

Abstract
Comparative genomic hybridizations have been used to examine genetic relationships among bacteria. The microarrays used in these experiments may have open reading frames from one or more reference strains (whole-genome microarrays), or they may be composed of random DNA fragments from a large number of strains (mixed-genome microarrays [MGMs]). In this work both experimental and virtual arrays are analyzed to assess the validity of genetic inferences from these experiments with a focus on MGMs. Empirical data are analyzed from an Enterococcus MGM, while a virtual MGM is constructed in silico using sequenced genomes (Streptococcus). On average, a small MGM is capable of correctly deriving phylogenetic relationships between seven species of Enterococcus with accuracies of 100% (n = 100 probes) and 95% (n = 46 probes); more probes are required for intraspecific differentiation. Compared to multilocus sequence methods and whole-genome microarrays, MGMs provide additional discrimination between closely related strains and offer the possibility of identifying unique strain or lineage markers. Representational bias can have mixed effects. Microarrays composed of probes from a single genome can be used to derive phylogenetic relationships, although branch length can be exaggerated for the reference strain. We describe a case where disproportional representation of different strains used to construct an MGM can result in inaccurate phylogenetic inferences, and we illustrate an algorithm that is capable of correcting this type of bias. The bias correction algorithm automatically provides bootstrap confidence values and can provide multiple bias-corrected trees with high confidence values.
Genetic discrimination at the genus, species, or strain level is important in bacterial phylogeny, epidemiology, and ecology studies (17, 27), and a practical discrimination method should quickly and accurately characterize unknown isolates and be relatively simple to implement in the lab. Although genetic discrimination can be accomplished by sequencing and alignment of complete genomes (8), cost and time make this approach impractical for many groups of bacteria, particularly when considering intraspecies comparisons. Common approaches to this problem include single locus (for example, 16S rRNA) and multilocus sequence alignments, but the inferences of these analyses can depend on which genes are selected for analysis (28). For example, the phylogenetic relationship between strains of Streptococcus and Enterococcus has been examined by sequence alignment of the single genes rnpB and atpA, respectively (19, 26). Multilocus sequence typing (MLST) includes data from multiple shared genes (11, 15, 16), and while this is clearly an improvement over potential biases generated from single-locus procedures, incorporating genome-wide information into phylogeny studies would be more objective and comprehensive given the complexity of evolutionary processes (29, p. 67).
With the availability of whole-genome sequences and the introduction of microarrays, comparative genomic hybridization (CGH) is being used to make phylogenetic inferences between bacteria (9, 10, 18, 21, 24). One common CGH method relies on a whole-genome microarray (18, 21) constructed from most of the open reading frames of one completely sequenced reference strain. Hybridization of sample strains onto the array identifies genes that are either present or absent or else are highly divergent from a sample strain. Genetic relatedness is determined based on a comparison of gene presence and absence patterns (or more specifically, accessory gene presence and absence patterns) among sample strains.
Using a whole-genome microarray and the CGH approach to infer phylogenetic relationships is more advantageous than single and multilocus methods, if only because much more information is incorporated into the analysis (10). Nevertheless, there are two inherent problems with using CGH data in this manner. First, only those genes harbored by the reference strain are available for analysis; genes specific to nonsequenced strains are not included. For example, genomes for the Escherichia coli strains CFT073, K-12, O157:H7, and H3110 share only 40% of their genetic content. Therefore, construction of a microarray using any one of these might not render sufficient information to discriminate between the other strains. Second, use of a single genome as the basis of comparison may introduce bias into the analysis. In an extreme case, all target strains that share few genes with the reference strain will appear closely related by CGH even if they are actually highly divergent from each other.
One alternative approach is to incorporate genetic information from multiple strains within a single microarray. This can be done by inclusion of specific genes from multiple whole-genome sequences (if available) or by using a “mixed-genome microarray” (MGM) that incorporates randomly selected gene fragments from many strains of bacteria (2-4, 25). An MGM is constructed from a shotgun library built from a pool of isolates belonging to the same species or genera of interest. Genomic DNA (gDNA) is fragmented by sonication (25) or by use of restriction enzymes (2-4) and size fractionated to isolate 500- to 600-bp fragments. The fragments are cloned, and a randomly selected collection of clones is used to construct a glass-based microarray. Genetic comparisons are made by hybridizing gDNA from test strains and assessing signal intensity across the multiple strains. For analysis, hybridization data can be converted to binary variables (present or absent), or relative intensities can be compared. In addition to its usefulness in CGH, inclusion of genetic variation from a number of different reference strains on the MGM enables the detection of lineage- or strain-specific genes that can serve as useful molecular markers or as targets for further functional analysis (3, 4, 25). Finally, MGMs have an advantage over conventional CGH arrays because no a priori information about the genome is required to construct these microarrays.
It would be ideal if all isolates were equally represented in the library used to construct the MGM. Nevertheless, depending on the scope of the project, this may not be practical, and it also assumes that the resultant shotgun library produces a truly random selection of clones (4) whereby all strains are equally represented. Thus, the question of library bias due to unequal strain representation arises as an important issue. Can phylogenetic relationships be correctly determined with the existence of library bias or incomplete representation with respect to the target strains? In this paper, we use both experimental and computational methods to assess the applicability of the MGM in determining phylogenetic relationships among strains of bacteria. The experimental method uses an Enterococcus MGM, and the computational method uses a virtual Streptococcus microarray to simulate MGM experiments, including construction, hybridization, imaging, and analysis. We show that MGM results can be used to accurately infer phylogenetic relationships among strains. We also analyze the effects of array size and library bias on the accuracy of the MGM, and we provide an easily applied method that effectively corrects for library bias.
MATERIALS AND METHODS
Enterococcus MGM.
An MGM was constructed as described by Soule et al. (25). Briefly, 413 Enterococcus isolates were collected from 5 animal hosts (cows, n = 200; dogs, n = 61; waterfowl, n = 43; humans, n = 54; and elk and deer, n = 55). Equal amounts of gDNA from 50 isolates per host (43 for waterfowl) were mixed to prepare 5 separate shotgun libraries. Probes (n = 4,320; 864 probes per library) were spotted onto glass slides to form 8 subarrays using a BioRobotics MicroGrid II arrayer (Genomic Solutions, Ann Arbor, MI). Four replicates of a fragment of a 16S rRNA gene from an Enterococcus sp. isolate were spotted on each subarray as a control for hybridization efficiency, and four replicates of an arbitrary biotinylated oligonucleotide were spotted as a control for detection chemistry.
Sample hybridization and detection.
Seven Enterococcus strains (generously donated by Rachel Noble, University of North Carolina, Chapel Hill) were used in this analysis: E. hirae, E. gallinarum, E. dispar, E. avium, E. faecalis, E. casseliflavus, and E. faecium. Isolates were retrieved from −80°C and recovered by streaking on M-Enterococcus agar plates and incubation for 48 to 72 h at 37°C. For each strain, one colony was placed into 3 ml brain heart infusion broth and grown overnight at 37°C. Genomic DNA was extracted using a DNeasy tissue kit (QIAGEN, Valencia, CA) and quantified using spectrophotometry. A segment of 16S rRNA was PCR amplified as described by Soule et al. (25) and sequenced to verify strain identity. Genomic DNA (1 μg) was fragmented and biotinylated using nick translation (Bio-Nick kit; Invitrogen, Carlsbad, CA), and after ethanol precipitation the labeled DNA was resuspended in 80 μl hybridization buffer (4× SSC [60 mM NaCl, 0.6 mM sodium citrate; pH 7.0) and 5× Denhardt's solution (0.1% [wt/vol] Ficoll, 0.1% polyvinylpyrrolidone, 0.1% bovine serum albumin). Slides were preblocked for 30 min at room temperature with 200 μl TNB buffer (100 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% blocking reagent [TSA biotin system; Perkin-Elmer, Boston, MA]). Nick-translated gDNAs (80 μl) were heat denatured (95°C for 2 min), applied to the slide, enclosed by a humidified, conical tube (50 ml), and incubated overnight at 60°C. After incubation, the slides were then incubated and washed as described previously (5), with 600 μl of the appropriate reagent applied to the slide at each step. After the final washing and drying, slides were imaged with an arrayWoRxe scanner (Applied Precision, Issaquah, WA). The resulting images were stored as TIFF files with pixel values ranging from 0 to 65,535. Each strain was hybridized on two independent slides. Images were segmented using SoftWoRx software (Applied Precision, Issaquah, WA), and median probe intensity values were exported to Microsoft (Redmond, WA) Excel.
Enterococcus MGM analysis.
Hybridization data sets (n = 14) were normalized with respect to the average intensity of the four 16S rRNA control spots on each subarray. Normalized intensities of spots from replicate slides were averaged. Data were converted to a binary format; probes having a normalized intensity of <0.5 were considered absent or highly divergent (“0”), and probes with intensity values of ≥0.5 were considered present (“1”). A Euclidean distance matrix was then calculated using
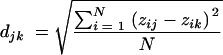 |
where N is the total number of probes on the microarray and zij is the normalized signal intensity of spot i for sample j. The distance matrix was used to construct a phylogenetic tree using the neighbor-joining method (Phylip 3.64) (12), and the output file was viewed using Treeview (version 1.6.6) (20).
MLST analysis.
Probes with intensities greater than 60,000 for all 14 Enterococcus hybridizations were retrieved from the clone library, PCR amplified with primers T3 and T7, and then sequenced (Amplicon Express, Pullman, WA). Probe sequences were trimmed for vector contamination, and PCR primer sets were designed for each probe sequence using primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) (22) with default settings except for product size, which was set to >400 bp, and primer length, which was constrained to 20 bp. Primer sets (Table 1) were commercially synthesized (Invitrogen) and used to amplify corresponding sequences from each Enterococcus strain. PCR was performed in 50-μl volumes with 10 μl of gDNA template (10 ng), 1× reaction buffer (Fisher Scientific, Pittsburgh, PA), 0.2 mM (each) deoxynucleoside triphosphate, 2 mM MgCl2, 1 U Taq (Fisher Scientific), and 0.4 μM of each primer. The cycling program consisted of 2 min of initial denaturation at 95°C, followed by 30 cycles of 95°C for 30 s, annealing at 56°C for 60 s, and 72°C for 60 s, and it concluded with a final extension at 72°C for 10 min. The annealing temperature for the EPE8 product was 48.6°C. PCR products were sequenced (Amplicon Express) and submitted to GenBank. Genetic sequences corresponding to different genetic fragments were concatenated in the same order and stored in FASTA format for alignment (ClustalW1.8.3 at http://www.ebi.ac.uk/clustalw/) (7). The alignment output file (.ph) was sent to Treeview for a tree plot, and the output file (.aln) was sent to Splitstree4 (14) for bootstrap analysis.
TABLE 1.
PCR primers used to generate MLST data for comparison of Enterococcus species
| Primer name | Primer sequence (5′ to 3′)b | Product length (bp) | Strain-specific GenBank accession no.a |
|---|---|---|---|
| EPW2 | ACGACTTCACCCCAATCATC (fwd) CAAAGTGACAGGTGGTGCAT (rvs) | 463 | DQ839477, DQ839478, DQ839479, DQ839480, DQ839481, DQ839482, DQ839483 |
| EPE1 | ACCGACTTCGGGTGTTACAA (fwd) GAAGCAAAATCGCGAAGAAC (rvs) | 480 | DQ839449, DQ839450, DQ839451, DQ839452, DQ839453, DQ839454, DQ839455 |
| EPH4 | GGTAGCGGAGAAATTCCAAA (fwd) CCCTTATACCGGCATTCTCA (rvs) | 490 | DQ839470, DQ839471, DQ839472, DQ839473, DQ839474, DQ839475, DQ839476 |
| EPE8 | CCCCGTACATGAAATTTGGA (fwd) GAATTTCTCCGCTACCCACA (rvs) | 456 | DQ839463, DQ839464, DQ839465, DQ839466, DQ839467, DQ839468, DQ839469 |
| EPE7 | GCGTTGGAAATTTGAGAGGA (fwd) TTCTGTGTTCGGCATGGTTA (rvs) | 421 | DQ839456, DQ839457, DQ839458, DQ839459, DQ839460, DQ839461, DQ839462 |
For each primer set, corresponding PCR products of seven Enterococcus species were sequenced and submitted to GenBank under the reported accession numbers.
fwd, forward; rvs, reverse.
Enterococcus MGM size analysis.
We sampled the Enterococcus microarray data to create data subsets representing 1/2, 1/5, 1/10, 1/20, 1/50, 1/100, 1/200, and 1/500 of the original data set, and these were used to analyze the effect of array size on interpretation of genetic relationships. A custom program was written to process the data as follows: for each data subset with size denoted by n, the program processed the data by randomly choosing 1,000 subsets of n probes with replacement from the microarray to find the percentage of times that each subset resulted in the same tree as, or a tree similar to, the one derived from the entire array. More specifically, for each selected subset of probes, normalization and quantification were applied, and a distance matrix was calculated to generate a phylogenetic tree file using the neighbor-joining method (23). All 1,000 tree files were then compared to the original tree.
Tree comparison was handled carefully, because different branch arrangements of phylogenetic trees can represent the same tree; what matters is how strains are clustered rather than the ordering of branches or clusters. In our case a cluster is defined as the set of all leaves that descend from a common, nonroot node of the tree (6). Each tree is first rerooted with E. faecium serving as the tree root, and then all the branches in a cluster are collected into a set (the order was not important), and all the sets (clusters) are collected into a bigger set. Each tree is finally represented by a set containing multiple sets of branches corresponding to all clusters of the tree. For example, for the rerooted tree shown in Fig. 1a, a complete set corresponding to the tree is as follows: {{E. gallinarum, E. avium}, {E. dispar, E. faecalis}, {E. gallinarum, E. avium, E. casseliflavus}, {E. dispar, E. faecalis, E. gallinarum, E. avium, E. casseliflavus}, {E. dispar, E. faecalis, E. gallinarum, E. avium, E. casseliflavus, E. hirae}}. For cases when we only want to compare trees to a limited “depth,” we delete the corresponding sets of no interest to us. For example, for the tree above, if we do not want to differentiate how E. gallinarum, E. avium, and E. casseliflavus are clustered, we simply delete the set {E. gallinarum, E. avium} from the tree's corresponding set. Finally, if a subsampled data set produces a set contained in the complete set that corresponds to the original tree derived from the entire data set, it is scored as a match, and we can calculate the percentage of matches for 1,000 subsamples. For each data subset size, the program was run for 10 iterations to find the average and standard deviation of the percentage agreement with the original tree. The custom program (available upon request) used for the calculations was implemented using Matlab 7.0 (MathWorks, Natick, MA) equipped with the Bioinformatics toolbox.
FIG. 1.

Phylogenetic tree for seven Enterococcus strains using (a) MGM analysis or (b) MLST with E. faecium as the root. Both data sets identified the same four clusters, A, B, C, and D, marked beside nodes.
Library bias correction.
A program was developed to compensate for representational bias—that is, unequal representation of species or genera used to construct the microarray. After data normalization and quantification for each hybridization hi, probes classified as “present” were collected into a set, Si. The size of each set was denoted by si, and the size of the largest set for all hybridization experiments was denoted by smax. For each hybridization hi, smax − si additional probes were added by randomly selecting smax − si probes with replacement from set Si. The resulting data were used as a representational bias-corrected microarray for distance matrix construction and neighbor-joining tree construction (all implemented in Matlab as described above). Representational bias-corrected microarrays (n = 1,000) were created, and corresponding trees were generated, with the most frequently reported phylogenetic tree serving as the “consensus tree.” To find the “consensus tree,” the count was set to 1 for each tree that did not exactly match any previous tree; if a tree matched a previous tree, the count of that tree type was increased by 1. The tree with the highest count was the consensus tree (see above for the tree matching procedure). The record of the counts for all the unique trees was used to calculate the percentage of the existence of each branch of a tree among all the bias-corrected trees, which was reported as a bootstrap value. The program (available upon request) for library bias correction was also written using Matlab 7.0.
Virtual MGM simulation.
Streptococcus genome files of 15 strains belonging to five different species were downloaded from PubMed in FASTA format. MGM construction was simulated by randomly choosing n (n is the microarray size) positions with replacement in the genome sequences and collecting n gene segments 600 bp long (as probe sequences) into FASTA-format files. To construct a virtual array with equal representation of the 5 species, 800 probes were randomly chosen for each species (n = 4,000). Hybridization was simulated using stand-alone BLAST 2.2.13 (1). The FASTA-format files were used to construct local libraries, and the 15 genomes were compared with local libraries to generate BLAST report files. The BLAST report files contained queries of all genomes for each probe. Imaging was simulated using a Perl program that determined the best score among all reported hits for a given genome against a probe, and the length of the matched sequence corresponding to the best score was divided by 600. The resulting value was used as the normalized hybridization intensity of that genome for the probe. Finally, Matlab 7.0 was used to read the intensity files and calculate the distance matrices for the 15 genomes. The neighbor-joining method and Treeview were used for phylogenetic tree construction as described previously.
For MLST analysis, six genes were selected, namely, 16S rRNA, cpn10, dnaK, groEL, hsp, and htpX. Gene sequences were retrieved from GenBank and were concatenated in the same order for each strain. The concatenated sequences were aligned using ClustalW, and a phylogenetic tree was constructed using Treeview as described above.
For construction of the virtual whole-genome microarray, 4,000 probes were randomly selected from a single species. To simulate unequal representation, a different proportion of probes was selected from each species. For both the equally represented and unequally represented MGM size study, 10,000 random subsets (1,000 per iteration, 10 iterations total) were generated for each desired array size, and the mean and standard deviation of percent correct identification were plotted. In addition, for the unequally represented microarray with library bias correction, 1,000 random subsets (100 per iteration, 10 iterations total) were generated, and the library bias correction method was applied to each subset to find the consensus tree among 50 randomly generated bias-corrected trees from each subset.
Nucleotide sequence accession numbers.
PCR product sequences determined in this work have been submitted to GenBank under the accession numbers listed in Table 1.
RESULTS AND DISCUSSION
Analyzing Enterococcus phylogeny using an MGM.
We hybridized seven Enterococcus species to an Enterococcus MGM (25) (each with replicate hybridizations) and used these data to study the validity of inferences from the MGM and the effects of library representation and the number of probes included on an MGM. Following normalization, we constructed a distance matrix for the seven species and built a phylogenetic tree using the neighbor-joining method (23). As shown in Fig. 1a, with E. faecium as the tree root, E. dispar and E. faecalis form one cluster (cluster A). E. gallinarum and E. avium are grouped together and further cluster with E. casseliflavus to form cluster B. Clusters A and B make up cluster C, which groups with E. hirae to form cluster D. We applied several common normalization methods to the microarray data, including global and local z scores, global and local normalization with respect to the average signal of bright spots and all spots, and other quantification methods (for example, bimodal classification [2, 13]), as well as direct application of raw intensity values (4). All methods identified clusters A and B; a minor difference in the positions of E. hirae and E. faecium affected clusters C and D. Because various normalization and quantification methods generated similar results, the phylogenetic relationships determined using the MGM appear to be robust regardless of the normalization method employed.
Enterococcus MGM versus MLST.
To compare the MGM results with a conventional MLST analysis, we first identified five probes that were positive for all seven Enterococcus species (by hybridization) and designed primer sets for the corresponding sequences (Table 1). These target sequences were then PCR amplified for each of the seven strains of Enterococcus and sequenced for MLST. We concatenated the five sequences for each species and generated a phylogenetic tree (Fig. 1b). The results closely reflect the MGM results by identifying all four clusters, A, B, C, and D, with differences related to how E. gallinarum, E. avium, and E. casseliflavus are grouped in cluster B. The bootstrapping values of all nodes were above 99.2%.
Effect of the number of probes from Enterococcus MGM.
We hypothesized that the accuracy of the MGM for phylogeny analysis depends on the number of probes used. Larger arrays provide more details on genetic differences, but the benefit is probably asymptotic and a function of the total variation between the strains being tested. For interspecies comparisons with the Enterococcus MGM, we randomly selected 10,000 probe subsets of different sizes (including 10 iterations; 1,000 for each iteration). The mean and standard deviation for the percent correct identification of either clusters A and B or clusters A, B, C, and D are shown in Fig. 2. The percent correct identification curves resemble a steep step function, which indicates that very few probes are necessary for robust cluster identification. Approximately 100 probes are sufficient to identify the two major clusters, A and B, with 100% accuracy, and on average, these two clusters could be identified with 95% accuracy using as few as 46 probes. Moreover, as expected, more probes are required to consistently identify all four clusters. Approximately 1,000 probes are necessary for 100% recovery of all 4 clusters, whereas 460 probes identify all 4 clusters in 96% of the sampling comparisons. This analysis indicates that robust comparisons between Enterococcus species can be obtained with a relatively small MGM.
FIG. 2.

Percent correct identification of two clusters (A and B; solid line) or all four clusters (A, B, C, and D; dashed line) as a function of the number of microarray probes included from the Enterococcus MGM analysis. The mean and standard deviation (bars) of the percentage correct identification for each probe subset for 10 iterations, each with 1,000 runs, is shown.
Enterococcus MGM library representation and bias correction.
The Enterococcus MGM shotgun library was constructed from equal numbers of isolates from five sources as part of a different study (25). The proportion of Enterococcus species included in the original libraries was not tracked, but we assessed this in a posthoc manner by examining the number of bright spots (intensity, >25,000) when each Enterococcus species was hybridized to the array (Table 2). There were obvious extremes, such as the case of E. faecium, which had more than 1,000 bright spots, while E. avium had slightly more than 200 bright spots. The average intensities for the probes included in this analysis were very similar (Table 2), indicating that differences between species did not arise from differential loading of gDNA on the slides. Consequently, it appears that the original shotgun libraries did not include an equal distribution of species. E. gallinarum, E. avium, and E. casseliflavus appear poorly represented compared to E. hirae, E. dispar, E. faecalis, and E. faecium. Representational bias may be responsible for the apparent long distance (branch length) between E. faecium and all other species in the MGM analysis (Fig. 1a). This is because the redundant probes for E. faecium contribute little to discrimination of the other target strains but do contribute to the estimated distance of E. faecium from the other strains.
TABLE 2.
Enterococcus microarray hybridization patterns
| Enterococcus sp. | No. of probes with intensity of >25,000a | Avg intensityb |
|---|---|---|
| E. hirae | 901 | 56,699 |
| E. gallinarum | 240 | 57,084 |
| E. dispar | 855 | 57,949 |
| E. avium | 221 | 57,212 |
| E. faecalis | 888 | 58,332 |
| E. casseliflavus | 282 | 56,935 |
| E. faecium | 1,406 | 55,259 |
Maximum intensity is 65,535.
Average probe intensity for probes having intensities of >25,000.
Using the library bias correction algorithm, we obtained a phylogenetic relationship (data not shown) that appears very similar to the original tree before the algorithm is applied (Fig. 1a). Thus, for this particular Enterococcus MGM, there is no significant change in interpretation based on our bias correction. Nevertheless, we hypothesized that in some cases library bias might yield misleading results. To further explore this issue, we used virtual MGM simulations for which all probe identities are known, permitting us to alter the degree of representational bias in the analysis.
MGM array versus MLST.
Using sequenced genomes, we can simulate MGM construction, hybridization, and imaging in silico. For this analysis, we used Streptococcus for the virtual simulations because of the availability of a relatively large number of sequenced strains (n = 15) and species (n = 5). We randomly chose a total of 4,000 gene segments (each 600 bp long) from the 15 Streptococcus genomes, 800 segments from each of the five species, to construct an equally represented, virtual MGM to use for virtual hybridization. In the virtual MGM (data not shown), the gray level of each spot is proportional to the normalized hybridization intensity of a target strain to that probe. Relative intensities were converted to binary scores, after which the phylogenetic tree for the 15 Streptococcus strains was constructed (Fig. 3a, with S. mutans as the tree root). This analysis correctly grouped the strains belonging to each species, forming four species clusters: J, K, L, and M. At the species level, Streptococcus agalactiae and Streptococcus pyogenes form cluster A, Streptococcus pneumoniae and Streptococcus thermophilus form cluster B, and clusters A and B form cluster C.
FIG. 3.

Phylogenetic tree of 15 Streptococcus strains based on (a) an equally represented virtual Streptococcus MGM, (b) MLST, (c) a virtual whole genome composed of sequences from S. pneumoniae only, or (d) a microarray composed of sequences from S. pyogenes only. Cluster labels are shown to the left of nodes, and the same clusters are denoted by the same label.
For comparison, a phylogenetic tree was constructed using conventional sequence comparisons for six loci (16S rRNA, cpn10, dnaK, groEL, hsp, and htpX) (Fig. 3b). This analysis demonstrates that the MGM is capable of identifying the same phylogenetic groups at the species level as a multilocus sequence analysis. Also, the MGM analysis provides greater genetic differentiation between strains within a species. For example, note the differentiation between S. pyogenes strains by the MGM, whereas little differentiation is indicated using sequence analysis.
MGM versus whole-genome microarray.
The MGM is also capable of identifying the same phylogenetic groups at the species level as a whole-genome microarray, such as that constructed from the S. pneumoniae genome (Fig. 3c) or from the S. pyogenes genome (Fig. 3d). At the strain level, the MGM results almost exactly match the whole-genome microarray result constructed using S. pyogenes as the probe reference (Fig. 3d). In contrast, the MGM clearly outperforms the whole-genome microarray constructed with S. pneumoniae as the probe reference (Fig. 3c), as can be seen from the latter's poor differentiation of S. pyogenes strains (E′′ and H′′ in Fig. 3c but E and H in Fig. 3a). This example illustrates how a whole-genome microarray constructed from one strain or species may not provide enough information to differentiate other strains. Furthermore, when the microarray is constructed using data from a single genome, we see exaggerated separation between the source strain/species and other strain/species used in the comparison. This is evident from the outlying separation of S. pneumoniae and S. pyogenes in the virtual array analysis (Fig. 3c and d) and the probable overrepresentation of E. faecium in the Enterococcus array (Fig. 1a; Table 2).
When only one species is used to construct the virtual microarray (for example, S. pneumoniae; Fig. 3c), most probe intensities are classified as “1” for the reference species, while few probes appear positive for most of the other strains and species hybridizations. Thus, out of 4,000 probes selected from S. pneumoniae (Fig. 3c), only a relatively small number of probes play a role in the genetic discrimination of other strains, and this produces an exaggerated branch distance for S. pneumoniae relative to other species. This bias does not usually affect the phylogeny, because all strains not used for library construction have similar hybridization strengths. According to the neighbor-joining algorithm (23), when searching for nearest neighbors to join, the long distance between S. pneumoniae and the other strains is offset and does not affect clustering. However, this may not be true for some extreme and complicated library biases for which hybridization strengths vary among all strains.
MGM library bias analysis and correction.
Although in most cases library bias does not affect phylogeny relationships because of the implementation of the neighbor-joining algorithm, this is not always true, as demonstrated by the example, shown in Fig. 4a. The phylogeny in Fig. 4a is obtained from virtual hybridization of 15 Streptococcus strains on a virtual MGM consisting of 1,500 probes from S. agalactiae, 1,500 probes from S. thermophilus, 420 probes from S. mutans, 420 probes from S. pyogenes, and 160 probes from S. pneumoniae. The number of bright spots for the 15 hybridizations varies considerably (Table 3). Library bias causes the formation of incorrect clusters at the species level, with clusters A′ and B′ differing from their counterparts in Fig. 3. However, the library bias correction algorithm compensates for the bias, and the phylogenetic tree (clusters A, B, and C) is correctly generated at the species level, as shown in Fig. 4b. The library bias correction algorithm has the advantage that it can provide a bootstrap confidence value for each node (Fig. 4b) and can also provide multiple bias-corrected trees with a high consensus frequency.
FIG. 4.

Phylogenetic tree of 15 Streptococcus strains based on a virtual unequally represented MGM before and after library bias correction analysis. Panel a shows an example for which representational bias produces an incorrect phylogenetic tree (clusters A′ and B′, instead of A and B). Panel b shows that after library bias correction, the correct phylogeny is retrieved. Bootstrapping values are adjacent to nodes.
TABLE 3.
Virtual Streptococcus microarray hybridization pattern
| Strain used for hybridizationb | No. of positive probes for different microarraysa,c
|
|||
|---|---|---|---|---|
| Equal representation | Only from S. pneumoniae | Only from S. pyogenes | Mixed | |
| S. agalactiae 1603V/R | 1,040 | 242 | 541 | 1,592 |
| S. agalactiae A909 | 1,019 | 234 | 540 | 1,589 |
| S. agalactiae NEM316 | 1,020 | 225 | 530 | 1,601 |
| S. mutans UA159 | 1,047 | 207 | 311 | 694 |
| S. pneumoniae R6 | 989 | 3,787 | 253 | 422 |
| S. pyogenes M1 GAS | 1,003 | 213 | 3,667 | 703 |
| S. pyogenes MGAS10394 | 996 | 214 | 3,672 | 707 |
| S. pyogenes MGAS315 | 1,006 | 218 | 3,738 | 716 |
| S. pyogenes MGAS5005 | 1,000 | 213 | 3,651 | 703 |
| S. pyogenes MGAS6180 | 999 | 219 | 3,618 | 714 |
| S. pyogenes MGAS8232 | 998 | 217 | 3,662 | 702 |
| S. pyogenes SSI-1 | 1,005 | 218 | 3,737 | 717 |
| S. thermophilus CNRZ1066 | 1,043 | 312 | 306 | 1,652 |
| S. thermophilus LMG 18311 | 1,048 | 313 | 305 | 1,653 |
Numbers of probes with normalized intensity of >0.5 are shown.
Fifteen Streptococcus species genomes were hybridized onto a microarray.
Each column represents one of the four microarrays: constructed from equal representation of five Streptococcus species, only from S. pneumoniae, only from S. pyogenes, or from an unequal representation of all five species.
In order to study the effectiveness of our library bias correction algorithm, we varied the sizes of both the equally represented and unequally represented virtual MGMs and compared the percentages of correct identification for phylogenetic relationships at the species level. For the unequally represented library, the resulting virtual hybridization experiments showed that the consensus tree would be detected with a frequency of less than 0.1% (Fig. 5). Application of our library bias correction increased the percent correct identification of the consensus tree to a level similar to that generated using the equally represented MGM.
FIG. 5.

Comparison of an unequally represented Streptococcus array before (dotted line) and after (dashed line) library bias correction with an equally represented Streptococcus array (solid line). For each subset size of the array, the mean and standard deviation of the percentage of correct identification of clusters A, B, and C are plotted as a function of the number of microarray probes.
Conclusions.
We used both experimental and computational methods to analyze the performance of the MGM for making inferences about the genetic relationships within and between bacterial species. Both methods verify that the MGM is a reliable genetic discrimination method when assessed relative to MLST and whole-genome microarray methods, with the added advantage that the MGM provides greater discrimination between strains, as demonstrated empirically elsewhere (25). Construction of an MGM does not require a fully sequenced genome; this makes it less restrictive and more readily applicable than a whole-genome microarray. Another advantage of the MGM is its ability to identify genetic markers specific to a sample strain or cluster.
The virtual MGM provides an effective method for analyzing an experimental MGM and, in fact, has potential as a tool for genetic analysis when sequenced genome information is available. Probes for the virtual MGM are selected randomly from sequenced genomes so that the virtual MGM consists of genetic information from multiple genomes. Rather than having to perform a cumbersome genome-wide comparison, the virtual MGM method permits a shotgun sequence comparison that provides reliable phylogeny information, as shown by this study. For cases when library bias exists, the proposed library bias correction method provides effective compensation with bootstrap confidence values.
Acknowledgments
We thank Stacey LaFrentz and Edward Kuhn for assistance with wet lab experiments and Min-Su Kang and Sonja Lloyd for helpful discussions. Rachel Noble provided the Enterococcus strains used in this study.
This project was partially funded by USDA NRI contract 2002-35102-12374, by the Agricultural Animal Health Program at the College of Veterinary Medicine, Washington State University, Pullman, and by the Carl M. Hansen Foundation.
Footnotes
Published ahead of print on 5 January 2007.
REFERENCES
- 1.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 2.Borucki, M. K., M. J. Krug, W. T. Muraoka, and D. R. Call. 2003. Discrimination among Listeria monocytogenes isolates using a mixed genome DNA microarray. Vet. Microbiol. 92:351-362. [DOI] [PubMed] [Google Scholar]
- 3.Borucki, M. K., S. H. Kim, D. R. Call, S. C. Smole, and F. Pagotto. 2004. Selective discrimination of Listeria monocytogenes epidemic strains by a mixed-genome DNA microarray compared to discrimination by pulse-field gel electrophoresis, ribotyping, and multilocus sequence typing. J. Clin. Microbiol. 42:5270-5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Call, D. R., M. K. Borucki, and T. E. Besser. 2003. Mixed-genome microarrays reveal multiple serotype and lineage-specific differences among strains of Listeria monocytogenes. J. Clin. Microbiol. 41:632-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Call, D. R., M. K. Bakko, M. J. Krug, and M. C. Roberts. 2003. Identifying antimicrobial resistance genes with DNA microarrays. Antimicrob. Agents Chemother. 47:3290-3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, D., O. Eulenstein, D. Fernández-Baca, and M. J. Sanderson. 2006. Minimum-flip supertrees: complexity and algorithms. IEEE/ACM Trans. Comput. Biol. Bioinform. 3:165-173. [DOI] [PubMed] [Google Scholar]
- 7.Chenna, R., H. Sugawara, T. Koike, R. Lopez, T. J. Gibson, D. G. Higgins, and J. D. Thompson. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31:3497-3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delcher, A. L., S. Kasif, R. D. Fleischmann, J. Peterson, O. White, and S. L. Salzberg. 1999. Alignment of whole genomes. Nucleic Acids Res. 27:2369-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorrell, N., J. A. Mangan, K. G. Laing, J. Hinds, D. Linton, H. Al-Ghusein, B. G. Barrell, J. Parkhill, N. G. Stoker, A. V. Karlyshev, P. D. Butcher, and B. W. Wren. 2001. Whole genome comparison of Campylobacter jejuni human isolates using a low-cost microarray reveals extensive genetic diversity. Genome Res. 11:1706-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards-Ingram, L. C., M. E. Gent, D. C. Hoyle, A. Hayes, L. I. Stateva, and S. G. Oliver. 2004. Comparative genomic hybridization provides new insights into the molecular taxonomy of the Saccharomyces sensu stricto complex. Genome Res. 14:1043-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feil, E. J., B. C. Li, D. M. Aanensen, W. P. Hanage, and B. G. Spratt. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186:1518-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felsenstein, J. 2004. Inferring phylogenies. Sinauer Associates, Sunderland, MA.
- 13.Gonzalez, S., M. J. Krug, M. E. Nielsen, Y. Santos, and D. R. Call. 2004. Simultaneous detection of marine fish pathogens by using multiplex PCR and a DNA microarray. J. Clin. Microbiol. 42:1414-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huson, D. H., and D. Bryant. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254-267. [DOI] [PubMed] [Google Scholar]
- 15.Liebert, C. A., J. Wireman, T. Smith, and A. O. Summers. 1997. Phylogeny of mercury resistance (mer) operons of gram-negative bacteria isolated from the fecal flora of primates. Appl. Environ. Microbiol. 63:1066-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maiden, M. C. J., J. A. Bygraves, E. Feil, G. Morelli, J. E. Russell, R. Urwin, Q. Zhang, J. Zhou, K. Zurth, D. A. Caugant, I. M. Feavers, M. Achtman, and B. G. Spratt. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 95:3140-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris, C. E., M. Bardin, O. Berge, P. Frey-Klett, N. Fromin, H. Girardin, M. Guinebretière, P. Lebaron, J. M. Thiéry, and M. Troussellier. 2002. Microbial biodiversity: approaches to experimental design and hypothesis testing in primary scientific literature from 1975 to 1999. Microbiol. Mol. Biol. Rev. 66:592-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray, A. E., D. Lies, G. Li, K. Nealson, J. Zhou, and J. M. Tiedje. 2001. DNA/DNA hybridization to microarrays reveals gene-specific differences between closely related microbial genomes. Proc. Natl. Acad. Sci. USA 98:9853-9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naser, S., F. L. Thompson, B. Hoste, D. Gevers, K. Vandermeulebroecke, I. Cleenwerck, C. C. Thompson, M. Vancanneyt, and J. Swings. 2005. Phylogeny and identification of enterococci by atpA gene sequence analysis. J. Clin. Microbiol. 43:2224-2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Page, R. D. M. 1996. TREEVIEW: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12:357-358. [DOI] [PubMed] [Google Scholar]
- 21.Porwollik, S., R. M. Y. Wong, and M. McClelland. 2002. Evolutionary genomics of Salmonella: gene acquisitions revealed by microarray analysis. Proc. Natl. Acad. Sci. USA 99:8956-8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rozen, S., and H. J. Skaletsky. 2000. Primer3 on the WWW for general users and for biologist programmers, p. 365-386. In S. Krawetz and S. Misener (ed.), Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, NJ. [DOI] [PubMed]
- 23.Saitou, M., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406-425. [DOI] [PubMed] [Google Scholar]
- 24.Salama, N., K. Guillemin, T. K. McDaniel, G. Sherlock, L. Tompkins, and S. Falkow. 2000. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc. Natl. Acad. Sci. USA 97:14668-14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soule, M., E. Kuhn, F. Loge, J. Gay, and D. R. Call. 2006. Using DNA microarrays to identify library-independent markers for bacterial source tracking. Appl. Environ. Microbiol. 72:1843-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Täpp, J., M. Thollesson, and B. Herrmann. 2003. Phylogenetic relationships and genotyping of the genus Streptococcus by sequence determination of the RNase P RNA gene, rnpB. Int. J. Syst. Evol. Microbiol. 53:1861-1871. [DOI] [PubMed] [Google Scholar]
- 27.van Belkum, A., M. Struelens, A. de Visser, H. Verbrugh, and M. Tibayrenc. 2001. Role of genomic typing in taxonomy, evolutionary genetics, and microbial epidemiology. Clin. Microbiol. Rev. 14:547-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Berkum, P., Z. Terefework, L. Paulin, S. Suomalainen, K. Lindström, and B. D. Eardly. 2003. Discordant phylogenies with the rrn loci of Rhizobia. J. Bacteriol. 185:2988-2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou, J., D. K. Thompson, Y. Xu, and J. M. Tiedje. 2004. Microbial functional genomics. Wiley-Liss, Hoboken, NJ.