

Abstract
The organization, distribution, and function of neural progenitor cells (NPCs) in the adult spinal cord during motor neuron degeneration in amyotrophic lateral sclerosis (ALS) remain largely unknown. Using nestin promoter controlled LacZ reporter transgenic mice and mutant G93A-SOD1 transgenic mice mimicking ALS, we showed that there was an increase of NPC proliferation, migration, and neurogenesis in the lumbar region of adult spinal cord in response to motor neuron degeneration. The proliferation of NPCs detected by BrdU incorporation and LacZ staining was restricted to the ependymal zone surrounding central canal (EZ). Once the NPCs moved out from the EZ, they lost the proliferative capability, but maintained migratory function vigorously. During ALS-like disease onset and progression, NPCs in the EZ migrated initially toward the dorsal horn direction, and then to the ventral horn regions, where motor neurons have degenerated. More significantly, there was an increased de novo neurogenesis from NPCs during ALS-like disease onset and progression. The enhanced proliferation, migration, and neurogenesis of (from) NPCs in the adult spinal cord of ALS-like mice may play an important role in attempting to repair the degenerated motor neurons and restore the dysfunctional circuitry which resulted from the pathogenesis of mutant SOD1 in ALS.
Keywords: Neural progenitor cells, radial glia, glial progenitor cells, motor neurons, nestin, mutant SOD1, ALS
Introduction
The mechanisms of motor neuron degeneration in ALS remain largely unknown and effective therapy for ALS is not yet available (1,2). Mutations of SOD1 cause 2–5% of human ALS (3–5). Transgenic rodents overexpressing mutant SOD1 develop motor neuron degeneration mimicking human ALS (6–9). Thus, these transgenic animals have provided the best model to date for ALS studies (10). The existence of NPCs and neurogenesis from NPCs in the adult CNS have been well-established (11–14). For example, neurogenesis generates functional neurons in vitro from adult human and primate brain (15–17). In addition, pathological processes promote neurogenesis as reported in human patients with Alzheimer’s disease (18), and Huntington’s disease (19). Brain and spinal cord injury facilitates neurogenesis in traumatic animal models (20–23). These findings suggest that newly generated neurons may be able to functionally replace degenerated (damaged) cells during pathogenesis or injury. In fact, neural stem cell-mediated neuronal regeneration has been proposed, and used in some cases, for therapy of ALS and other neurodegenerative diseases (24–27). Nevertheless, the role of adult NPCs in response to motor neuron degeneration in ALS remains unknown. More significantly, the status and the differences of NPCs in the normal and ALS disease have not been defined. To this end, we utilized the animal model mimicking human ALS to identify and characterize NPCs associated with disease onset and progression. Expression of nestin in the CNS is generally considered a reliable NPC marker and has been extensively used for the characterization of NPCs in vitro and in vivo (28–32). For these reasons, we have combined the nestin promoter driven LacZ reporter transgenic (pNes-Tg) mouse model (33,34) with the mutant SOD1-transgenic (G93A-SOD1-Tg) mouse model (9) to analyze the responses of NPCs to motor neuron degeneration in the ALS-like mice.
Materials and Methods
Transgenic mouse lines
Nestin promoter driven LacZ repoter transgenic mice (pNes-Tg) and mutant SOD1 mimicking human ALS transgenic mice (G93A-SOD1-Tg) (Jackson Laboratory, Bar Harbor, ME) were used to generate bi-transgenic mice containing both LacZ and G93A-SOD1 (Bi-Tg) through heterozygous breeding. Transgenic progeny were identified by regular PCR amplification of tail DNA using specific primers. Like mutant G93A-SOD1 transgenic mice, the Bi-Tg mice had ALS-like disease onset and disease progression at 70–90 and 100–130 days of age respectively. Because mutant SOD1-mediated motor neuron degeneration in ALS is inherited in an autosomal dominant fashion, the transgenic mice used in this study were heterozygous. Age-matched littermates of pNes-Tg mice were used as controls. The experimental protocols were approved by the Institutional Animal Use and Care Committee and are in close agreement with the National Institutes of Health guideline for the care and use of laboratory animals.
In vivo 5-bromodeoxyuridine (BrdU) labeling
BrdU at 50 mg/kg/day was intraperitoneally (IP) administrated for 4, 9 and 14 days respectively to adult Bi-Tg and age-matched pNes-Tg mice. Spinal cords were dissected out 1 day after the last injection of BrdU and processed for BrdU immunohistochemical staining as described in the following section. To identify and define the increased number of NPCs in the dorsal and ventral regions was derived from the EZ of the ALS-like spinal cords, mice were pulsed with 50 mg/kg/day of BrdU by IP for 25 days following by 5 days of chasing. The percentage of BrdU labeled cells in LacZ positive cells were determined after LacZ staining and BrdU immunohistochemical staining.
LacZ staining, immunohistochemical staining, image analysis and quantification
The lumbar region of the spinal cord was used to analyze the organization and distribution of NPCs in response to motor neuron degeneration. For LacZ staining, sections (12 μm) were incubated in 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) solution for 16 h at room temperature as previously described. For immunohistochemical staining, sections were incubated in blocking buffer (10% goat serum/0.2% triton X-100 in 1×PBS, pH 7.5) for 1 h at room temperature. Primary antibody was then added to the blocking buffer (1:250) and the section was incubated at 4 °C overnight. The next day, sections were washed 5 times (5 min each) in 1×PBS (pH 7.5) containing 0.5% triton X-100, followed by incubation with specific fluorescein-conjugated secondary antibody for 2 h at room temperature. After extensive washes, sections were covered with anti-fade medium and sealed for fluorescent microscopic analysis. For negative control staining, sections were incubated without primary antibody.
All images were collected and analyzed with a Nikon fluorescent microscope E800 equipped with the Spot digital camera and Photoshop software.
Quantifications of LacZ staining intensity at the ependymal zone surrounding central canal (EZ) of Bi-Tg mouse spinal cord during disease free, disease onset and disease progression compared with that of age-matched littermate control pNes-Tg mice were performed with the NIH software Image J. At least 3 sections/mouse, and 3 mice at specific ages were analyzed. The arbitrary units were used to express the LacZ staining intensity of EZ. To quantify the distribution of NPCs in the adult ALS-like mice compared to that of age-matched controls, all LacZ positive cells (nuclei) in the dorsal and ventral horn regions (5 sections/region/mouse and 3 mice/group) were manually counted. Statistical analysis was performed using the paired Student t test. P < 0.05 was considered significant.
Results
Organization and distribution of NPCs in the normal and ALS-like adult mouse spinal cord
A combination of LacZ reporter (LacZ), nestin expression (Nestin) and BrdU labeling (BrdU) were used to identify NPCs in the adult ALS-like mouse spinal cord (35,36). Adult (40 to 130 days of age) control mice (pNes-Tg) and ALS-like bi-transgenic mice (Bi-Tg) containing both LacZ and G93A-SOD1 transgenes were injected with BrdU to characterize the organization and distribution of NPCs in the spinal cord. We and others have defined the ALS-like mouse lifespan into clinical disease free (before 60 days of age), disease onset (70–90 days of age) and disease progression (100–130 day of age) stages (9,10). We focused on the lumbar regions (L1 to L5) of the spinal cord to identify and characterize NPCs, because the ALS-like mice have clinical manifestation of hindlimb paralysis and spinal cord motor neuron degeneration (9,10). Figures 1A to 1C show that the EZ contains proliferative NPCs positively stained with LacZ, nestin and BrdU antibodies. Systematic analyses of the organization of NPCs in the different ages of pNes-Tg and Bi-Tg mice (n= 3 in each age group) demonstrated that the BrdU, LacZ and nestin positive NPCs were primarily localized in the EZ of the adult spinal cord (12). More significantly, there was an increase of LacZ staining intensity (Figs. 2A–2C) in the EZ of the ALS-like mice (Bi-Tg) compared to the age-matched control mice (pNes-Tg), suggesting that there is an increase of NPC proliferation.
Figure 1. Identification and characterization of NPCs in the adult ALS-like (Bi-Tg) and normal (pNes-Tg) mouse spinal cord.
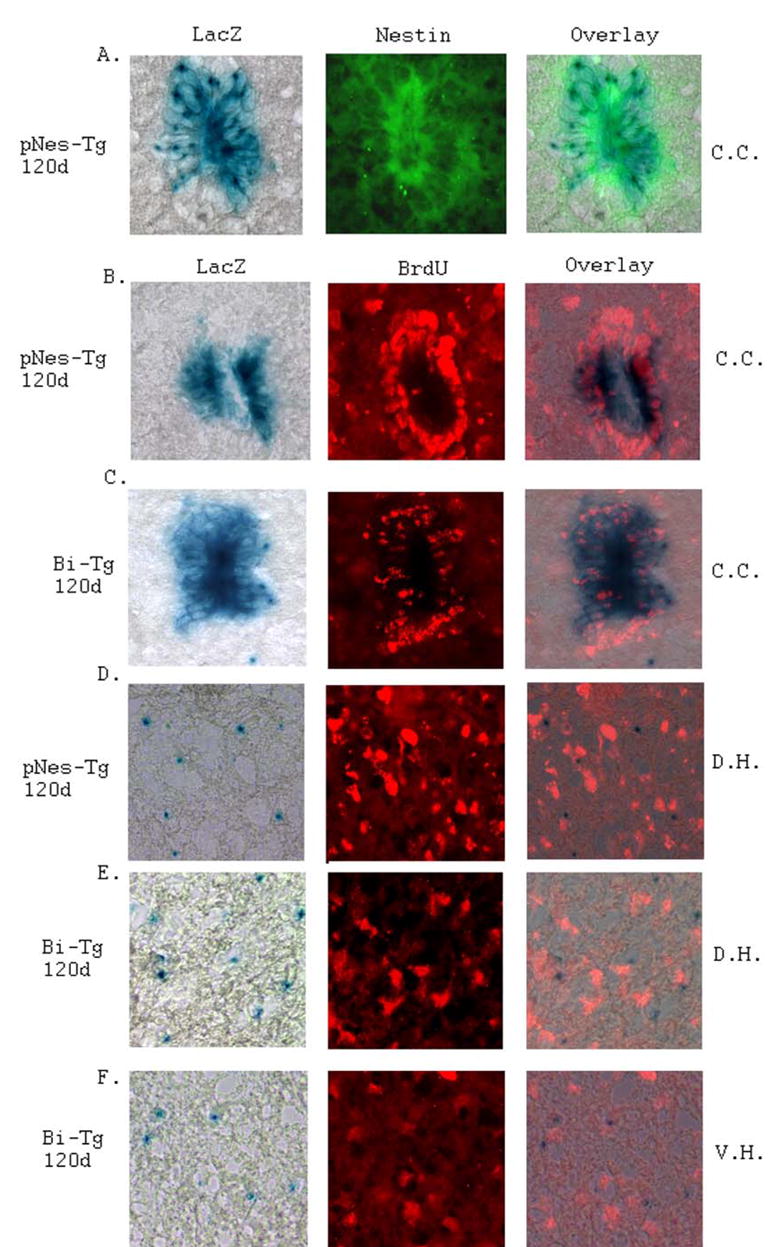
Identification of NPCs in the ependymal zone surrounding central canal region (C.C.) of adult mouse spinal cord was performed by LacZ staining and nestin immunostaining (A). Characterization of NPCs in the ependymal zone surrounding central canal (C.C.: B and C), dorsal horn region (D.H.: D and E) and ventral horn region (V.H.: F) of pNes-Tg (B and D) and Bi-Tg (C, E and F) mouse spinal cord was conducted by LacZ staining and BrdU labeling. Notably, some of the LacZ positively stained NPCs in the ependymal zone were labeled with BrdU. In contrast, the NPCs from dorsal and ventral horn regions of the pNes-Tg and Bi-Tg mouse spinal cord were not labeled with BrdU.
Figure 2. LacZ staining intensity in the ependymal zone surrounding central canal of the pNes-Tg and Bi-Tg mouse spinal cord.
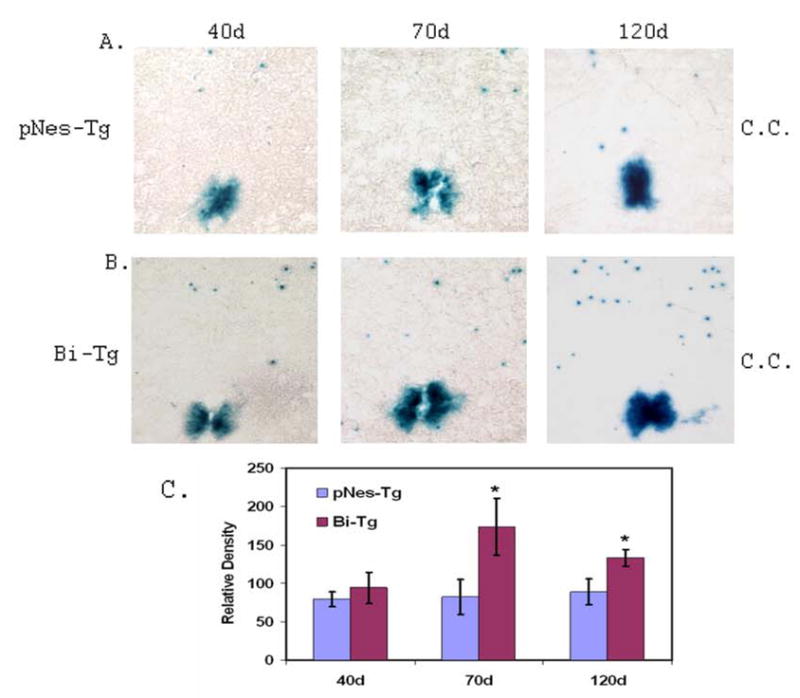
The LacZ staining intensity in the ependymal zone surrounding central canal regions (C.C.) of ALS-like (B: Bi-Tg) mouse spinal cord during disease free (40d), disease onset (70d) and progression (120d) stages was compared with that of age-matched littermate control mice (A: pNes-Tg). The relative LacZ staining intensity in the ependymal zone surrounding central canal regions was presented in C (5 sections/mouse, n=3 mice; *p< 0.05).
In addition, we have also identified the LacZ stained NPCs distributed in other areas of the adult mouse spinal cord (Figs. 1D–1F, and Figs. 3A–3E). The majority of the NPCs were found in the Lamina I–III of the dorsal horns, although there were a few NPCs sparsely distributed in other regions of the normal control spinal cord. Notably, NPCs outside of the EZ were not labeled with BrdU (Figs. 1D–1F) in contrast to the NPCs in the EZ (Figs. 1B–1C), suggesting that the NPCs outside of the EZ were not proliferative. Systematic analyses with different ages of animals showed that there were more NPCs in the dorsal horn region than in the ventral horn region of the spinal cord (Fig. 3). In particular, there were more NPCs in the ALS-like (Bi-Tg) mouse spinal cord than that of the age-matched littermate control mice (pNes-Tg), suggesting that the increased number of NPCs was associated with the motor neuron degeneration in the ALS-like mouse model (Figs. 1D–1F and Fig. 3).
Figure 3. Organization and distribution of NPCs in the lumbar regions of the pNes-Tg and Bi-Tg mouse spinal cord.
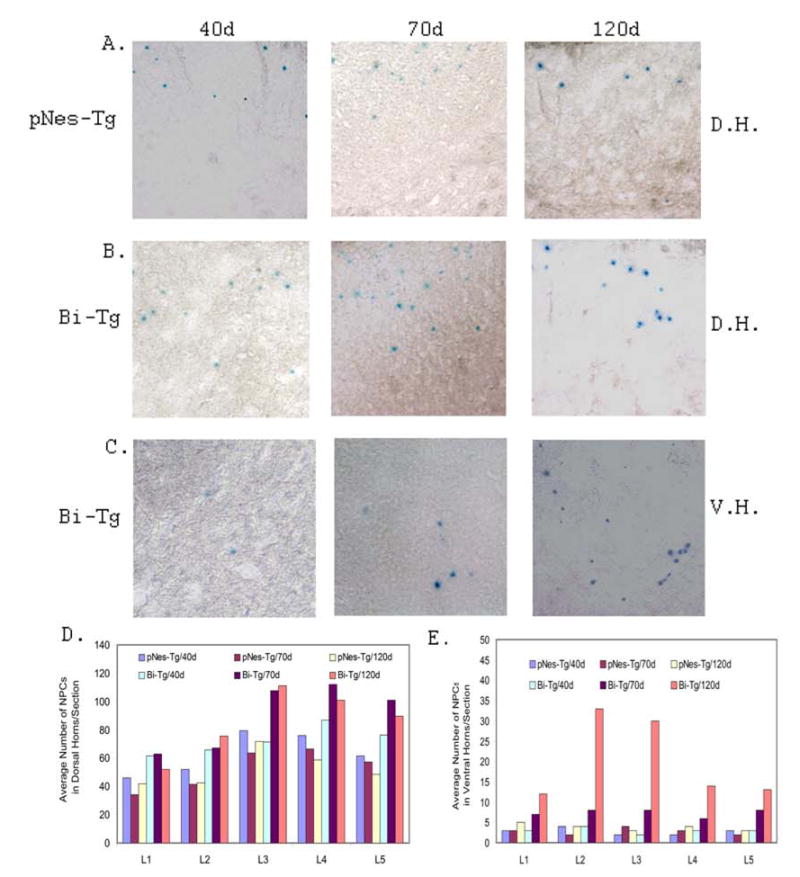
The representative distribution of NPCs in the dorsal horn region (D.H.: A and B) and ventral horn region (V.H.: C) of the pNes-Tg (A) and Bi-Tg (B and C) mouse spinal cord corresponding to clinical disease free (40d), disease onset (70d) and disease progression (120d) was characterized by LacZ staining. The number of NPCs in the dorsal horn region (D) and ventral horn region (E) of the adult mouse spinal cord was counted and statistically analyzed from 5 sections/lumbar region/mouse (n=3 mice).
Based on the well-established clinical and pathological manifestations, we have analyzed the distribution of NPCs in the spinal cord of the Bi-Tg mice at 40, 70 and 120 days of age, which are correspondent to disease free, disease onset and disease progression (9,10). Notably, there were dynamic changes of NPCs in the spinal cord of the Bi-Tg mice during the ALS-like disease onset and progression as compared to the control pNes-Tg mice and Bi-Tg mice at the clinical disease free stage (Fig. 3). The distribution of NPCs in the dorsal horn and ventral horn regions at different time points was shown in Figs. 3D and 3E respectively. The increased number of NPCs in the dorsal and ventral horn areas during the ALS-like disease onset and progression was not from de novo proliferation, because these NPCs were not labeled with BrdU even up to 15 days of pulsing (Fig. 1). However, these cells were highly migratory and could mobilize an immediate response to spinal cord injury (manuscript in preparation) and motor neuron degeneration in the ALS-like mice. This is the first report on the identification of the dormant NPCs in the dorsal and ventral horn regions of the adult spinal cord. The increased number of NPCs in these areas of the Bi-Tg mouse spinal cord was largely attributed to the migration of pre-existing NPCs from the EZ (Figs. 4 to 6).
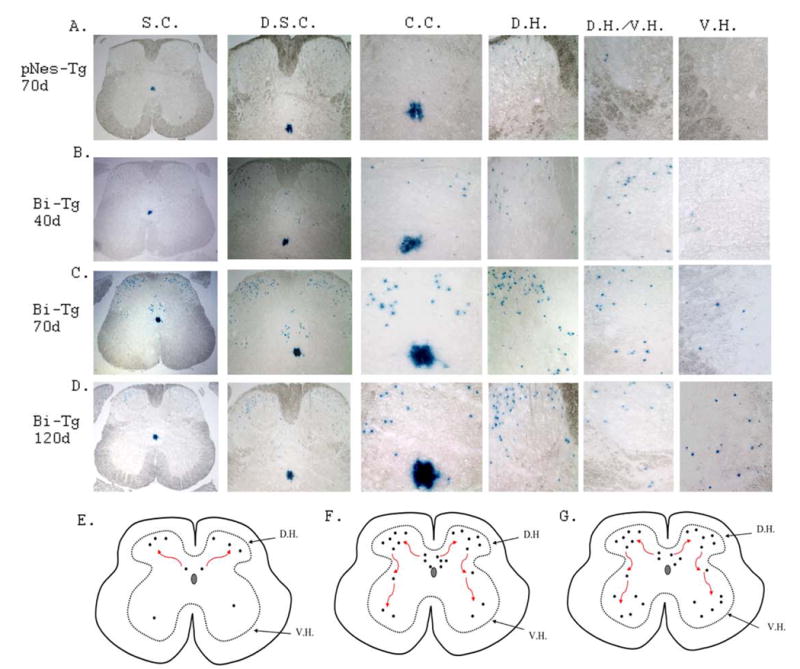
Figure 4. LacZ staining images demonstrating NPC migration patterns.
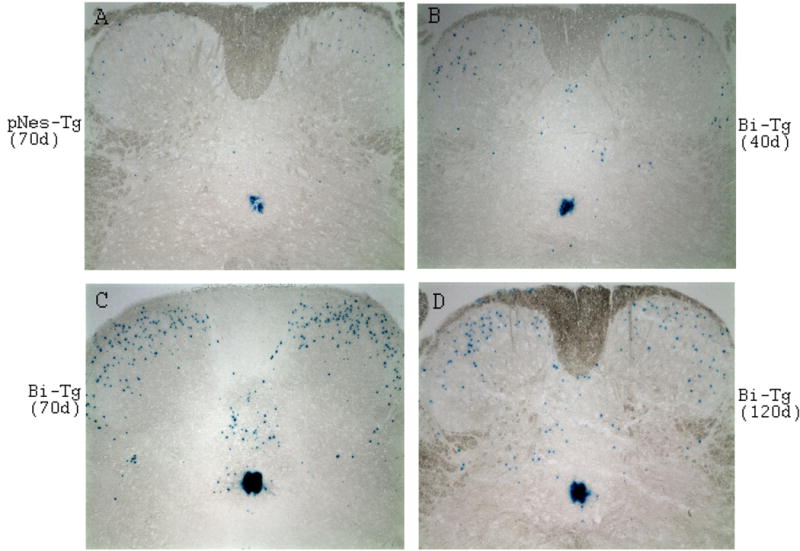
A few NPCs in control pNes-Tg mice at age of 70 days of age (A), while increased number of NPCs in Bi-Tg mice during disease free (B: 40 days of age), disease onset (C: 70 days of age) and disease progression (D: 120 days of age) migrated out from the ependymal zone surrounding central canal toward dorsal direction, and subsequently to ventral regions.
Figure 6. Identification and determination of the source of increased NPCs in the dorsal and ventral horn regions of the adult Bi-Tg mouse spinal cords in response to motor neuron degeneration in ALS-like mice.
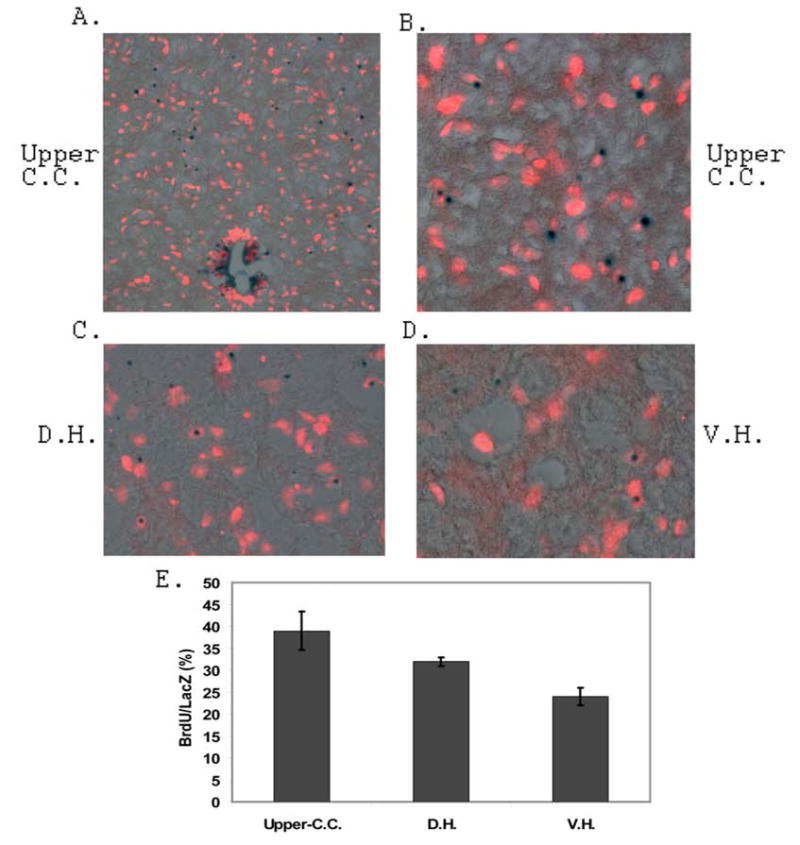
Analysis of the source of increased NPCs in the dorsal and ventral horn regions of the ALS-like mouse spinal cords was carried out by 25 days of BrdU pulse labeling and 5 days of chasing experiments. The BrdU labeled and LacZ stained NPCs in the upper region of central canal, dorsal horn and ventral horn regions of the 115 days of age Bi-Tg mice were shown in A, B, C and D respectively. The ratio of BrdU labeled LacZ positive cells in the 115 days of age Bi-Tg mice were shown in E (3 sections/mouse, n=3 mice).
Migration and migratory paths of NPCs in the adult ALS-like mouse spinal cord during clinical disease free, disease onset and disease progression stages
In a separate study, we showed that spinal cord injury promoted migration of pre-existing NPCs from the central canal toward the dorsal region, where the lesion occurs (manuscript in preparation). We hypothesized that NPCs generated in the EZ of ALS-like mouse spinal cord might migrate to the ventral direction directly in response to motor neuron degeneration. To this end, Bi-Tg mice were assessed to characterize NPC migration and migratory paths during ALS-like disease onset and progression compared to age-matched littermate control (pNes-Tg) mice. In contrast with our original assumption, NPCs from the EZ did not migrate directly toward the degenerated ventral motor neuron domain in the ALS-like mice. Instead, NPCs initially migrated from the EZ toward the dorsal horn direction and then, some of them migrated to the ventral horn regions (Figs. 4A–4D and Figs. 5A–5D). More significantly, the number of NPCs migrating out from the EZ to the dorsal, and from dorsal to ventral regions was dramatically increased during ALS-like disease onset and progression (Figs. 4C–4D and Figs. 5C–5D). In fact, even in the disease-free stage (40 days of age), the number of NPCs migrating in the dorsal region in ALS-like mice was also increased compared to age-matched control mice (Figs. 4B and 5B, and data not shown). In contrast, there are only a few NPCs migrating out from the EZ to the dorsal horn, and no NPCs were detected migrating further towards the ventral horn in normal adult (pNes-Tg) mouse spinal cord (Figs 4A and 5A).
Figure 5. Migration and migratory paths of NPCs in the adult mouse spinal cord in response to motor neuron degeneration in ALS-like mice.
The migration of NPCs characterized by LacZ staining from the ependymal zone surrounding central canal region to the dorsal direction, and subsequently to ventral direction in control pNes-Tg mice (A: at 70 days of age), in ALS-like Bi-Tg mice during disease free (B: at 40 days of age), disease onset (C: at 70 days of age) and disease progression (D: at 120 days of age). The migratory paths of NPCs at different stages were presented in E (for normal and clinical disease free stages), F (for disease onset) and G (for disease progression) respectively.
The migratory paths of NPCs in normal, clinical disease free, disease onset and disease progression were presented in Figs. 5E–5G based on the analyses of NPC organization and distribution in different stages of ALS-like mice (Figs. 4A–4D, and Figs. 5A–5D). To identify and confirm vigorously the increased number of NPCs in the dorsal and ventral regions was derived from the EZ of the central canal, we further carried out experiments with 25 days of BrdU pulse labeling and 5 days of chasing in the ALS-like (Bi-Tg) mice. We demonstrated that there was an increase of BrdU labeling in the LacZ positive cells located in the upper central canal (Figs. 6A, 6B and 6E), dorsal horn (Figs. 6C and 6E) and ventral horn (Figs. 6D and 6E) regions. Thus, the increased migration of NPCs from the EZ led to the increased numbers of NPCs in the dorsal and ventral areas of the spinal cord during the ALS-like disease onset and progression (Fig. 3, and Fig. 4). More interestingly, several NPCs migrated to the ventral horn regions were in the vicinity of degenerated motor neurons in the adult Bi-Tg mouse spinal cord (Fig.8F). Though the specific mechanisms of NPC migration and NPC migratory paths in response to motor neuron degeneration remain to be determined, it is likely that these NPCs were mobilized to attempt to functionally replace (repair) the degenerated motor neurons.
Figure 8. De Novo neurogenesis from NPCs in the Bi-Tg mouse spinal cord compared to that of pNes-Tg mice.
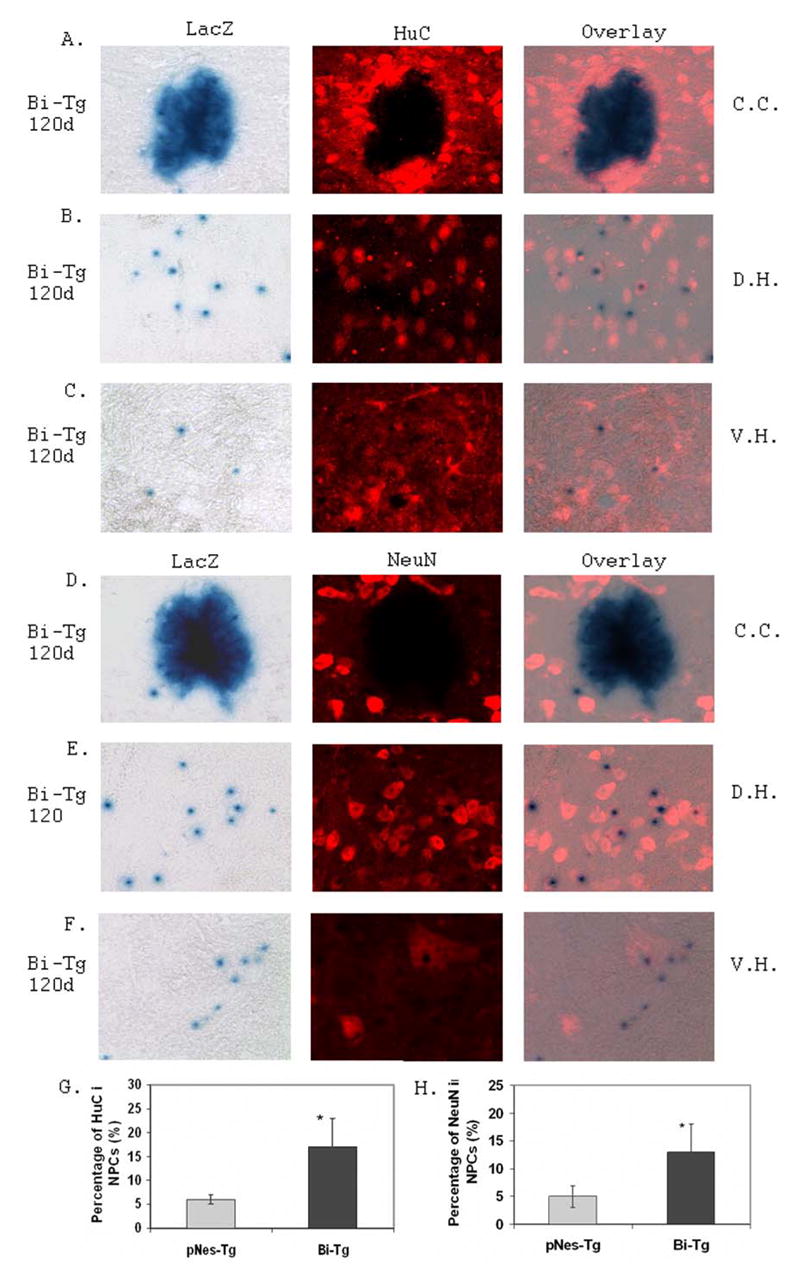
Analysis of neurogenesis from NPCs was carried out by immunostaining of spinal cord sections with pre-neuronal marker, HuC (A, B, and C), and neuronal marker, NeuN (D, E, and F) in the ependymal zone surrounding central canal region (C.C.: A and D), dorsal horn region (D.H.: B and E) and ventral horn region (V.H.: C and F). There was an increased of neuronal differentiation as ALS-like disease onset (data not shown) and progression (G and H) were advanced in the Bi-Tg mice compared to that of the age-matched control pNes-Tg mice (*p< 0.05).
Expression of CXCR4, a SDF-1 receptor, was demonstrated to associate with neural stem cell migration (37,38). For this reason, we analyzed the expression of CXCR4 to test its potential role in adult NPC migration in response to motor neuron degeneration in the ALS-like mouse model (Bi-Tg) compared to that of age-matched littermate control mice (pNes-Tg). We demonstrated that the expression of CXCR4 was dramatically increased in the adult spinal cords of ALS-like mice during disease onset at the age of 70 days old (Figs. 7B and 7D) and progression (data not shown) compared to the age-matched control normal mice (Figs. 7A and 7C). There was almost no detection of CXCR4 staining in age-matched littermate control and disease free stage of ALS mice (40 days of age) (data not shown). More importantly, some NPCs were shown to express CXCR4 as ALS-like disease onset (Figs. 7B and 7D) and progression (Fig. 7E, and data not shown). The percentage of CXCR4 positive staining in the NPCs outside of the EZ in the spinal cords is shown in Fig. 7E.
Figure 7. CXCR4 expression and NPC migration in the Bi-Tg mouse spinal cords compared to that of control pNes-Tg mice.
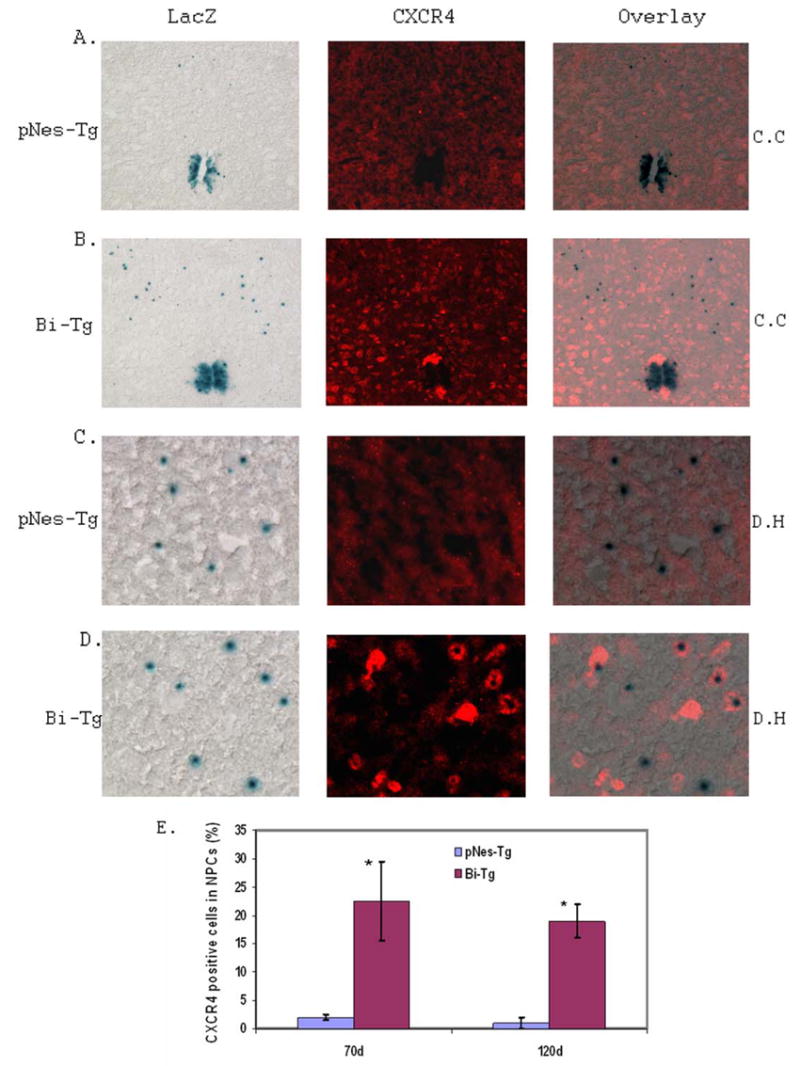
Analysis of the potential role of CXCR4 expression in NPC migration was carried out by immunostaining of spinal cord sections with anti-CXCR4 antibody. There was an increase of CXCR4 staining as ALS-like disease onset (B and D: at 70 days of age) compared to that of the age-matched control pNes-Tg mice (A and C: at 70 days of age). The representative staining in the central canal and adjacent areas (A and B) and dorsal horn regions (C and D) were shown. The percentage of CXCR4 positive cells out of NPCs in the spinal cord sections during disease onset (at 70 days of age) and progression (at 120 days of age) was shown in F (*p< 0.05).
De Novo neurogenesis from NPCs in response to motor neuron degeneration during the ALS-like disease onset and progression
To further study the potential functionality of the NPCs in response to motor neuron degeneration, we have analyzed the possibility of neurogenesis, astrogenesis and oligogenesis from the NPCs in the ALS-like mouse model. An increase of de novo neurogenesis from NPCs outside of the EZ emerged during the ALS-like disease onset and progression as determined with pre-neuronal and neuronal markers, HuC (Figs. 8A–8C and 7G), TuJ1 (data not shown) and NeuN (Figs. 8D–8F and 8H) respectively. No neurogenesis was detected in the EZ with these neuronal markers. Similarly, we also analyzed astrogenesis and oligogenesis using the specific astrocyte and oligodendrocyte markers respectively (Fig. 9). To a large extent, there was no astrogenesis and oligogenesis detected from the NPCs using GFAP (Fig. 9A), NG-2 (Fig. 9B) and Olig-2 (Fig. 9C) immunostaining respectively. In addition, there was no microgliogenesis from the NPCs as detected with OX-42 immunostaining (Fig. 9D). These results collectively support an increase of de novo neurogenesis from the NPCs in the adult mouse spinal cord, as motor neuron degeneration advanced in the ALS-like mice.
Figure 9. Gliogenesis and NPCs in the Bi-Tg mouse spinal cords compared to that of pNes-Tg mice.
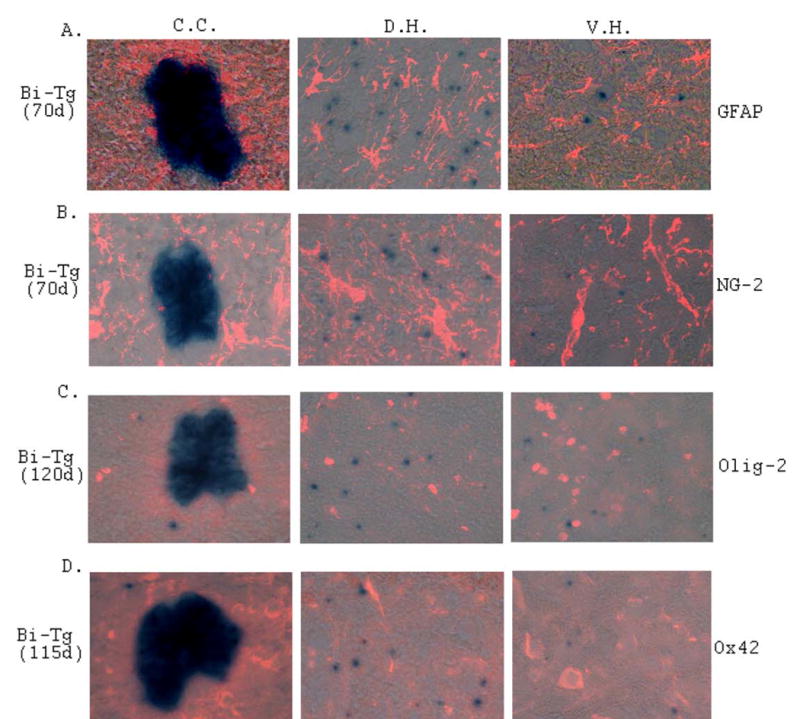
Analyses of astrogenesis (A and B), oligogenesis (C) and microgaliogenesis (D) in the spinal cord of ALS-like (Bi-Tg) mice with specific cell type markers. No astrogenesis, oligogenesis and microgaliogenesis were detected from NPCs at the disease onset (A and B: 70 days of age) and disease progression (C and D: 115–120 days of age) stages.
Discussion
Although many lines of evidence have demonstrated that NPCs are present in the adult CNS, the dynamic responses of NPCs to neurodegeneration at disease onset and progression across lifespan remain largely unexplored. Animal models mimicking human degenerative diseases would be particularly useful to gain the significant insights in this respect (39–41). To this end, we have focused our effort to analyze the organization and distribution of NPCs in the ALS-like transgenic mouse model (Bi-Tg) compared with the age-matched littermate pNes-Tg mouse controls. Selection of the Bi-Tg mouse model for this study was primarily based on two major characteristics of the newly generated mouse line through breeding: First, the Bi-Tg mice express mutant G93A-SOD1 and develop ALS-like disease mimicking human ALS. More importantly, the phenotype and pathology of the Bi-Tg mice across their lifespan are very similar to the parental mutant G93A-SOD1 transgenic mouse line previously reported (9,10); Second, the Bi-Tg mice contain nestin promoter controlled LacZ reporter gene, which specifically expresses LacZ protein product in NPCs (33,34). Together, the unique features of the Bi-Tg mice allow us to identify and characterize the responses of NPCs in the adult CNS to motor neuron degeneration in the ALS-like model.
The present study using the Bi-Tg mice demonstrates three major findings of NPCs in the adult CNS in relation to ALS-like disease: 1). Mutant SOD1-mediated motor neuron degeneration enhances NPC proliferation in the EZ of the Bi-Tg mouse spinal cord; 2). Motor neuron degeneration promotes NPC migration from the EZ toward the dorsal horn, and subsequently to the ventral horn during ALS disease onset and progression; 3). Motor neuron degeneration increases the generation of neuron-like cells from NPCs in the spinal cord compared with the basal levels of neurogenesis. Thus, this study provides compelling evidence that the pathological processes of motor neuron degeneration stimulate NPC proliferation, migration and neurogenesis in ALS-like mouse model.
In light of these findings, we have also identified and characterized two closely related populations of NPCs in the adult mouse spinal cord. One population of NPCs was proliferative as determined with BrdU incorporation. These NPCs were at an undifferentiated state and were primarily localized in the EZ (Figs. 1, 2 and 4). Notably, the LacZ staining intensity was evidently increased in the EZ of ALS-like mice (Bi-Tg mice), particularly during the disease onset and progression, compared to that of age-matched normal littermate controls (Figs. 2 and 4). The LacZ staining and BrdU labeling together suggested that there was an increase of NPC proliferation in response to motor neuron degeneration (Figs. 1 and 2). Another population of NPCs was distributed sparsely, but unevenly across spinal cord, majority of which were in the dorsal horn regions. These NPCs were not proliferative, because they were not labeled with BrdU pulsing for 15 days (Fig. 1). Interestingly, the number of NPCs distributed in the dorsal horn region (Lamina I–III areas) was much more than in the ventral horn and other regions (Figs. 1 and 3). Most significantly, there was a dramatic increase in the number of NPCs in both the dorsal and ventral horn regions in the ALS-like mice compared to normal control mice (Fig. 3). To the best of our knowledge, this is the very first report on the organization and distribution of NPCs in the normal and ALS-like adult mouse spinal cords. The significance of the predominant distribution of NPCs in dorsal horn region compared to the ventral horn region remains unknown. Based on the distribution (Fig. 3), migratory patterns (Figs. 4 and 5), and long-term of BrdU pulse and chase labeling (Fig. 6), we concluded that the NPCs in the dorsal and ventral regions were derived from the proliferative NPCs in the EZ. Once migrated out of the EZ, the NPCs lost proliferative ability but maintained migratory function. The increased NPC proliferation in the EZ and the increased number of NPCs in the dorsal and ventral horn regions are the specific responses to motor neuron degeneration in the ALS-like mice.
Compared to the age-matched littermate control pNes-Tg mice, the migration of NPCs from the EZ was greatly enhanced in Bi-Tg mice, even at the clinical disease free stage (Figs. 4 and 5). The maximum migration of NPCs in the Bi-Tg mice across their lifespan occurred at disease onset stage, during which there was a maximum of NPC distribution in the dorsal horn region (Figs. 4 and 5). At disease progression stage, more NPCs migrated to the ventral horn region. The migratory pattern of NPCs in response to motor neuron degeneration during disease onset and progression is intriguing. Because motor neuron degeneration is in the ventral motor domain, we initially hypothesized that NPCs from the EZ may migrate directly to the ventral horn direction. Different from our hypothesis, NPCs from the EZ migrated to dorsal region first, and then, some of which migrated to the ventral region subsequently. Such a temporospatially regulated migration pattern of NPCs in the adult spinal cord in response to disease and/or traumatic injury has not been reported. However, during embryonic spinal cord developmental or early postnatal stages, the oligodendrocyte precursor cells (OPCs) defined by NKX6.1, NKX2.2, or Olig1/2 were shown to have specific migratory pattern (42–46). Nevertheless, the OPCs initiated from ventral domain, moved down in ventral direction briefly, and then migrated toward the dorsal direction along the peripheral area of spinal cord. The migratory patterns of OPCs at embryonic and early postnatal stages, and NPCs at adult stages, particularly during ALS-like disease onset and progression were quite different. The intrinsic mechanism(s) governing NPC migration and migratory patterns in the spinal cord of the ALS-like mice remain elusive. Further analysis with immunohistochemical staining suggested that the expression of CXCR4 in NPCs may contribute, at least partially to the NPC migration (Fig. 7). Several reports have demonstrated that chemokine receptors participate in NPC migration. For example, expression of CXCR4 receptor directed the migration of neural stem cells to the lesioned sites in the CNS injury model (47). However, the increased CXCR4 receptor expression in the spinal cords of ALS-like mice appears to be the global response of neurons and glia to motor neuron degeneration by mutant SOD1 effects. With respect to the current finding in NPC migration and migratory pathways, what and how chemokine(s)/chemokine receptor(s) or other molecule(s) participate in the directionality of NPC migration remain to be defined. Functionally, the temporospatially organized migratory pathways may have advantages in repairing the dysfunctional circuitry involved in not only motor neurons, but other cell types as well.
One important, but has not been unambiguously resolved issue of the current study is the lineage of the adult NPCs we have identified with the LacZ positive staining. Apparently, the adult NPCs defined by nestin promoter controlled LacZ reporter staining are different from glial precursor cells (GPCs) identified from embryonic stages in that the GPCs do, while NPCs do not express immature and mature markers of astrocyte and oligodendrocytes (48–50). Thus, the NPCs and GPCs represent two distinguished cell type populations. However, the NPCs and GPCs may be developmentally derived from the same or similar origin (progenitor cells), because both are positive in nestin staining. We showed that some of the NPCs differentiated into neurons, but not into astrocytes and oligodendrocytes in adult brain (data not shown) and spinal cord (Figs. 8 and 9, and additional discussion in next section). More importantly, the percentage of NPCs that differentiated into neurons was increased in ALS-like mice as disease onset and progression were advanced. To a large extent, the NPCs identified in this study may be derived from radial glia, that have been demonstrated to give rise to adult neural stem cells and adult neuronal cells in vivo (51,52). Because the adult NPCs in this study differentiate primarily into neurons, not astrocytes or oligodendrocytes, we think that these adult NPCs represent a population of radial glia derivatives with a default characteristic of differentiation potential toward neuronal direction.
During the ALS-like disease onset and progression, there was an increase of neurogenesis, but not astrogenesis, oligogenesis or microgliogeneis from NPCs as detected with specific cell type markers respectively (Figs. 8 and 9). Enhancement of neurogenesis has been observed in animal models of Alzheimer’s disease (53) and ischemic stroke (54). In addition, increased neurogenesis has been reported in patients with Alzheimer’s disease (18) and Huntington’s disease (19). In the ALS-like mouse model, we demonstrated that motor neuron degeneration promoted neurogenesis from NPCs in the mouse spinal cord. Interestingly, there were NPCs in the vicinity of some dying motor neurons (Fig. 8F), suggesting that some factor(s) from degenerated motor neurons may induce NPC migration and differentiation. Thus, identifying the factors that promote NPC migration and differentiation may contribute to delay or prevent ALS disease onset and progression and enhance survival. Though the molecular mechanisms governing the proliferation, migration and neurogenesis of adult NPCs in the ALS-like mice remain to be defined, the present study will allow us to explore the therapeutic potential of stimulating de novo neurogenesis for functional replacement of degenerated neurons in ALS and other neuron degenerative diseases.
Acknowledgments
This study was supported in part by U.S. Public Health Service Grants AG23923, NS45829, HL75034 and Muscular Dystrophy Association grant 3334.
Reference List
- 1.Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996–2000. J Neurol Neurosurg Psychiatry. 2003;74:1258–1261. doi: 10.1136/jnnp.74.9.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks BR. Diagnostic dilemmas in amyotrophic lateral sclerosis. J Neurol Sci. 1999;165 :S1–S9. doi: 10.1016/s0022-510x(99)00019-2. Suppl 1. [DOI] [PubMed] [Google Scholar]
- 3.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 4.Rosen DR, Sapp P, O'Regan J, et al. Genetic linkage analysis of familial amyotrophic lateral sclerosis using human chromosome 21 microsatellite DNA markers. Am J Med Genet. 1994;51:61–69. doi: 10.1002/ajmg.1320510114. [DOI] [PubMed] [Google Scholar]
- 5.Cleveland DW. From Charcot to SOD1: mechanisms of selective motor neuron death in ALS. Neuron. 1999;24:515–520. doi: 10.1016/s0896-6273(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 6.Nagai M, Aoki M, Miyoshi I, et al. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruijn LI, Becher MW, Lee MK, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 8.Dunlop J, Beal MH, She Y, Howland DS. Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis. J Neurosci. 2003;23:1688–1696. doi: 10.1523/JNEUROSCI.23-05-01688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 10.Liu R, Althaus JS, Ellerbrock BR, Becker DA, Gurney ME. Enhanced oxygen radical production in a transgenic mouse model of familial amyotrophic lateral sclerosis. Ann Neurol. 1998;44:763–770. doi: 10.1002/ana.410440510. [DOI] [PubMed] [Google Scholar]
- 11.Clarke D, Frisen J. Differentiation potential of adult stem cells. Curr Opin Genet Dev. 2001;11:575–580. doi: 10.1016/s0959-437x(00)00235-5. [DOI] [PubMed] [Google Scholar]
- 12.Clarke DL, Johansson CB, Wilbertz J, et al. Generalized potential of adult neural stem cells. Science. 2000;288:1660–1663. doi: 10.1126/science.288.5471.1660. [DOI] [PubMed] [Google Scholar]
- 13.Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisen J. Identification of a neural stem cell in the adult mammalian central nervous system. Cell. 1999;96:25–34. doi: 10.1016/s0092-8674(00)80956-3. [DOI] [PubMed] [Google Scholar]
- 14.Lie DC, Song H, Colamarino SA, Ming GL, Gage FH. Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu Rev Pharmacol Toxicol. 2004;44:399–421. doi: 10.1146/annurev.pharmtox.44.101802.121631. [DOI] [PubMed] [Google Scholar]
- 15.Roy NS, Wang S, Jiang L, et al. In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nat Med. 2000;6:271–277. doi: 10.1038/73119. [DOI] [PubMed] [Google Scholar]
- 16.Bedard A, Parent A. Evidence of newly generated neurons in the human olfactory bulb. Brain Res Dev Brain Res. 2004;151:159–168. doi: 10.1016/j.devbrainres.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 17.Bernier PJ, Bedard A, Vinet J, Levesque M, Parent A. Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc Natl Acad Sci U S A. 2002;99:11464–11469. doi: 10.1073/pnas.172403999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin K, Peel AL, Mao XO, et al. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curtis MA, Penney EB, Pearson AG, et al. Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc Natl Acad Sci U S A. 2003;100:9023–9027. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tattersfield AS, Croon RJ, Liu YW, Kells AP, Faull RL, Connor B. Neurogenesis in the striatum of the quinolinic acid lesion model of Huntington's disease. Neuroscience. 2004;127:319–332. doi: 10.1016/j.neuroscience.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 21.Rice AC, Khaldi A, Harvey HB, et al. Proliferation and neuronal differentiation of mitotically active cells following traumatic brain injury. Exp Neurol. 2003;183:406–417. doi: 10.1016/s0014-4886(03)00241-3. [DOI] [PubMed] [Google Scholar]
- 22.Jin K, Sun Y, Xie L, et al. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci. 2003;24:171–189. doi: 10.1016/s1044-7431(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 23.Yagita Y, Kitagawa K, Ohtsuki T, et al. Neurogenesis by progenitor cells in the ischemic adult rat hippocampus. Stroke. 2001;32:1890–1896. doi: 10.1161/01.str.32.8.1890. [DOI] [PubMed] [Google Scholar]
- 24.Silani V, Cova L, Corbo M, Ciammola A, Polli E. Stem-cell therapy for amyotrophic lateral sclerosis. Lancet. 2004;364:200–202. doi: 10.1016/S0140-6736(04)16634-8. [DOI] [PubMed] [Google Scholar]
- 25.Silani V, Fogh I, Ratti A, Sassone J, Ciammola A, Cova L. Stem cells in the treatment of amyotrophic lateral sclerosis (ALS) Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:173–181. doi: 10.1080/146608202760839001. [DOI] [PubMed] [Google Scholar]
- 26.Goh EL, Ma D, Ming GL, Song H. Adult neural stem cells and repair of the adult central nervous system. J Hematother Stem Cell Res. 2003;12:671–679. doi: 10.1089/15258160360732696. [DOI] [PubMed] [Google Scholar]
- 27.Lindvall O, Kokaia Z, Martinez-Serrano A. Stem cell therapy for human neurodegenerative disorders-how to make it work. Nat Med. 2004;10 :S42–S50. doi: 10.1038/nm1064. Suppl. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz PH, Bryant PJ, Fuja TJ, Su H, O'Dowd DK, Klassen H. Isolation and characterization of neural progenitor cells from post-mortem human cortex. J Neurosci Res. 2003;74:838–851. doi: 10.1002/jnr.10854. [DOI] [PubMed] [Google Scholar]
- 29.Yaworsky PJ, Kappen C. Heterogeneity of neural progenitor cells revealed by enhancers in the nestin gene. Dev Biol. 1999;205:309–321. doi: 10.1006/dbio.1998.9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujiwara Y, Tanaka N, Ishida O, et al. Intravenously injected neural progenitor cells of transgenic rats can migrate to the injured spinal cord and differentiate into neurons, astrocytes and oligodendrocytes. Neurosci Lett. 2004;366:287–291. doi: 10.1016/j.neulet.2004.05.080. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi M, Arai Y, Kurosawa H, Sueyoshi N, Shirai S. Ependymal cell reactions in spinal cord segments after compression injury in adult rat. J Neuropathol Exp Neurol. 2003;62:185–194. doi: 10.1093/jnen/62.2.185. [DOI] [PubMed] [Google Scholar]
- 32.Englund U, Bjorklund A, Wictorin K. Migration patterns and phenotypic differentiation of long-term expanded human neural progenitor cells after transplantation into the adult rat brain. Brain Res Dev Brain Res. 2002;134:123–141. doi: 10.1016/s0165-3806(01)00330-3. [DOI] [PubMed] [Google Scholar]
- 33.Mitsuhashi T, Aoki Y, Eksioglu YZ, et al. Overexpression of p27Kip1 lengthens the G1 phase in a mouse model that targets inducible gene expression to central nervous system progenitor cells. Proc Natl Acad Sci U S A. 2001;98:6435–6440. doi: 10.1073/pnas.111051398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aoki Y, Huang Z, Thomas SS, et al. Increased susceptibility to ischemia-induced brain damage in transgenic mice overexpressing a dominant negative form of SHP2. FASEB J. 2000;14:1965–1973. doi: 10.1096/fj.00-0105com. [DOI] [PubMed] [Google Scholar]
- 35.Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24:1726–1733. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lie DC, Dziewczapolski G, Willhoite AR, Kaspar BK, Shults CW, Gage FH. The adult substantia nigra contains progenitor cells with neurogenic potential. J Neurosci. 2002;22:6639–6649. doi: 10.1523/JNEUROSCI.22-15-06639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imitola J, Raddassi K, Park KI, et al. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci U S A. 2004;101:18117–18122. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng H, Huang Y, Rose J, et al. Stromal cell-derived factor 1-mediated CXCR4 signaling in rat and human cortical neural progenitor cells. J Neurosci Res. 2004;76:35–50. doi: 10.1002/jnr.20045. [DOI] [PubMed] [Google Scholar]
- 39.Hsia AY, Masliah E, McConlogue L, et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li B, Ryder J, Su Y, et al. Overexpression of GSK3betaS9A resulted in tau hyperphosphorylation and morphology reminiscent of pretangle-like neurons in the brain of PDGSK3beta transgenic mice. Transgenic Res. 2004;13:385–396. doi: 10.1023/b:trag.0000040039.44899.6f. [DOI] [PubMed] [Google Scholar]
- 41.Kahle PJ, Neumann M, Ozmen L, et al. Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am J Pathol. 2001;159:2215–2225. doi: 10.1016/s0002-9440(10)63072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu R, Cai J, Hu X, et al. Region-specific and stage-dependent regulation of Olig gene expression and oligodendrogenesis by Nkx6.1 homeodomain transcription factor. Development. 2003;130:6221–6231. doi: 10.1242/dev.00868. [DOI] [PubMed] [Google Scholar]
- 43.Miller RH. Regulation of oligodendrocyte development in the vertebrate CNS. Prog Neurobiol. 2002;67:451–467. doi: 10.1016/s0301-0082(02)00058-8. [DOI] [PubMed] [Google Scholar]
- 44.Miller RH, Payne J, Milner L, Zhang H, Orentas DM. Spinal cord oligodendrocytes develop from a limited number of migratory highly proliferative precursors. J Neurosci Res. 1997;50:157–168. doi: 10.1002/(SICI)1097-4547(19971015)50:2<157::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 45.Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Q, Choi G, Anderson DJ. The bHLH transcription factor Olig2 promotes oligodendrocyte differentiation in collaboration with Nkx2.2. Neuron. 2001;31:791–807. doi: 10.1016/s0896-6273(01)00414-7. [DOI] [PubMed] [Google Scholar]
- 47.Imitola J, Raddassi K, Park KI, et al. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci U S A. 2004;101:18117–18122. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Rao MS. Glial progenitors in the CNS and possible lineage relationships among them. Biol Cell. 2004;96:279–290. doi: 10.1016/j.biolcel.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 49.Cai J, Wu Y, Mirua T, et al. Properties of a fetal multipotent neural stem cell (NEP cell) Dev Biol. 2002;251:221–240. doi: 10.1006/dbio.2002.0828. [DOI] [PubMed] [Google Scholar]
- 50.Rao MS. Multipotent and restricted precursors in the central nervous system. Anat Rec. 1999;257:137–148. doi: 10.1002/(SICI)1097-0185(19990815)257:4<137::AID-AR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 51.Anthony TE, Klein C, Fishell G, Heintz N. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron. 2004;41:881–890. doi: 10.1016/s0896-6273(04)00140-0. [DOI] [PubMed] [Google Scholar]
- 52.Merkle FT, Tramontin AD, Garcia-Verdugo JM, varez-Buylla A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci U S A. 2004;101:17528–17532. doi: 10.1073/pnas.0407893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin K, Galvan V, Xie L, et al. Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A. 2004;101:13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin K, Minami M, Lan JQ, et al. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci U S A. 2001;98:4710–4715. doi: 10.1073/pnas.081011098. [DOI] [PMC free article] [PubMed] [Google Scholar]