubMed]
- Mahy NL, Perry PE, Bickmore WA (2002a) Gene density and transcription influence the localization of chromatin outside of chromosome territories detectable by FISH. J Cell Biol 159:753–763 [DOI] [PMC free article] [PubMed]
- Mahy NL, Perry PE, Gilchrist S, Baldock RA, Bickmore WA (2002b) Spatial organization of active and inactive genes and noncoding DNA within chromosome territories. J Cell Biol 157:579–589 [DOI] [PMC free article] [PubMed]
- Mayer R, Brero A, von Hase J, Schroeder T, Cremer T, Dietzel S (2005) Common themes and cell type specific variations of higher order chromatin arrangements in the mouse. BMC Cell Biol 6:44 [DOI] [PMC free article] [PubMed]
- Mongelard F, Vourc’h C, Robert-Nicoud M, Usson Y (1999) Quantitative assessment of the alteration of chromatin during the course of FISH procedures. Fluorescent in situ hybridization. Cytometry 36:96–101 [DOI] [PubMed]
- Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE (2000) Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science 290:138–141 [DOI] [PubMed]
- Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, Fraser P (2004) Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet 36:1065–1071 [DOI] [PubMed]
- Raap AK, Marijnen JG, Vrolijk J, van der Ploeg M (1986) Denaturation, renaturation, and loss of DNA during in situ hybridization procedures. Cytometry 7:235–242 [DOI] [PubMed]
- Rabbitts P, Impey H, Heppell-Parton A, Langford C, Tease C, Lowe N, Bailey D, Ferguson-Smith M, Carter N (1995) Chromosome specific paints from a high resolution flow karyotype of the mouse. Nat Genet 9:369–375 [DOI] [PubMed]
- Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, Belmont AS (1996) In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol 135:1685–1700 [DOI] [PMC free article] [PubMed]
- Roix JJ, McQueen PG, Munson PJ, Parada LA, Misteli T (2003) Spatial proximity of translocation-prone gene loci in human lymphomas. Nat Genet 34:287–291 [DOI] [PubMed]
- Shelby RD, Hahn KM, Sullivan KF (1996) Dynamic elastic behavior of alpha-satellite DNA domains visualized in situ in living human cells. J Cell Biol 135:545–557 [DOI] [PMC free article] [PubMed]
- Solovei I, Cavallo A, Schermelleh L, Jaunin F, Scasselati C, Cmarko D, Cremer C, Fakan S, Cremer T (2002) Spatial preservation of nuclear chromatin architecture during three- dimensional fluorescence in situ hybridization (3D-FISH). Exp Cell Res 276:10–23 [DOI] [PubMed]
- Spector DL (2003) The dynamics of chromosome organization and gene regulation. Annu Rev Biochem 72:573–608 [DOI] [PubMed]
- Sproul D, Gilbert N, Bickmore WA (2005) The role of chromatin structure in regulating the expression of clustered genes. Nat Rev Genet 6:775–781 [DOI] [PubMed]
- Stadler S, Schnapp V, Mayer R, Stein S, Cremer C, Bonifer C, Cremer T, Dietzel S (2004) The architecture of chicken chromosome territories changes during differentiation. BMC Cell Biol 5:44 [DOI] [PMC free article] [PubMed]
- Sumner AT, Evans HJ, Buckland RA (1973) Mechanisms involved in the banding of chromosomes with quinacrine and Giemsa. I. The effects of fixation in methanol–acetic acid. Exp Cell Res 81:214–222 [DOI] [PubMed]
- Telenius H, Pelmear AH, Tunnacliffe A, Carter NP, Behmel A, Ferguson-Smith MA, Nordenskjold M, Pfragner R, Ponder BA (1992) Cytogenetic analysis by chromosome painting using DOP-PCR amplified flow-sorted chromosomes. Genes Chromosomes Cancer 4:257–263 [DOI] [PubMed]
- Verschure PJ, van Der Kraan I, Manders EM, van Driel R (1999) Spatial relationship between transcription sites and chromosome territories. J Cell Biol 147:13–24 [DOI] [PMC free article] [PubMed]
- Volpi EV, Chevret E, Jones T, Vatcheva R, Williamson J, Beck S, Campbell RD, Goldsworthy M, Powis SH, Ragoussis J, Trowsdale J, Sheer D (2000) Large-scale chromatin organization of the major histocompatibility complex and other regions of human chromosome 6 and its response to interferon in interphase nuclei. J Cell Sci 113:1565–1576 [DOI] [PubMed]
- Williams RR, Broad S, Sheer D, Ragoussis J (2002) Subchromosomal positioning of the epidermal differentiation complex (EDC) in keratinocyte and lymphoblast interphase nuclei. Exp Cell Res 272:163–175 [DOI] [PubMed]
- Yokota H, van den Engh G, Hearst JE, Sachs RK, Trask BJ (1995) Evidence for the organization of chromatin in megabase pair sized loops arranged along a random walk path in the human G0/G1 interphase nucleus. J Cell Biol 130:1239–1249 [DOI] [PMC free article] [PubMed]
- Young IT (1977) Proof without prejudice:use of the Kolmogorov–Smirnov test for the analysis of histograms from flow systems and other sources. J Histochem Cytochem 25:935–941 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
Parameters of nuclear shape. Nuclei were evaluated in 3 independent series of experiments directly after fixation or after FISH. Given are mean values (mv), standard deviation (sd), medians of maximal nuclear width (w) and height (h), the ratio (r) of width/height and the number of examined nuclei (n) in each experiment (DOC 44 kb)
Below is the image is a link to a high resolution version.
(JPEG 147 kb)
High resolution image file (TIFF 658 kb)