

Abstract
The photoisomerization of the retinal in bacteriorhodopsin is selective and efficient and yields perturbation of the protein structure within femtoseconds. The stored light energy in the primary intermediate is then used for the net translocation of a proton across the membrane in the microsecond to millisecond regime. This study is aimed at identifying how the protein changes on photoisomerization by using the O-H groups of threonines as internal probes. Polarized Fourier-transform IR spectroscopy of [3-18O]threonine-labeled and unlabeled bacteriorhodopsin indicates that 3 of the threonines (of a total of 18) change their hydrogen bonding. One is exchangeable in D2O, but two are not. A comprehensive mutation study indicates that the residues involved are Thr-89, Thr-17, and Thr-121 (or Thr-90). The perturbation of only three threonine side chains suggests that the structural alteration at this stage of the photocycle is local and specific. Furthermore, the structural change of Thr-17, which is located >11 Å from the retinal chromophore, implicates a specific perturbation channel in the protein that accompanies the retinal motion.
Bacteriorhodopsin (BR) is a light-driven proton pump in Halobacterium salinarum that contains all-trans retinal as chromophore (1–4). Its tertiary structure has been determined recently by cryoelectron microscopy (5–7) and x-ray crystallography (8–13). The retinal binds covalently to Lys-216 through a protonated Schiff base linkage. Absorption of light triggers a cyclic reaction that comprises a series of intermediates, designated as the J, K, KL, L, M, N, and O states (1–4). Protein structural changes in these intermediate states cause proton translocation across the protein, and their mechanism is the central question in current studies of BR (14).
The all-trans to 13-cis photoisomerization leads to the formation of the primary K intermediate (15–17). It is well known that photoisomerization in BR is highly selective and efficient. In solution, the photoproduct of all-trans retinal with protonated Schiff base is mainly 11-cis [82% (vol/vol) 11-cis/6% (vol/vol) 13-cis/12% (vol/vol) 9-cis in methanol; ref. 18], whereas in BR, the photoproduct is 100% (vol/vol) 13-cis. The quantum efficiency for isomerization in BR (≈0.6; refs. 19 and 20) is much higher than for retinal in solution (0.13 in methanol; ref. 18). This efficiency is correlated closely with the rate constant of the isomerization, because it occurs on the femtosecond time scale. The protein environment thus certainly facilitates the specificity of the reaction of the retinal chromophore.
How does the highly selective and efficient isomerization occur in BR? How does the protein respond to the chromophore motion? To address these questions, structural analysis of the primary intermediates is essential. Cryoelectron microscopy of the K intermediate reported that the structural change at this stage is not resolved with 3.5-Å resolution (21). More recent analysis of the K intermediate by x-ray crystallography identified few changes around the retinal chromophore (22). These results suggest that nuclear motions of the chromophore and surrounding protein would be minimal at the earliest stages. According to our low-temperature polarized Fourier-transform IR (FTIR) spectroscopy, however, a number of bands were observed from the protein that changed between the BR and the K intermediate (23). Because the formation and the relaxation of the K intermediate are coupled to structural rearrangement for efficient photoisomerization and proton translocation, respectively, a detailed analysis is required.
One potentially useful approach is to assign and study the vibrational bands in FTIR difference spectra. This approach is applicable for side chains, peptide backbone, and water molecules (24). For instance, BR has 18 threonines, with 11 in the trans-membrane region. These are Thr-17 and Thr-24 in helix A; Thr-46, Thr-47, and Thr-55 in helix B; Thr-89 and Thr-90 in helix C; Thr-121 in helix D; Thr-142 in helix E; Thr-178 in helix F; and Thr-205 in helix G (Fig. 1). Thus, threonine residues are distributed over all of the helices, in both their extracellular and cytoplasmic regions, and can be good internal probes. Furthermore, in view of their propensity to form hydrogen bonds, which and how threonines change on photoisomerization are themselves intriguing questions. The distances from the side-chain oxygen of threonines to the nearest atom of the retinal chromophore (Scheme S1) are listed in Table 1 on the basis of the six structures in the protein data bank. Although independently refined, the two models with the highest resolutions (9, 11) yielded virtually identical values. Three residues, Thr-89, Thr-90, and Thr-142, are in direct van der Waals contact with the retinal. On the other hand, in four residues, Thr-17, Thr-47, Thr-55, and Thr-205, the side-chain oxygen atoms are more than 10 Å from the chromophore, although they are located in the intramembrane region. We expect that, when the retinal moves on all-trans to 13-cis isomerization, neighboring threonines can change their structure through direct steric interaction; however, the influence of distant threonines would occur indirectly.
Figure 1.

Structure of BR (1BRX; ref. 10). (a) View from the cytoplasmic side along the membrane normal. (b) View from the C helix side along the membrane. The retinal chromophore is colored yellow in the space-filling model. Backbones of A–D helices (residues < 131) and E–G helices (residues ≥ 131) are colored blue and red, respectively. The side chains of the 11 threonines inside the membrane, at positions 17, 24, 46, 47, 55, 89, 90, 121, 142, 178, and 205, are represented by space-filling models. The other threonines are at positions 5, 67, 107, 128, 157, 170, and 247 in the loop and terminal regions.
Scheme 1.

Structure of the retinal chromophore.
Table 1.
The distance from the oxygen atom of each threonine residue to the nearest atom of the retinal chromophore (identified with the numbering convention in Scheme S1)
| Residue | Average distance, Å* | Distance, Å
|
|||||
|---|---|---|---|---|---|---|---|
| 2BRD† [5] | 2AT9† [7] | 1QHJ‡ [9] | 1C3W‡ [11] | 1BRR‡ [12] | 1BM1‡ [13] | ||
| Thr-17 | 11.74 ± 0.63 | Cɛ: 11.52 | Cδ: 12.90 | Cδ: 11.70 | Cδ: 11.56 | Cγ: 11.72 | Cɛ: 11.01 |
| Thr-24 | 8.97 ± 1.32 | Cδ: 11.52 | Cγ: 9.25 | Cδ: 8.28 | Cδ: 8.42 | Cδ: 8.31 | Cδ: 8.04 |
| Thr-46 | 7.25 ± 0.95 | Cγ: 7.25 | Cγ: 9.29 | Cγ: 9.88 | Cγ: 9.76 | Cγ: 9.13 | Cγ: 9.32 |
| Thr-47 | 11.39 ± 0.42 | Cγ: 10.68 | Cγ: 11.65 | Cγ: 11.69 | Cγ: 11.66 | Cγ: 11.59 | Cγ: 11.04 |
| Thr-55 | 10.50 ± 0.69 | Cδ: 10.03 | Cɛ: 10.93 | Cɛ: 9.70 | Cɛ: 9.94 | Cδ: 11.36 | Cδ: 11.02 |
| Thr-89 | 3.62 ± 0.30 | Cδ: 3.53 | C15: 3.64 | Cɛ: 3.32 | Cɛ: 3.33 | Cɛ: 4.12 | Cδ: 3.75 |
| Thr-90 | 5.09 ± 1.01 | C11: 3.65 | C12: 6.00 | C11: 5.67 | C11: 5.58 | C11: 5.68 | C12: 3.95 |
| Thr-121 | 7.73 ± 1.43 | C4: 9.28 | C4: 6.50 | C2: 6.52 | C3: 6.30 | C2: 8.66 | C4: 9.12 |
| Thr-142 | 4.20 ± 0.71 | 5-Me: 5.14 | 5-Me: 3.90 | 5-Me: 3.67 | C4: 3.50 | C4: 3.96 | 5-Me: 5.05 |
| Thr-178 | 7.64 ± 0.58 | 13-Me: 7.64 | 9-Me: 7.50 | 13-Me: 7.35 | 13-Me: 7.35 | 13-Me: 8.79 | 13-Me: 7.23 |
| Thr-205 | 12.72 ± 0.60 | 1-Me: 13.40 | N: 11.79 | 1-Me: 12.96 | 1-Me: 12.67 | 1-Me: 13.20 | 1-Me: 12.28 |
*Average distances are shown ± SD and are taken from the six structures.
†Cryoelectron microscopy.
‡X-ray crystallography.
For the present study, we prepared [3-18O]threonine-labeled BR and used polarized FTIR spectroscopy to investigate the possible involvement of hydrogen-bonding alterations in the photoisomerization of the retinal chromophore. As a result of the accurate detection that we have developed throughout the mid-IR region (23), we found that three threonines change their hydrogen bonding. Further mutation studies allowed determination of the identities of the altered sites. The results are unexpected and not simply distance dependent.
Materials and Methods
Preparation of Samples.
l-[3-18O]threonine was synthesized as described (25). [3-18O]threonine-labeled BR was prepared by growing Halobacterium salinarum (JW-3) in a defined medium similar to that described by Gochnauer and Kushner (26), except that the d-amino acids and NH4Cl were omitted and the l-threonine was replaced by 0.25 g/liter l-[3-18O]threonine. Under these conditions, lipid extraction and amino acid analysis with radiotracers show that typically about two-thirds of the threonine residues are labeled, with no scrambling of the label (27). The mutated genes for T17V, T24V, T46V, T47V, T55V, T89S, T90V, T121V, T142N, T178N, and T205V were constructed and introduced into Halobacterium salinarum as described (28). The purple membrane was isolated by the method of Oesterhelt and Stoeckenius (29).
Polarized FTIR Spectroscopy.
Polarized FTIR spectroscopy was applied as described (23, 25). A 120-μl aliquot of the sample in 2 mM phosphate buffer (pH 7.0) was dried on a BaF2 window with diameter of 18 mm. After hydration by 1 μl of D2O, the sample was placed in a cell and then mounted in an Oxford DN-1704 cryostat. The film was illuminated with >500-nm light for 1 min at 273 K to obtain the light-adapted state of BR.
Illumination with 501-nm light (Toshiba interference filter; full width of half maximum = 4 nm; ref. 30) at 77 K for 2 min converted BR to the K intermediate. Because the K intermediate completely reverted to BR on illumination with >660-nm light for 1 min, as evidenced by the same but inverted spectral shape, the cycles of alternative illuminations with a 501-nm light and >660-nm light were repeated a number of times. The difference spectrum was calculated from the spectra constructed with 128 interferograms before and after the illumination, and 24 spectra obtained in this way were averaged for each K minus BR spectrum under various conditions.
The details of polarized FTIR spectroscopy are described elsewhere (31, 32). Briefly, a BaF2 polarizer in the vertical xy plane is placed in front of a mercury-cadmium-technetium detector in a Bio-Rad FTS-40 FTIR spectrometer. The IR probe light travels along the z axis to the window with the vertical and horizontal polarizations, AV and AH, in the xz and yz planes, respectively. The window in the xy plane was tilted around the vertical x axis by rotation of the rod holding the window. The tilt angles (φ0) were 0°, 17.8°, 35.7°, and 53.5°. The dichroic ratio R is defined as
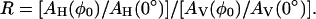 |
1 |
Increase in the intensity of the AV component from the increased effective number of BR molecules absorbing light with tilting was corrected in Eq. 1. R is related to the angle between the dipole moment and the membrane normal θ0 by the equation described previously,
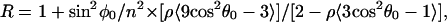 |
2 |
where n, the refractive index of the film in the IR region, was assumed to be 1.7 as used previously, and ρ, the degree of orientation of the membrane, was taken to be 0.95 as reported before (31, 32). The latter assumption is reasonable, as judged from the intensity ratio of the amide II/amide I in our unhydrated film of 1.02 in comparison with the earlier value of 0.98.
Results and Discussion
Assignment of the O-H and O-D Stretch Bands to Threonine Side Chains.
The dotted lines in Fig. 2 show K minus BR difference spectra in D2O at sample tilt angles of 0° (Fig. 2a) and at 53.5° (Fig. 2b), which reproduced previous results (23). The solid lines show the spectra of [3-18O]threonine-labeled BR. The solid and dotted lines coincide well, except for some regions that identify threonine as the origin of the bands. The difference is more prominent at 53.5°, where three peak pairs show spectral downshift. These bands are thus assigned to the stretching vibrations of threonines. Among three peak pairs, two are O-H stretches, and one is an O-D stretch, as judged from their frequencies.
Figure 2.

The K minus BR difference spectra of unlabeled (dotted lines) and [3-18O]threonine-labeled (solid lines) BR in the 3,700- to 2,000-cm−1 region with the window-tilting angles (φ0) of 0° (a) and 53.5° (b). The sample was hydrated with D2O, and spectra were measured at 77 K. These frequencies cover the entire O-H and O-D stretches. One division of the y axis corresponds to 0.001 absorbance unit. Both dotted and solid lines overlap well except for three frequency region shown by arrows, indicating that three threonines change the stretching frequencies of their O-H (O-D) groups on photoisomerization. Diff. Abs., difference absorbance.
The D2O-sensitive bands at 2,506 (−) and 2,466 (+) cm−1 have been assigned recently (25) to the O-D stretch of Thr-89. The corresponding O-H stretching vibrations are at 3,378 cm−1 in BR and at 3,317 cm−1 in the K intermediate (23). The latter value is at a lower frequency than the values of neat secondary alcohol (3,340–3,355 cm−1), indicating strong interaction with a hydrogen-bond acceptor. As determined by both crystallographic structures and spectroscopic evidence, this acceptor is the negatively charged side chain of Asp-85. Strengthened hydrogen bonding on photoisomerization implies that the distance between Thr-89 and Asp-85 becomes shorter. Thr-89 is in direct contact with the chromophore near the Schiff base (5–13) such that retinal isomerization should affect the hydrogen-bonding structure of Thr-89. No other changes were observed for D2O-exchangeable threonines.
Perturbations of threonine residues are observed in the O-H stretching region (3,500–3,400 cm−1) also (Fig. 2). The detailed spectral changes of these D2O-insensitive bands are shown in Fig. 3. The measurement at 0° tilt (Fig. 3a) shows that a peak pair at 3,480 (+) and 3,462 (−) cm−1 of unlabeled BR (dotted line) has a clear downshift to 3,474 (+) and 3,452 (−) cm−1, respectively, in the measurement of [3-18O]threonine-labeled BR (solid line). Although no other changes are visible at 0° (Fig. 3a), another change is observed on tilting of the sample window. Fig. 3d measured at 53.5° clearly shows that a peak pair at 3,415 (+) and 3,402 (−) cm−1 of unlabeled BR (dotted line) has a downshift to 3,399 (+) and 3,387 (−) cm−1, respectively, in the measurement of [3-18O]threonine-labeled BR (solid line).
Figure 3.

The K minus BR difference spectra of unlabeled (dotted lines) and [3-18O]threonine-labeled (solid lines) BR in the 3,530- to 3,350-cm−1 region. The sample was hydrated with D2O. The window-tilting angles (φ0) are 0° (a), 17.8° (b), 35.7° (c), and 53.5° (d). One division of the y axis corresponds to 0.001 absorbance unit. Two peak pairs, 3,480 (+)/3,462 (−) cm−1 and 3415 (+)/3402 (−) cm−1, are able to be assigned for the O-H stretching vibrations of threonine side chains. Diff. Abs., difference absorbance.
When the sample window is tilted, the positive 3,480-cm−1 band becomes larger, whereas the negative 3,462-cm−1 band slightly loses its intensity. Particularly remarkable enhancement is observed for the 3,415 (+)/3,402 (−) cm−1 bands. These variations originate from the orientations of the dipole moments of the O-H stretching modes relative to the membrane normal. From Eqs. 1 and 2, the angles were determined as follows: 45° for the 3,480-cm−1 (+) band, 65° for the 3,462-cm−1 (−) band, 32° for the 3,415-cm−1 (+) band, and 26° for the 3,402-cm−1 (−) band. It is noted that the corresponding bands are not determined uniquely; because of possible overlap, three peaks do not necessarily indicate that only three threonines change.
Location of the Threonine Side Chains: Mutation Studies.
As shown in Fig. 1, threonine residues are distributed over all seven helices, with 11 residues in the intramembrane region. In addition to the previously identified D2O-exchangeable O-H group of Thr-89, which threonines change on photoisomerization? Table 1 shows the distance between the oxygen atom of each threonine and the nearest atom of the retinal chromophore. Thr-89 is indeed the closest one, at about 3.6 Å from the Schiff base region. Which are the other perturbed threonines? Although the most direct and unambiguous method of identifying these bands would be site-directed isotope labeling, we attempted to assign them from FTIR spectra of threonine mutants. We prepared mutants that replace each of the 11 threonines in the intramembrane region (Table 1) and performed polarized FTIR spectroscopy at 77 K.
In all cases, the chromophore bands indicated that the photoproduct was the K intermediate (data not shown). Fig. 4 shows the K minus BR difference spectra in the 3,530- to 3,350-cm−1 region of various mutants (Fig. 4 a–k) in D2O as well as in that of the wild type (dotted line). As can be clearly seen, the 3,480 (+)/3,462 (−) cm−1 band disappears in T17V (Fig. 4a), whereas the bilobic spectral feature is preserved in all other threonine mutants (Fig. 4 b–k). Thus, the O-H stretching vibration at 3,462 cm−1 in BR and at 3,480 cm−1 in K is assigned uniquely to that of Thr-17.
Figure 4.

The K minus BR difference spectra in the 3,530- to 3,350-cm−1 region for the wild type (dotted lines) and the mutants (solid lines) T17V (a), T24V (b), T46V (c), T47V (d), T55V (e), T89S (f), T90V (g), T121V (h), T142N (i), T178N (j), and T205V (k). The sample was hydrated with D2O, and the window-tilting angle (φ0) is 53.5°. One division of the y axis corresponds to 0.001 absorbance unit.
On the other hand, the 3,415 (+)/3,402 (−) cm−1 bands diminished in both T90V (Fig. 4g) and T121V (Fig. 4h). If Thr-90 and Thr-121 contribute to this band independently, mutation of either residue must reduce the amplitude of this band by half. The nearly complete abolition of this band in both T90V and T121V mutants strongly suggests that one of them possesses the O-H stretching vibration and that the mutation of the other causes loss of the change in the O-H stretching vibration by a secondary effect. Therefore, the O-H stretching vibration originates from either Thr-90 or Thr-121.
Structure of Thr-90 and Thr-121 and Their Changes.
Thus, we could not decide whether Thr-90 or Thr-121 has its O-H stretching vibration at 3,402 cm−1. However, the two possibilities might be discriminated by comparing the orientation of the measured dipole moment with the orientations in the BR structural models. The present study showed that the orientation of the dipole moment of the O-H stretch in question is 26° from the membrane normal. Although crystallographic structures do not possess the coordinates of hydrogen atoms, it is most likely that the O-H group of threonine forms hydrogen bonding with neighboring acceptors. The angle between the right threonine oxygen and the hydrogen-bonding acceptor should be about 26°. Table 2 shows that the oxygen atom of the peptide carbonyl of Trp-86 is within 3 Å from the oxygen atom of the side chain of Thr-90 in all six structures. The angle of this band is in the range of 50–65°, except in the model by Mitsuoka et al. (23.0°; ref. 7). The carboxylic oxygen of Asp-115 is also within 3 Å of Thr-90 in three of the structures (1QHJ, 1C3W, and 1BRR), but the angle is greater than the magic angle. For Thr-121, the oxygen atom of the peptide carbonyl of Ile-117 is located near the oxygen atom of its side chain in all six structures, making it the likely hydrogen-bonding acceptor. However, the angle is in the range of 45–50°, except in the model of Mitsuoka et al. (31.7°; ref. 7). The angle of the O-H group (26°) in the present study is therefore coincident with both Thr-90 and Thr-121 according to the structure by Mitsuoka et al. (7) and corresponds to neither Thr-90 nor Thr-121 in the other five structures (5, 9, 11–13). Thus, comparison with the BR structures does not allow identification of the threonine band at 3,402 cm−1.
Table 2.
The distance from the threonine oxygen to neighboring oxygen atoms and the angle between the O-O vector and the membrane normal
| Residue | Distance, Å; angle, degree
|
IR, degree | |||||
|---|---|---|---|---|---|---|---|
| 2BRD* [5] | 2AT9* [7] | 1QHJ† [9] | 1C3W† [11] | 1BRR† [12] | 1BM1† [13] | ||
| Thr-17 | 65 | ||||||
| O of Leu-13 | 2.86; 73.5 | 3.54; 51.0 | 2.72; 67.9 | 2.72; 70.4 | 2.56; 70.5 | 3.10; 70.6 | |
| 26 | |||||||
| Thr-90 | |||||||
| O of Trp-86 | 2.83; 64.6 | 2.57; 23.0 | 2.55; 53.0 | 2.54; 57.4 | 2.50; 50.1 | 2.84; 64.1 | |
| S.C. O of Asp-115‡ | 2.72; 67.9 | 2.40; 75.8 | 2.50; 80.9 | ||||
| Thr-121 | |||||||
| O of Ile-117 | 2.84; 50.9 | 2.74; 31.7 | 2.59; 45.6 | 2.59; 45.8 | 2.94; 48.1 | 3.17; 48.4 | |
*Cryoelectron microscopy.
†X-ray crystallography.
‡One of the side-chain (S.C.) oxygens of Asp-115.
On the other hand, the FTIR spectra of T90V and T121V in other frequency regions do provide some clues for the location of the threonine residue. The spectrum of T121V is similar to that of the wild type, but many differences are seen between T90V and the wild type. These are most significant in the chromophore bands. This observation is consistent with contact of Thr-90 with the retinal. In contrast, Thr-121 faces the β-ionone ring of the retinal near helix E (Table 1) but is not in direct contact with it. Normal proton pumping has been reported for T121V, whereas the pumping activity was reduced to 60–70% in T90V (33). On the basis of these facts, it is more likely that the 3,402-cm−1 band originates from Thr-121, whereas the mutation at position 90 causes global structural changes that could include the region of Thr-121. It is also supported by the fact that the O-H group of Asp-115 is sensitive to D2O. Because two side-chain oxygens form a hydrogen bond between Thr-90 and Asp-115, the D2O-insensitive O-H group is unlikely to originate from Thr-90.
If the 3,402-cm−1 band originates from Thr-121, as we believe, then Thr-89 is the only one of the three perturbed threonines that change their hydrogen bonding in the K state (Thr-17, Thr-89, and Thr-121) that is in direct van der Waals contact with the retinal chromophore (Table 1). The retinal is located in a restricted binding pocket, and the ultrafast isomerization reaction must occur without greatly changing the surrounding protein structure (21, 22).
Structure of Thr-17 and Its Change.
Of the three threonines, Thr-17 is the farthest from the retinal chromophore. In fact, in all six structures, even the closest atom of the retinal chromophore is more than 11 Å from the oxygen atom of Thr-17 (Table 1). In all crystallographic structures, the hydrogen-bonding acceptor of the O-H group of Thr-17 is the oxygen atom of the peptide carbonyl of Leu-13 (Table 2). The angle of the dipole moment of the O-H stretch in all structures (50–75°) agrees with the results for the 3,402-cm−1 band (65°). The spectral features of the Thr-17 band are influenced in frequency by mutations of Thr-46, Thr-55, Thr-89, and Thr-178 (Fig. 4), indicating that these mutations affect the fine structure of the threonine at position 17.
The space between the retinal chromophore and Thr-17 is occupied by two bulky groups, Tyr-57 and Met-20, which are connected with Asp-212 in the Schiff base region by van der Waals contact and hydrogen bonding (8–13). Thus, it is likely that retinal motion in the active center forces some movements in these bulky groups such that hydrogen bonding of Thr-17 is altered. Motions of Tyr-57 and Met-20 were indeed observed in the recent x-ray crystallographic study of the K intermediate (22). The shift of the O-H stretch to higher frequency indicates a weakened hydrogen bond at position 17. Thr-17 is located in the extracellular side; however, it is not connected to a hydrogen-bonding network, because the O-H group of Thr-17 is not exchangeable for D2O.
The all-trans to 13-cis photoisomerization occurs in the femtosecond regime (15–17). The primary photoproduct is the J intermediate, and it converts to the K intermediate in 3 ps. Therefore, it is unexpected for a conformation change to be transmitted over 11 Å on formation of the K intermediate. However, the selective and efficient nature of the photoisomerization in BR has been attributed to the surrounding protein (23). We therefore suggest that the specific protein environment involving Thr-17 may work as the specific reaction field of photoisomerization. Tyr-57 and Asp-212 are all conserved among archaeal rhodopsins, whereas Thr-17 and Met-20 are conserved in the proton-pumping BR family (34). Thus, there may be common features in their structure and dynamics. In structure-function studies of BR, amino acid residues in the A helix have never been taken into account. Although no change on the proton-pumping activity has been reported for T17V (33), the possible steric contribution of Thr-17 to the primary photoisomerization processes has to be examined.
Conclusions
Of the 18 threonines in BR, 3 change their hydrogen-bonding strengths in the K state formed on photoisomerization. One is Thr-89, which is in direct contact with the retinal chromophore; its hydrogen bond becomes strengthened. The others are Thr-17 and Thr-121 (or Thr-90), and their hydrogen bonds become weaker. The observation of no changes in the other 15 threonines suggests that, at this stage, the protein structural alteration is local and specific. This result is consistent with the need for the protein to mediate selective and efficient photoisomerization. The involvement of Thr-17, >11 Å from the chromophore, in the protein structural change implicates the presence of a specific perturbation channel in protein accompanying the retinal motion.
Acknowledgments
This work was supported by Japanese Ministry of Education, Culture, Sports, and Science, Japan, Grants 10206206, 10358016, and 11480193 (to H.K.) and by National Institutes of Health Grant GM 36810 (to J.H.). Y.Y. is supported by a research fellowship from the Japan Society for the Promotion of Science for Young Scientists.
Abbreviations
- BR
bacteriorhodopsin
- FTIR
Fourier-transform IR
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.080064797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.080064797
References
- 1.Mathies R A, Lin S W, Ames J B, Pollard W T. Annu Rev Biophys Biophys Chem. 1991;20:491–518. doi: 10.1146/annurev.bb.20.060191.002423. [DOI] [PubMed] [Google Scholar]
- 2.Ebrey T G. In: Thermodynamics of Membranes, Receptors and Channels. Jackson M, editor. New York: CRC; 1993. pp. 353–378. [Google Scholar]
- 3.Lanyi J K. Biochim Biophys Acta. 1993;1183:241–261. doi: 10.1016/0005-2728(93)90226-6. [DOI] [PubMed] [Google Scholar]
- 4.Haupts U, Tittor J, Oesterhelt D. Annu Rev Biophys Biomol Struct. 1999;28:367–399. doi: 10.1146/annurev.biophys.28.1.367. [DOI] [PubMed] [Google Scholar]
- 5.Grigorieff N, Ceska T A, Downing K H, Baldwin J M, Henderson R. J Mol Biol. 1996;259:393–421. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 6.Kimura Y, Vassylyev D G, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T, Fujiyoshi Y. Nature (London) 1997;389:206–211. doi: 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- 7.Mitsuoka K, Hirai T, Murata K, Miyazawa A, Kidera A, Kimura Y, Fujiyoshi Y. J Mol Biol. 1999;286:861–882. doi: 10.1006/jmbi.1998.2529. [DOI] [PubMed] [Google Scholar]
- 8.Pebay-Peyroula E, Rummel G, Rosenbusch J P, Landau E M. Science. 1997;277:1676–1681. doi: 10.1126/science.277.5332.1676. [DOI] [PubMed] [Google Scholar]
- 9.Belrhali H, Nollert P, Royant A, Menzel C, Rosenbusch J P, Landau E M, Pebay-Peyroula E. Structure (London) 1999;7:909–917. doi: 10.1016/s0969-2126(99)80118-x. [DOI] [PubMed] [Google Scholar]
- 10.Luecke H, Richter H-T, Lanyi J K. Science. 1998;280:1934–1937. doi: 10.1126/science.280.5371.1934. [DOI] [PubMed] [Google Scholar]
- 11.Luecke H, Schobert B, Richter H-T, Cartailler J P, Lanyi J K. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 12.Essen L, Siegent R, Lehmann W D, Oesterhelt D. Proc Natl Acad Sci USA. 1998;95:11673–11678. doi: 10.1073/pnas.95.20.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato H, Takeda K, Tani K, Hino T, Okada T, Nakasako M, Kamiya N, Kouyama T. Acta Crystallogr D. 1999;55:1251–1256. doi: 10.1107/s090744499900503x. [DOI] [PubMed] [Google Scholar]
- 14.Lanyi J K. J Biol Chem. 1997;272:31209–31212. doi: 10.1074/jbc.272.50.31209. [DOI] [PubMed] [Google Scholar]
- 15.Dobler J, Zinth W, Kaiser W, Oesterhelt D. Chem Phys Lett. 1988;144:215–220. [Google Scholar]
- 16.Kandori H, Yoshihara K, Tomioka H, Sasabe H, Shichida Y. Chem Phys Lett. 1993;211:385–391. [Google Scholar]
- 17.Hasson K C, Gai F, Anfinrud P A. Proc Natl Acad Sci USA. 1996;93:15124–15129. doi: 10.1073/pnas.93.26.15124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koyama Y, Kubo K, Komori M, Yasuda H, Mukai Y. Photochem Photobiol. 1991;54:433–443. doi: 10.1111/j.1751-1097.1991.tb02038.x. [DOI] [PubMed] [Google Scholar]
- 19.Govindjee R, Balashov S P, Ebrey T G. Biophys J. 1990;58:597–608. doi: 10.1016/S0006-3495(90)82403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tittor J, Oesterhelt D. FEBS Lett. 1990;263:269–273. [Google Scholar]
- 21.Bullough P A, Henderson R. J Mol Biol. 1999;286:1663–1671. doi: 10.1006/jmbi.1999.2570. [DOI] [PubMed] [Google Scholar]
- 22.Edman K, Nollert P, Royant A, Belrhali H, Pebay-Peyroula E, Hajdu J, Neutze R, Landau E M. Nature (London) 1999;401:822–826. doi: 10.1038/44623. [DOI] [PubMed] [Google Scholar]
- 23.Kandori H, Kinoshita N, Maeda A, Shichida Y. J Phys Chem B. 1998;102:7899–7905. [Google Scholar]
- 24.Maeda A, Kandori H, Yamazaki Y, Nishimura S, Hatanaka M, Chon Y-S, Sasaki J, Needleman R, Lanyi J K. J Biochem (Tokyo) 1997;121:399–406. doi: 10.1093/oxfordjournals.jbchem.a021602. [DOI] [PubMed] [Google Scholar]
- 25.Kandori H, Kinoshita N, Yamazaki Y, Maeda A, Shichida Y, Needleman R, Lanyi J K, Bizounok M, Herzfeld J, Raap J, et al. Biochemistry. 1999;38:9676–9683. doi: 10.1021/bi990713y. [DOI] [PubMed] [Google Scholar]
- 26.Gochnauer M B, Kushner D J. Can J Microbiol. 1969;15:1157–1165. doi: 10.1139/m69-211. [DOI] [PubMed] [Google Scholar]
- 27.Karstens W F J, Berger H J F F, van Haren E R, Lugtenburg J, Raap J. J Labelled Compd Radiopharm. 1995;36:1077–1096. [Google Scholar]
- 28.Needleman R, Chang M, Ni B, Váró G, Fornes J, White S H, Lanyi J K. J Biol Chem. 1991;266:11478–11484. [PubMed] [Google Scholar]
- 29.Oesterhelt D, Stoeckenius W. Methods Enzymol. 1973;31:667–678. doi: 10.1016/0076-6879(74)31072-5. [DOI] [PubMed] [Google Scholar]
- 30.Kandori H, Maeda A. Biochemistry. 1995;34:14220–14229. doi: 10.1021/bi00043a029. [DOI] [PubMed] [Google Scholar]
- 31.Hatanaka M, Kandori H, Maeda A. Biophys J. 1997;73:1001–1006. doi: 10.1016/S0006-3495(97)78133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kandori H. J Am Chem Soc. 1998;120:4546–4547. [Google Scholar]
- 33.Marti T, Otto H, Mogi T, Rösselet S J, Heyn M P, Khorana H G. J Biol Chem. 1991;266:6919–6927. [PubMed] [Google Scholar]
- 34.Ihara K, Umemura T, Katagiri I, Kitajima-Ihara T, Sugiyama Y, Kimura Y, Mukohata Y. J Mol Biol. 1999;285:163–174. doi: 10.1006/jmbi.1998.2286. [DOI] [PubMed] [Google Scholar]