

Abstract
Persistent colonization of the human stomach by Helicobacter pylori is a risk factor for the development of gastric cancer and peptic ulcer disease. H. pylori secretes a toxin, VacA, that targets human gastric epithelial cells and T lymphocytes and enhances the ability of H. pylori to colonize the stomach in a mouse model. To examine how VacA contributes to H. pylori colonization of the mouse stomach, we investigated whether murine T lymphocytes were susceptible to VacA activity. VacA inhibited interleukin-2 (IL-2) production by a murine T-cell line (LBRM-33), similar to its effects on a human T-cell line (Jurkat), but did not inhibit IL-2 production by primary murine splenocytes or CD4+ T cells. VacA inhibited activation-induced proliferation of primary human CD4+ T cells but did not inhibit the proliferation of primary murine CD4+ T cells. Flow cytometry studies indicated that the levels of VacA binding to primary murine CD4+ T cells were significantly lower than levels of VacA binding to human CD4+ T cells. This suggests that the resistance of primary murine CD4+ T cells to VacA is attributable, at least in part, to impaired VacA binding to these cells.
Helicobacter pylori persistently colonizes the stomach in about 50% of the global human population. In response to H. pylori infection, the human host mounts a mucosal inflammatory response, termed superficial gastritis. H. pylori-induced gastric inflammation does not cause symptoms in most infected persons. However, H. pylori infection is a risk factor for development of duodenal ulcer disease, gastric ulcer disease, gastric adenocarcinoma, and gastric lymphoma (reviewed in reference 26).
An important virulence factor produced by H. pylori is a secreted toxin, VacA. When added to epithelial cells in vitro, VacA causes swelling of late endosomal compartments and can increase mitochondrial membrane permeability (reviewed in reference 3). Many different types of epithelial cells are susceptible to VacA (6, 14, 23). VacA can also have effects on human T cells, B cells, macrophages, and mast cells (2, 11, 19, 28, 29). VacA inhibits the activation and nuclear translocation of NFAT (nuclear factor of activated T cells) in Jurkat cells (a transformed human T-cell line) (2, 11), an effect similar to that of cyclosporine A, a drug that inhibits the NFAT phosphatase calcineurin. Treatment of Jurkat cells with VacA results in the altered expression of many proteins, including interleukin-2 (IL-2), CD25, and CD69 (2, 11). VacA can also induce the activation of p38 mitogen-activated protein kinase in Jurkat cells (2). In comparison to the inhibitory effect of VacA on IL-2 production in Jurkat cells, VacA has a relatively weak inhibitory effect on IL-2 production by primary human CD4+ T cells (28). However, VacA inhibits the activation-induced proliferation of primary human CD4+ T cells (28). The process by which VacA inhibits the proliferation of primary human CD4+ T cells is somewhat different from what is observed in Jurkat cells and is not dependent on the inhibition of NFAT activation (22, 28). Inhibition of T-cell function by VacA may be one of the mechanisms by which H. pylori resists immune clearance.
The vacA alleles found in different strains of H. pylori are characterized by a high level of genetic diversity, and several different families of vacA alleles have been recognized. The regions of maximum diversity are located near the 5′ end of vacA (type s1 and s2) and in the midregion of the gene (types m1 and m2) (1). Type s1m1, s1m2, and s2m2 vacA alleles are all found commonly in H. pylori isolates, whereas type s2m1 alleles are relatively rare. Type s1 forms of VacA are active in many in vitro assays of toxin activity, whereas type s2 forms of VacA lack detectable activity in most in vitro assays (1, 4, 16). Type s1m1 and type s1m2 forms of VacA exhibit different cell type specificities (1, 5, 13, 23). Infection with H. pylori strains containing type s1 forms of vacA is associated with an increased risk of gastric cancer or peptic ulcer disease compared to infection with strains containing type s2 forms of vacA (1, 9). Similarly, infection with H. pylori strains containing type m1 forms of vacA is associated with an increased risk of gastric cancer compared to infection with strains containing type m2 forms of vacA (9).
Animal models have been utilized to gain further insight into the role of VacA in vivo. H. pylori vacA null mutant strains can successfully colonize the stomachs of several animal models, including gnotobiotic piglets, Mongolian gerbils, and mice (8, 21, 32). However, one study reported that a VacA-expressing wild-type strain had a competitive advantage over a vacA null mutant strain in the colonization of the mouse stomach (25). Several studies have reported that VacA can cause gastric injury in animal models (10, 21, 30).
To gain further insight into how VacA contributes to H. pylori colonization of the mouse stomach and the genesis of gastric damage in this model, we investigated the effects of VacA on murine T lymphocytes. We show that in comparison to human and murine T-cell lines and primary human T cells, primary murine T cells are relatively resistant to VacA.
MATERIALS AND METHODS
Cell lines.
Jurkat lymphocytes (clone E6-1) were obtained from the American Type Culture Collection (ATCC TIB-152) and were cultured in RPMI 1640 medium containing 2 mM l-glutamine, 1.5 g/liter sodium bicarbonate, 4.5 g/liter glucose, 10 mM HEPES, and 1.0 mM sodium pyruvate supplemented with 10% fetal bovine serum (FBS). LBRM-33 lymphocytes (clone 4A2) were obtained from the American Type Culture Collection (ATCC TIB-155) and were cultured in RPMI 1640 medium containing 0.05 mM 2-mercaptoethanol, 2 mM l-glutamine, 1.5 g/liter sodium bicarbonate, 4.5 g/liter glucose, 10 mM HEPES, and 1.0 mM sodium pyruvate supplemented with 10% FBS.
H. pylori strains and purification of VacA.
The H. pylori strains used in this study included strain 60190, which secretes a type s1m1 VacA protein (5), and three strains derived from strain 60190. Strain AV452 secretes a mutant VacA protein known as VacAΔ(6-27), which lacks 22 amino acids near the amino terminus of VacA (31). This mutant protein [VacA(Δ6-27)] is defective in membrane channel-forming activity in planar lipid bilayer assays, does not cause vacuolation of HeLa cells, and does not inhibit activation-induced proliferation of primary human T cells (28, 31). H. pylori strains G119 and VM083 were engineered to secrete chimeric toxins (16). Strain G119 secretes a type s1m2 VacA protein, and strain VM083 secretes a type s2m1 VacA protein. H. pylori strains were cultured in broth, and oligomeric forms of each VacA protein were purified from culture supernatants as described previously (17, 31). All experiments were performed using acid-activated preparations of VacA (7, 18) or acidified buffer control (phosphate-buffered saline [PBS]) unless otherwise stated. The final VacA concentrations were 10 μg/ml for all experiments unless otherwise stated. In all experiments, cells were treated with VacA or control additives for 30 min prior to activation, and the additives remained in place throughout the experiment unless otherwise stated.
Analysis of IL-2 production by Jurkat cells and LBRM-33 cells.
Jurkat T cells were stimulated with anti-human CD3 (OKT3; American Type Culture Collection) and anti-human CD28 (BD Biosciences, Franklin Lakes, NJ) antibodies (hereafter termed T-cell-receptor/CD28 stimulation) (27) or were stimulated with phorbol myristate acetate (PMA) (50 ng/ml; Sigma) and ionomycin (500 ng/ml; Sigma) and maintained in RPMI medium 1640 containing 10% FBS for 24 h. LBRM-33 cells were stimulated with PMA and ionomycin. To inhibit activation, Jurkat or LBRM-33 cells were treated with cyclosporine (50 nM; Alexis Biochemicals, Lausen, Switzerland). IL-2 was measured using a human or mouse IL-2 Quantikine enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.
Immunoblot analysis of p38 activation.
LBRM-33 cells (1 × 106 cells per condition) were cultured in serum-free medium for 8 h and then treated with medium alone, acid buffer, VacA, or PMA-ionomycin for the specified times. The cells were then pelleted, washed, and lysed in 8% sodium dodecyl sulfate in 0.25 M Tris base in the presence of complete mini protease inhibitors (Roche), sodium fluoride (10 mM) and sodium orthovanadate (2 mM). Samples were boiled for 10 min with intermittent vortexing and were stored at −80°C prior to subsequent analysis. Protein extracts were then mixed with 6× sodium dodecyl sulfate loading buffer, electrophoresed on a 4 to 20% gradient precast acrylamide gel (Bio-Rad, Hercules, CA), and transferred onto polyvinylidene difluoride membranes. Membranes were immunoblotted with antibodies against phosphorylated p38 (Thr180/Tyr182) (Cell signaling Technology, Beverly, MA) or Stat5 (Cell signaling), followed by horseradish peroxidase-conjugated secondary antibodies (Bio-Rad). Immune complexes were revealed by using SuperSignal West Pico chemiluminescent substrate (Pierce Biotechnology, Rockford, IL).
Primary mouse splenocyte preparation and CD4+ splenocyte purification.
Spleens were harvested from FVB/N, C57BL/6, or BALB/c mice, and splenocytes were obtained by mechanical cell dissociation of the spleens using a 70-μm cell strainer (BD Falcon, Fisher, Pittsburgh, PA). Red blood cells were lysed by treatment of the cell suspension with red blood cell lysis buffer (0.17 M Tris-0.16 M ammonium chloride). CD4+ T cells were purified from mouse splenocytes using a FACSAria cell-sorting system (BD, San Alto, CA). Briefly, mouse splenocytes were stained with anti-CD4 phycoerythrin (PE)-Cy5 (BD) for 30 min at 4°C. Cells were washed and resuspended in 5% FBS-1 mM HEPES in PBS and then purified by cell sorting. Purification led to the isolation of approximately 20 million CD4+ cells per spleen with a purity of greater than 95%. Following overnight incubation in RPMI 1640 medium containing 5% FBS, 1 mM HEPES, and 20 U/ml IL-2, CD4+ T cells were washed prior to use in subsequent assays.
Activation of mouse splenocytes for studies of IL-2 secretion.
For assays designed to monitor IL-2 production, mouse splenocytes were plated at a concentration of 1 × 106 cells/ml (1 × 105 cells/well). Cells were then stimulated with either PMA-ionomycin or anti-mouse CD3/anti-mouse CD28 antibodies for 24 h. At 24 h after stimulation, cell supernatants were collected for analysis of IL-2 by using a mouse IL-2 Quantikine ELISA kit.
Analysis of primary murine splenocytes and CD4+ T cells in proliferation assays.
To monitor the proliferation of cells, mouse splenocytes or purified mouse CD4+ T cells were labeled with 5 μM carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes) prior to activation. The CFSE-labeled cells were activated by one of two different methods. In one approach, cells were activated with plate-bound anti-mouse CD3/anti-mouse CD28 antibodies (clones 500A2 and 37.51; BD) for 48 h, and the cells were then removed from stimulation for an additional 48 h while cultured in the presence of 20 U/ml recombinant murine IL-2 (Peprotech, Rocky Hill, NJ). In the second approach, cells were treated for 8 h with PMA (50 ng/ml; Sigma) and ionomycin (500 ng/ml; Sigma), washed, and then incubated for an additional 3 days. Whole splenocytes were costained with anti-CD4 PE-Cy5 and anti-CD3 PE antibodies (BD) and fixed, and purified CD4+ splenocytes were simply fixed prior to subsequent flow cytometry analysis (FACSCalibur system; BD, San Alto, CA).
Analysis of primary human CD4+ T cells.
Resting human CD4+ T cells were purified from healthy adult donors as described previously (27). The purified cells were 99% CD3+ CD4+ as assessed by flow cytometric analysis. Cell proliferation was monitored by labeling T cells with 5 μM CFSE (Molecular Probes) before stimulation with anti-human CD3/anti-human CD28 antibodies. Activation of primary human T cells was accomplished by using anti-human CD3 (OKT3; American Type Culture Collection) and anti-human CD28 (BD Biosciences, Franklin Lakes, NJ) antibodies (27). T cells were removed from the activation signals after 48 h, expanded in medium supplemented with recombinant human IL-2 (200 units/ml; Chiron), and then cultured and analyzed by flow cytometry as described previously (27).
Analysis of VacA binding to cells.
VacA binding was assessed using a flow cytometric assay. Cells (100,000 cells in a concentration of 1 × 106 cells/ml) were treated with acid-activated VacA (at various concentrations), acidified buffer, or no additives for 1 h at 4°C. Cells were then washed three times in cold PBS containing 2% FBS. Cells were incubated with a polyclonal rabbit anti-VacA antibody (958, at a 1:2,000 dilution) for 0.5 h at 4°C and were then washed twice in cold PBS containing 2% FBS. The secondary antibody, anti-rabbit immunoglobulin G-fluorescein isothiocyanate (Sigma), was added at a final dilution of 1:250 for 0.5 h at 4°C. Cells were washed twice and then fixed in 2% paraformaldehyde. The cells were collected using a flow cytometer (LSR II system; BD, San Alta, CA), and the flow cytometric analysis was performed using FCS Express 2.0 (DeNovo Software, Ontario, Canada).
Statistics.
For analysis of IL-2, proliferation, and binding data, an analysis of variance test was conducted using the SAS Statistical Software package (SAS Institute Inc., Cary, NC). The Student Neuman Keuls method for multiple comparisons was then applied.
RESULTS
VacA inhibits IL-2 production by Jurkat cells and LBRM-33 cells.
Previous studies have shown that VacA can inhibit IL-2 expression in Jurkat cells, a transformed human T-cell line (11). We sought to determine whether VacA had similar effects on LBRM-33 cells, a murine T-cell line. In initial studies, we tested the effects of type s1m1 VacA (purified from H. pylori strain 60190) on these two cell lines. Cells were pretreated with VacA, cyclosporine, or buffer control and stimulated for 24 h with PMA-ionomycin, and levels of IL-2 in the cell supernatants were then measured by ELISA. As expected, treatment of Jurkat cells or LBRM-33 cells with cyclosporine suppressed IL-2 production. Similarly, intoxication of Jurkat cells with s1m1 VacA also suppressed IL-2 production (Fig. 1A). Intoxication of LBRM-33 cells with s1m1 VacA resulted in a modest reduction in IL-2 production (Fig. 1B). The ability of VacA to suppress IL-2 secretion by Jurkat and LBRM-33 cells was dependent on the acid activation of VacA prior to the addition to cells and was dose dependent. In Jurkat cells, 5 μg/ml VacA inhibited IL-2 secretion by 50%, whereas in LBRM-33 cells, 10 μg/ml VacA inhibited IL-2 secretion by 50% (data not shown). A mutant form of VacA [VacA(Δ6-27)] had no detectable effects on IL-2 production by either cell line (Fig. 1).
FIG. 1.
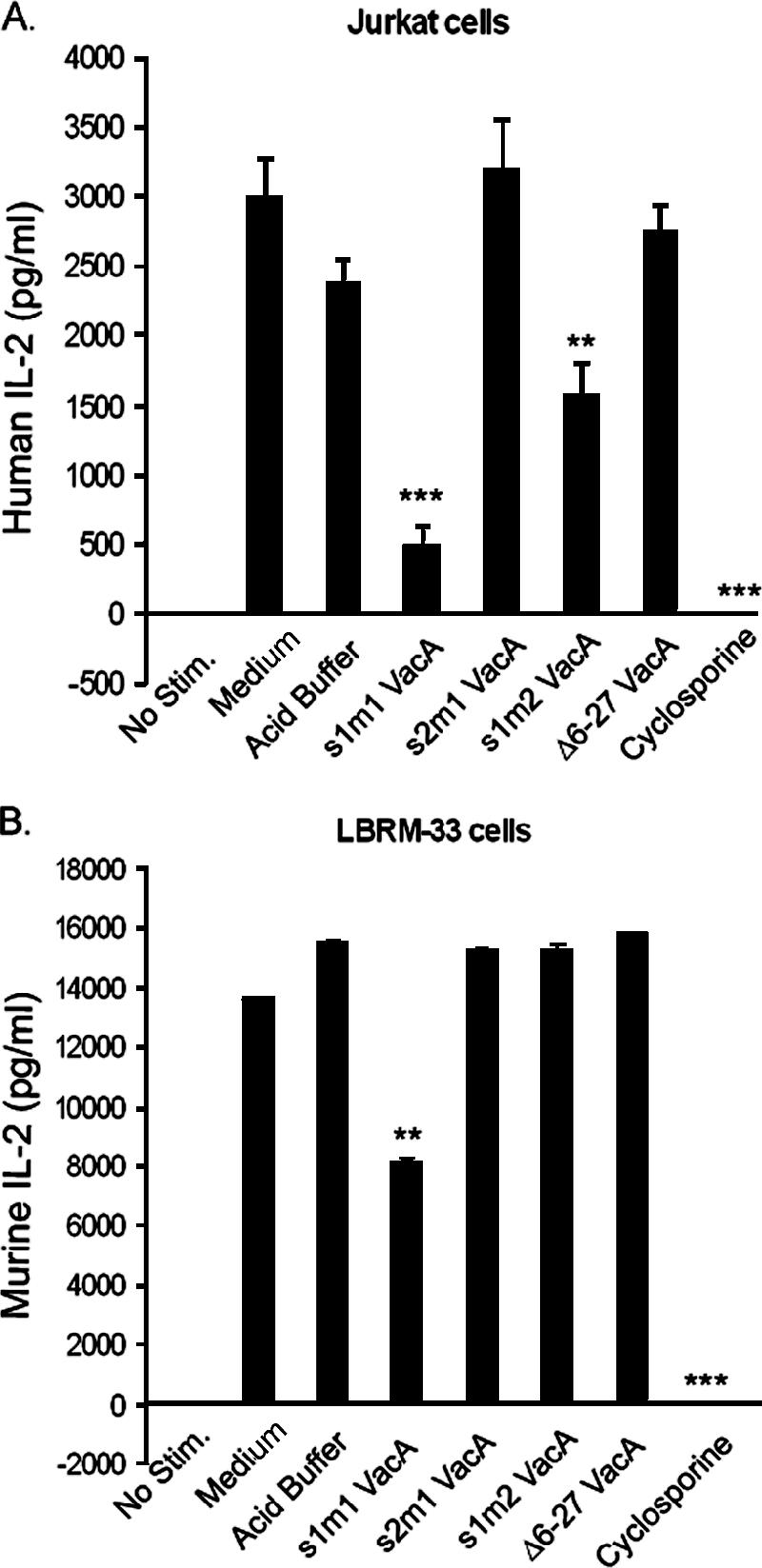
Effect of VacA on IL-2 secretion by Jurkat and LBRM-33 T cell lines. Jurkat cells (A) or LBRM-33 (B) cells were pretreated with medium alone, acidified buffer, several different types of acid-activated VacA (10 μg/ml), or cyclosporine and then stimulated with PMA-ionomycin. After 24 h, the cells were pelleted, and the supernatants were analyzed in a human or mouse IL-2 ELISA. Results represent the means ± standard deviations of triplicate determinations and are representative of three independent experiments. ***, P ≤ 0.001; **, P ≤ 0.01 (compared to acidified buffer). No Stim., no stimulation.
We next compared the effects of three different forms of VacA (s1m1, s1m2, and s2m1) on Jurkat and LBRM-33 cells. Type s1m1 VacA inhibited the production of IL-2 by both Jurkat and LBRM-33 cells. Type s1m2 VacA had a modest inhibitory effect on IL-2 secretion by Jurkat cells but had no detectable effect on LBRM-33 cells (Fig. 1). Type s2m1 VacA did not have any detectable effect on either cell type. Thus, of the three different forms of VacA tested, the s1m1 form caused the most pronounced inhibition of IL-2 production.
VacA does not inhibit IL-2 production by primary murine T cells.
We next investigated the susceptibility of primary murine T cells to VacA. The data presented in Fig. 1 indicated that type s1m1 VacA had the most pronounced inhibitory effects on IL-2 production by mouse LBRM-33 cells, and therefore, we initially selected type s1m1 VacA for use in these studies. As a first step, we tested the effects of s1m1 VacA on IL-2 production by splenocytes from FVB/N mice. In contrast to what was observed in experiments with Jurkat and LBRM-33 cells (Fig. 1), type s1m1 VacA did not have any detectable inhibitory effect on IL-2 production by mouse splenocytes from FVB/N mice (Fig. 2). The primary murine splenocytes intoxicated with s1m1 VacA secreted high levels of IL-2 regardless of whether they were stimulated with PMA-ionomycin or with anti-CD3 and anti-CD28 antibodies (Fig. 2A and B). We also tested the effects of s1m1 VacA on a preparation of CD4+ cells purified from FVB/N mouse spleens, and we did not detect any strong inhibitory effects of VacA on IL-2 secretion by these cells (Fig. 2A and B).
FIG. 2.

Effect of VacA on IL-2 secretion by primary murine T cells. Splenocytes or purified murine CD4+ T cells from FVB/N mice were pretreated with medium alone, acidified buffer, acid-activated s1m1 VacA (10 μg/ml), or cyclosporine and then stimulated with PMA-ionomycin (A) or anti-CD3/anti-CD28 antibodies (B). After 24 h, the cells were pelleted, and the supernatants were analyzed in a mouse IL-2 ELISA. Results represent the means ± standard deviations of triplicate samples from a single experiment and are representative of three independent experiments. (C). Splenocytes from BALB/c, C57BL/6, or FVB/N mice were pretreated with medium alone, acidified buffer, several different types of acid-activated VacA (10 μg/ml), or cyclosporine and then stimulated with PMA-ionomycin. IL-2 secretion was analyzed as described above. No Stim., no stimulation.
To determine whether the results obtained in experiments with splenocytes from FVB/N mice were generalizable to splenocytes from other strains of mice, we tested the ability of s1m1 VacA to inhibit IL-2 production by splenocytes from BALB/c and C57BL/6 mice. VacA did not have any detectable inhibitory effect on IL-2 production by PMA-ionomycin-stimulated splenocytes from these mouse strains (Fig. 2C). We also tested the effects of the s2m1 and s1m2 isoforms of VacA on PMA-ionomycin-stimulated splenocytes from BALB/c, C57BL/6, and FVB/N mice and did not detect any inhibitory effects on IL-2 secretion (Fig. 2C). Thus, VacA inhibited the production of IL-2 by Jurkat cells and LBRM-33 cells but did not inhibit IL-2 production by primary murine T cells.
VacA induces p38 activation in LBRM-33 cells but not primary murine T cells.
It was previously reported that VacA could induce tyrosine phosphorylation of p38 MAP kinase in several cell types, including AZ-521 epithelial cells (20), Jurkat cells, human peripheral neutrophils, and macrophages (2). Therefore, we investigated whether VacA was able to activate p38 in LBRM-33 cells. LBRM-33 cells were treated for 5 or 30 min with wild-type s1m1 VacA, a mutant form of VacA [VacA(Δ6-27)], or various controls. Western blot analyses showed that, as expected, PMA-ionomycin induced the activation of p38 (Fig. 3A). Both wild-type VacA and VacA(Δ6-27) induced p38 activation (Fig. 3A). As shown in Fig. 3B, the ability of wild-type VacA and VacA(Δ6-27) to activate p38 was dependent on the acid activation of the VacA protein prior to cell intoxication.
FIG. 3.

Effect of VacA on p38 MAP kinase activation. (A) Serum-starved LBRM-33 T cells were treated with PMA-ionomycin (PMA/Iono) (positive control), acidified buffer, acid-activated wild-type (WT) s1m1 VacA, or acid-activated VacA(Δ6-27) for 5 or 30 min. (B) Serum-starved LBRM-33 T cells were treated with wild-type s1m1 VacA or VacA(Δ6-27) (acid activated or nonactivated) for 5 min. (C) Serum-starved LBRM-33 T cells were treated with the indicated acid-activated VacA proteins for 5 min. After treatment, cells were lysed, and protein samples were then analyzed by immunoblotting to detect phosphorylated p38 (p-p38). Stat5 was used as a loading control. This blot is representative of three independent experiments.
We next compared the abilities of three different forms of VacA to induce the activation of p38. Both s1m1 and s2m1 VacA proteins induced the activation of p38 in LBRM-33 cells (Fig. 3C). In contrast, we did not detect p38 activation when cells were intoxicated with type s1m2 VacA (Fig. 3C). Thus, VacA stimulated the activation of p38 MAP kinase in a murine T-cell line, similar to its reported effects on Jurkat cells (2), and VacA-induced activation of p38 was observed with the type m1 form of VacA but not type m2 VacA.
To investigate whether VacA was able to activate p38 in primary murine T cells, CD4+ T cells were isolated from BALB/c, C57BL/6, and FVB/N mice. The purified CD4+ T cells were treated for 5 min with s1m1 VacA, s1m2 VacA, or s2m1 VacA; a mutant form of VacA [VacA(Δ6-27)]; or various controls. Western blot analyses showed that, as expected, PMA-ionomycin induced the activation of p38 in these cells. In contrast, we did not detect p38 activation when the cells were intoxicated with the VacA preparations (data not shown). Thus, in comparison to LBRM-33 cells, primary murine CD4+ T cells were resistant to the p38-activating effects of VacA.
VacA inhibits proliferation of primary human T lymphocytes but not primary murine T lymphocytes.
We previously showed that VacA can inhibit the activation-induced proliferation of primary human CD4+ T cells (28). Therefore, we next investigated whether VacA was able to inhibit the activation-induced proliferation of primary murine splenocytes or primary murine CD4+ T cells. Primary human CD4+ cells were tested as controls. Cells were isolated and labeled with CFSE as described in Materials and Methods. CFSE-labeled cells were pretreated with wild-type s1m1 VacA, a mutant form of VacA [VacA(Δ6-27)], or buffer control, followed by stimulation using anti-CD3 and anti-CD28 antibodies for 48 h. After stimulation, cells were expanded in IL-2-containing medium for an additional 3 days. As expected, wild-type VacA inhibited the proliferation of primary human CD4+ T cells (Fig. 4A) (28). In contrast, VacA did not inhibit the proliferation of splenocytes or CD4+ T cells from FVB/N mice (Fig. 4B and C). Similar results were obtained regardless of whether the murine cells were stimulated with anti-CD3 and anti-CD28 antibodies or with PMA-ionomycin (data not shown). Moreover, VacA did not inhibit the proliferation of splenocytes derived from BALB/c or C57BL/6 mice (data not shown). Thus, VacA inhibited the activation-induced proliferation of primary human CD4+ T cells but did not have any detectable effect on the proliferation of primary murine CD4+ T cells.
FIG. 4.

Effect of VacA on activation-induced proliferation of primary CD4+ human T lymphocytes and primary murine T lymphocytes. Cells were isolated, labeled with CFSE, and then treated with acid-activated s1m1 VacA (10 μg/ml), VacA(Δ6-27), or acidified buffer (PBS), followed by T-cell-receptor/CD28 stimulation. Activated T cells were expanded in IL-2-containing medium, and T-cell proliferation was analyzed at day 5 postactivation by flow cytometry. Panels show experiments with primary human CD4+ T cells (A), mouse splenocytes (B), and primary CD4+ T cells purified from mouse spleens (C). PBMCs, peripheral blood mononuclear cells.
Analysis of VacA binding to human and murine lymphocytes.
To investigate a possible mechanistic basis for the resistance of primary murine lymphocytes to VacA, we analyzed the ability of s1m1 VacA to bind to primary murine lymphocytes versus several susceptible types of T cells. Cells were incubated with VacA for 1 h at 4°C, and the binding of VacA to cells was quantified using a flow cytometry-based assay. Representative histograms illustrating the results of VacA binding assays are presented in Fig. 5A, and quantification of the levels of VacA binding to different types of lymphocytes is shown in Fig. 5B. VacA bound to Jurkat cells, LBRM-33 cells, and primary human CD4+ T cells, and the binding of VacA to these cells was acid and dose dependent (Fig. 5B). Notably, the levels of VacA binding to these cell types were significantly higher than levels of VacA binding to primary murine splenocytes and purified murine CD4+ T cells from FVB/N mice (Fig. 5B). The relatively weak binding of VacA to primary murine splenocytes and purified murine CD4+ T cells was observed regardless of whether the cells were isolated from BALB/c, C57BL/6, or FVB/N mice (Fig. 5C). These data suggest that the resistance of primary murine lymphocytes to VacA activity is due at least in part to the impaired binding of VacA to these cells.
FIG. 5.

VacA binding to human and murine T cells. (A) T cells were treated with either acid-activated or nonacidified s1m1 VacA for 1 h. After washing, cells were incubated with an anti-VacA primary antibody followed by a secondary anti-rabbit immunoglobulin G conjugated to fluorescein isothiocyanate (FITC). Cells were then analyzed by flow cytometry. Representative histograms of VacA binding to LBRM-33 T cells and primary murine CD4+ T cells are shown. (B) Graphical representation of VacA binding to different lymphocyte populations. Results represent the means ± standard deviations of triplicate samples from a single experiment, which was representative of two independent experiments. (C) Graphical representation of VacA binding to lymphocyte populations from different strains of mice. Results represent the means ± standard deviations of triplicate samples from a single experiment.
DISCUSSION
Several previous studies have reported that H. pylori VacA can inhibit the activation and proliferation of human T lymphocytes (2, 11, 28). In the current study, we investigated the effects of VacA on murine T cells. VacA inhibited the production of IL-2 by a murine T-cell line (LBRM-33 cells), similar to its effects on a human T-cell line (Jurkat), and the binding of VacA to LBRM-33 cells induced the activation of p38 MAP kinase, similar to its previously reported effects on Jurkat cells, AZ-521 cells, and primary human neutrophils and macrophages (2, 20). A striking finding in the current study is that primary murine T cells were relatively resistant to VacA immunomodulatory activity. Specifically, VacA did not inhibit IL-2 production by mouse splenocytes or primary murine CD4+ T cells, and VacA did not inhibit the activation-induced proliferation of primary murine splenocytes or CD4+ T cells. Flow cytometry studies indicated that VacA bound weakly to primary murine CD4+ T cells compared to primary human CD4+ T cells, and this provides a possible explanation for the different susceptibilities of these cells to VacA activity. We speculate that primary human CD4+ T cells express one or more cell surface receptors that can mediate VacA binding and that primary murine T cells either fail to express these receptors or express forms of the receptors that have a low affinity for VacA.
VacA is capable of causing vacuolation of many different types of cells (6, 14, 23). Previous studies have shown that VacA can induce vacuolation of primary murine gastric epithelial cells (10), can bind to a murine epithelial cell line (NIH 3T3 cells) (15), and can increase the permeability of monolayers formed by polarized murine epithelial cells (epH4) (24). In addition, VacA can cause gastric mucosal damage if it is administered directly into the stomachs of mice (10, 30). These previous findings, as well as our current results using LBRM-33 cells, suggest that mouse cells are not universally resistant to VacA, but instead, there seems to be a specific resistance exhibited by primary murine CD4+ T cells.
Several different isoforms of VacA have been described, and these isoforms differ in their cell type specificities (1, 5, 13, 23). Most epithelial cell lines are sensitive to the vacuolating cytotoxicity of type s1m1 VacA toxins (1, 5, 13, 23). RK-13 and primary human gastric epithelial cells are susceptible to the vacuolating activity of type s1m2 toxins, whereas several other cell lines (including HeLa) are relatively resistant to type s1m2 toxins (23). Data presented in the current study suggest that different isoforms of VacA differ in their abilities to modulate T-lymphocyte function. Specifically, by testing standardized concentrations of purified VacA toxins, we found that type s1m1 VacA was more potent than s1m2 VacA in the ability to inhibit IL-2 secretion by Jurkat or LBRM-33 T-cell lines, and a type s2m1 form of VacA lacked detectable inhibitory activity. These results are consistent with results of a previous study, which concluded that H. pylori strains producing m1 forms of VacA had a stronger inhibitory effect on IL-2 secretion by Jurkat cells than did strains producing m2 forms of VacA (11). Another finding in the current study was that VacA toxins containing an m1 region (s1m1 or s2m1) were able to induce p38 activation in LBRM-33 cells, whereas a type s1m2 form of VacA was not. We speculate that type m1 VacA and type m2 VacA toxins might bind to different receptors on the surface of these cells. Finally, the current results indicate that primary murine T cells are relatively resistant to all forms of VacA tested.
VacA has been reported to enhance the ability of H. pylori to colonize the mouse stomach (25). Since VacA is a multifunctional toxin that can affect the activities of many different types of cells, there are potentially many mechanisms by which VacA could contribute to gastric colonization (3). For example, VacA on the surface of H. pylori (12) could contribute to bacterial adherence to gastric epithelial cells in the mouse stomach. The secreted VacA protein is likely to cause multiple alterations in the function of gastric epithelial cells and several types of immune cells. Our current results, demonstrating resistance of primary murine CD4+ T cells to VacA, suggest that VacA enhances the ability of H. pylori to colonize the mouse stomach by a mechanism other than intoxication of murine CD4+ T cells. This conclusion is consistent with the observation that VacA expression contributes to gastric colonization during the first 2 weeks of infection (25), before a strong adaptive immune response to H. pylori would be elicited. It has been suggested that immunosuppressive effects of VacA on human T cells may be one of the factors that account for the persistence of H. pylori infection and its evasion of host immune responses (2, 3, 11, 28). We speculate that this may indeed be true in the setting of persistent human colonization that occurs over a period of decades. However, based on the current results, we hypothesize that the persistence of H. pylori infection in the mouse model occurs by mechanisms that do not require VacA effects on murine T cells.
The mouse model and other animal models can provide valuable insights into mechanisms by which H. pylori colonizes the stomach. However, as illustrated by the results of this study, there are important differences between H. pylori-mouse interactions and H. pylori-human interactions. In future studies, it will be important to define the molecular basis for the resistance of primary murine T cells to VacA activity.
Acknowledgments
This work was supported by NIH grants T32 AI-07474, R01AI39657, and R01DK53623, a Vanderbilt Discovery grant, and the Department of Veterans Affairs.
We are grateful to Jim Higginbotham of the Immunology Core Laboratory at Vanderbilt University (funded through MO1 RR-0095 from NCRR/NIH) and Mark Sundrud for assistance with flow cytometry. We thank the members of the Cover, Peek, and Wilson laboratories for helpful discussions.
Editor: D. L. Burns
Footnotes
Published ahead of print on 30 October 2006.
REFERENCES
- 1.Atherton, J. C., P. Cao, R. M. Peek, Jr., M. K. Tummuru, M. J. Blaser, and T. L. Cover. 1995. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J. Biol. Chem. 270:17771-17777. [DOI] [PubMed] [Google Scholar]
- 2.Boncristiano, M., S. R. Paccani, S. Barone, C. Ulivieri, L. Patrussi, D. Ilver, A. Amedei, M. M. D'Elios, J. L. Telford, and C. T. Baldari. 2003. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 198:1887-1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cover, T. L., and S. R. Blanke. 2005. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 3:320-332. [DOI] [PubMed] [Google Scholar]
- 4.Cover, T. L., U. S. Krishna, D. A. Israel, and R. M. Peek, Jr. 2003. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 63:951-957. [PubMed] [Google Scholar]
- 5.Cover, T. L., M. K. Tummuru, P. Cao, S. A. Thompson, and M. J. Blaser. 1994. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem. 269:10566-10573. [PubMed] [Google Scholar]
- 6.de Bernard, M., M. Moschioni, E. Papini, J. Telford, R. Rappuoli, and C. Montecucco. 1998. Cell vacuolization induced by Helicobacter pylori VacA toxin: cell line sensitivity and quantitative estimation. Toxicol. Lett. 99:109-115. [DOI] [PubMed] [Google Scholar]
- 7.de Bernard, M., E. Papini, V. de Filippis, E. Gottardi, J. Telford, R. Manetti, A. Fontana, R. Rappuoli, and C. Montecucco. 1995. Low pH activates the vacuolating toxin of Helicobacter pylori, which becomes acid and pepsin resistant. J. Biol. Chem. 270:23937-23940. [DOI] [PubMed] [Google Scholar]
- 8.Eaton, K. A., T. L. Cover, M. K. Tummuru, M. J. Blaser, and S. Krakowka. 1997. Role of vacuolating cytotoxin in gastritis due to Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 65:3462-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Figueiredo, C., J. C. Machado, P. Pharoah, R. Seruca, S. Sousa, R. Carvalho, A. F. Capelinha, W. Quint, C. Caldas, L. J. van Doorn, F. Carneiro, and M. Sobrinho-Simoes. 2002. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J. Natl. Cancer Inst. 94:1680-1687. [DOI] [PubMed] [Google Scholar]
- 10.Fujikawa, A., D. Shirasaka, S. Yamamoto, H. Ota, K. Yahiro, M. Fukada, T. Shintani, A. Wada, N. Aoyama, T. Hirayama, H. Fukamachi, and M. Noda. 2003. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat. Genet. 33:375-381. [DOI] [PubMed] [Google Scholar]
- 11.Gebert, B., W. Fischer, E. Weiss, R. Hoffmann, and R. Haas. 2003. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301:1099-1102. [DOI] [PubMed] [Google Scholar]
- 12.Ilver, D., S. Barone, D. Mercati, P. Lupetti, and J. L. Telford. 2004. Helicobacter pylori toxin VacA is transferred to host cells via a novel contact-dependent mechanism. Cell. Microbiol. 6:167-174. [DOI] [PubMed] [Google Scholar]
- 13.Ji, X., T. Fernandez, D. Burroni, C. Pagliaccia, J. C. Atherton, J. M. Reyrat, R. Rappuoli, and J. L. Telford. 2000. Cell specificity of Helicobacter pylori cytotoxin is determined by a short region in the polymorphic midregion. Infect. Immun. 68:3754-3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leunk, R. D., P. T. Johnson, B. C. David, W. G. Kraft, and D. R. Morgan. 1988. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J. Med. Microbiol. 26:93-99. [DOI] [PubMed] [Google Scholar]
- 15.Massari, P., R. Manetti, D. Burroni, S. Nuti, N. Norais, R. Rappuoli, and J. L. Telford. 1998. Binding of the Helicobacter pylori vacuolating cytotoxin to target cells. Infect. Immun. 66:3981-3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McClain, M., P. Cao, H. Iwamoto, A. D. Vinion-Dubiel, G. Szabo, Z. Shao, and T. Cover. 2001. A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J. Bacteriol. 183:6499-6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McClain, M. S., H. Iwamoto, P. Cao, A. D. Vinion-Dubiel, Y. Li, G. Szabo, Z. Shao, and T. L. Cover. 2003. Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J. Biol. Chem. 278:12101-12108. [DOI] [PubMed] [Google Scholar]
- 18.McClain, M. S., W. Schraw, V. Ricci, P. Boquet, and T. L. Cover. 2000. Acid activation of Helicobacter pylori vacuolating cytotoxin (VacA) results in toxin internalization by eukaryotic cells. Mol. Microbiol. 37:433-442. [DOI] [PubMed] [Google Scholar]
- 19.Molinari, M., M. Salio, C. Galli, N. Norais, R. Rappuoli, A. Lanzavecchia, and C. Montecucco. 1998. Selective inhibition of Ii-dependent antigen presentation by Helicobacter pylori toxin VacA. J. Exp. Med. 187:135-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakayama, M., M. Kimura, A. Wada, K. Yahiro, K. Ogushi, T. Niidome, A. Fujikawa, D. Shirasaka, N. Aoyama, H. Kurazono, M. Noda, J. Moss, and T. Hirayama. 2004. Helicobacter pylori VacA activates the p38/activating transcription factor 2-mediated signal pathway in AZ-521 cells. J. Biol. Chem. 279:7024-7028. [DOI] [PubMed] [Google Scholar]
- 21.Ogura, K., S. Maeda, M. Nakao, T. Watanabe, M. Tada, T. Kyutoku, H. Yoshida, Y. Shiratori, and M. Omata. 2000. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J. Exp. Med. 192:1601-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oswald-Richter, K., V. J. Torres, M. S. Sundrud, S. E. VanCompernolle, T. L. Cover, and D. Unutmaz. 2006. Helicobacter pylori VacA toxin inhibits human immunodeficiency virus infection of primary human T cells. J. Virol. 80:11767-11775. (First published 27 September 2006; doi: 10.1128/JVI.00213-06.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pagliaccia, C., M. de Bernard, P. Lupetti, X. Ji, D. Burroni, T. L. Cover, E. Papini, R. Rappuoli, J. L. Telford, and J. M. Reyrat. 1998. The m2 form of the Helicobacter pylori cytotoxin has cell type-specific vacuolating activity. Proc. Natl. Acad. Sci. USA 95:10212-10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papini, E., B. Satin, N. Norais, M. de Bernard, J. L. Telford, R. Rappuoli, and C. Montecucco. 1998. Selective increase of the permeability of polarized epithelial cell monolayers by Helicobacter pylori vacuolating toxin. J. Clin. Investig. 102:813-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salama, N. R., G. Otto, L. Tompkins, and S. Falkow. 2001. Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect. Immun. 69:730-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suerbaum, S., and P. Michetti. 2002. Helicobacter pylori infection. N. Engl. J. Med. 347:1175-1186. [DOI] [PubMed] [Google Scholar]
- 27.Sundrud, M. S., S. M. Grill, D. Ni, K. Nagata, S. S. Alkan, A. Subramaniam, and D. Unutmaz. 2003. Genetic reprogramming of primary human T cells reveals functional plasticity in Th cell differentiation. J. Immunol. 171:3542-3549. [DOI] [PubMed] [Google Scholar]
- 28.Sundrud, M. S., V. J. Torres, D. Unutmaz, and T. L. Cover. 2004. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc. Natl. Acad. Sci. USA 101:7727-7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Supajatura, V., H. Ushio, A. Wada, K. Yahiro, K. Okumura, H. Ogawa, T. Hirayama, and C. Ra. 2002. Cutting edge: VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J. Immunol. 168:2603-2607. [DOI] [PubMed] [Google Scholar]
- 30.Telford, J. L., P. Ghiara, M. Dell'Orco, M. Comanducci, D. Burroni, M. Bugnoli, M. F. Tecce, S. Censini, A. Covacci, Z. Xiang, et al. 1994. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 179:1653-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vinion-Dubiel, A. D., M. S. McClain, D. M. Czajkowsky, H. Iwamoto, D. Ye, P. Cao, W. Schraw, G. Szabo, S. R. Blanke, Z. Shao, and T. L. Cover. 1999. A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J. Biol. Chem. 274:37736-37742. [DOI] [PubMed] [Google Scholar]
- 32.Wirth, H. P., M. H. Beins, M. Yang, K. T. Tham, and M. J. Blaser. 1998. Experimental infection of Mongolian gerbils with wild-type and mutant Helicobacter pylori strains. Infect. Immun. 66:4856-4866. [DOI] [PMC free article] [PubMed] [Google Scholar]