

Clinical Evidence Linking Insulin Resistance, Hyperinsulinemia, and Cardiovascular Disease
Based on the recent evidence that patients with type 2 diabetes have the same risk of myocardial infarction as nondiabetic subjects with a history of infarction, diabetes has been designated as an atherosclerosis equivalent.1 Insulin resistance plays a primary role in the development of type 2 diabetes and considerable evidence supports the association between insulin resistance, hyperinsulinemia, and vascular disease.2,3 Although the molecular mechanisms are incompletely understood, this association is supported by several large clinical studies showing a direct relationship between insulin levels and cardiovascular risk. The Paris Prospective Study4 and the Multiple Risk Factor Intervention Trial (MRFIT)5 reported positive relationships between insulin levels and atherosclerotic events. In addition, the Veterans Affairs High Density Lipoprotein Intervention Trial (VA-HIT)6 demonstrated the highest incidence of cardiovascular events in the subgroups with highest levels of insulin. Finally, the landmark Insulin Resistance Atherosclerosis Study (IRAS) provided further evidence for an inverse relationship between carotid intima-medial thickness and insulin sensitivity.7
Insulin Resistance and Smooth Muscle Cell Proliferation
Controversy exists regarding the cellular mechanisms leading to atherosclerosis in insulin resistance and type 2 diabetes. Because of the observed numerical increases and functional abnormalities in intimal smooth muscle cells (SMC) in diabetes, this cell type has received intensive attention. In advanced lesions, SMC and their secreted products are a major component of the lesion comprising up to 70 to 80% of the total content of advanced human lesions.8 In particular, diabetes accelerates SMC accumulation in atheroscle-rotic lesions and SMC proliferation directly correlates with insulin levels.9,10 SMC proliferation is also the primary mechanism leading to the failure of procedures used to treat occlusive atherosclerotic diseases in patients with diabetes.11 Restenosis following coronary revascularization in patients with type 2 diabetes results from excessive neointima formation because of SMC proliferation.12 In addition, compensatory hyperinsulinemia associated with insulin resistance in patients with type 2 diabetes strongly predicts neoin-timal SMC proliferation.13
Despite more than 3 decades of intensive investigations, the detailed molecular mechanisms underlying the association between insulin resistance, SMC proliferation and accelerated atherosclerosis are still unclear. Hyperglycemia, hyperinsulinemia, advanced gly-cation endproducts, and dyslipidemia have each been suggested to stimulate SMC proliferation. Although hyperglycemia has been demonstrated to stimulate SMC proliferation in vitro and is thought to contribute to neointima formation, this concept has been challenged by recent reports demonstrating no mitogenic activity of high glucose.9,14 In addition, hyperglycemia in streptozotocin-induced diabetic rats is not associated with increased neointima formation.15 Instead, it is becoming increasingly evident that insulin resistance and resulting hyperinsulinemia play key roles in promoting SMC proliferation and vascular neointima formation.14,15 Insulin signaling in SMC results in phosphorylation of tyrosine residues on the insulin receptor substrates which activates downstream PI-3 kinase/Akt or ERK1/2-MAPK signaling pathways. However, insulin resistance and compensatory hyperinsulinemia result in a selective impairment of the PI 3-kinase pathway with intact signaling along the ERK1/2-MAPK pathway.3,16 Consistent with this concept, the proliferative ERK1/2-MAP kinase signaling pathway is activated in the arterial wall under insulin resistant conditions and induces the expression of c-Fos, Egr-1 and other early growth response genes that control the transition from SMC quiescence to proliferation and migration.17 Insulin alone is a rather weak mitogen and because insulin potentates the effects of other mitogens such PDGF, angiotensin II, and thrombin, increased neointima formation in insulin resistance may not be exclusively explained by hyperin-sulinemia but rather by a complex interplay between several mitogens and their downstream activation of the ERK1/2-MAPK signaling pathway.
C-Peptide: The Old Dog With a New Trick
In this issue of Circulation Research, Walcher and colleagues extend our current knowledge on mechanisms promoting SMC proliferation under conditions of hyperinsulinemia by adding C-peptide to the list of mitogens.18 C-peptide, the 31 amino-acid residue formed during cleavage of insulin from proinsulin, is released by the pancreatic β cell in equimolar amounts with insulin and has long been thought to be biologically inert. Recent evidence indicates that C-peptide may not merely be an inactive by-product of insulin biosynthesis but act as hormonally active peptide.19 Early studies have suggested that C-peptide may have beneficial vascular effects by attenuating vascular and neural dysfunction in tissues of diabetic rats.20 However, recent experiments identified the presence of C-peptide specifically in atherosclerotic lesions from diabetic patients and revealed that C-peptide may also have proathergenic effects such as stimulation of monocytes and T-cell chemotaxis.21,22 In their present study, Walcher and colleagues demonstrate colocalization of C-peptide with SMC in early human atherosclerotic lesions of diabetic subjects. In vitro, stimulation of SMC with C-peptide resulted in a dose-dependent induction of cell proliferation evidenced by [3H] thymidine incorporation and nuclear KI-67 staining. These studies outline a previously unrecognized role for C-peptide to act as mitogen for SMC.
Of course, these results require further characterization of the related mechanisms by which C-peptide exerts its effect to induce SMC proliferation. Notably, C-peptide induces several signaling pathways including the ERK1/2-MAPK and PI-3 kinase pathways.23 Although C-peptide specifically binds to the plasma membrane24 and activation of these signaling pathways suggest the involvement of a G-protein-coupled receptor, a specific receptor has yet to be identified. In their studies, Walcher and coauthors demonstrate that C-peptide–induced SMC proliferation is mediated through phosphorylation of the protein tyrosine kinase Src which has been implicated as intermediate in signaling networks that couple G-protein-coupled receptors with downstream signaling cascades such as the PI-3 kinase/Akt and the Ras/MAP kinase pathway.25 Consistent with this concept, both the PI-3 kinase/Akt and the ERK1/2-MAPK are considered to be important signaling pathways regulating SMC proliferation and pharmacological inhibition of these pathways prevented C-peptide–induced SMC proliferation. As the final common pathway activated by PI-3 kinase and ERK1/2 MAPK signaling is the cell cycle, C-peptide increased cyclin D1 expression and subsequently phosphorylation of the retinoblastoma protein as the gatekeeper of G1→S phase cell cycle progression. Based on these observations, C-peptide stimulates SMC proliferation through a Src→PI-3 kinase/ERK1/2-MAPK-dependent progression of the cell cycle (Figure 1).
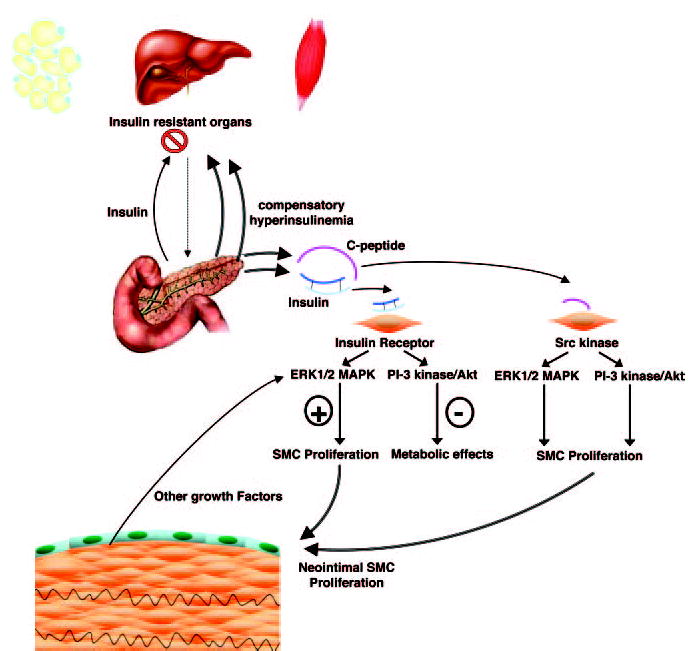
Regulation of SMC Proliferation in Insulin Resistance. The sine qua non of insulin resistance in adipose tissue, liver, and muscle is compensatory hyperinsulin-emia. In the insulin resistant state, tyrosine phosphorylation of the insulin receptor and signaling via the insulin receptor substrate (IRS)-1/2/PI-3 kinase/Akt pathway is impaired resulting in diminished metabolic effects. In contrast, tyrosine phosphorylation of ERK1/2 MAPK by insulin is maintained and perpetuated by other growth factors resulting in SMC proliferation and migration. As demonstrated by Walcher et al18 C-peptide, secreted in equimolar amounts to insulin, activates both the PI-3 kinase/Akt and ERK1/2 MAPK pathways via upstream activation of the Src kinase. Activation of these pathways results in SMC proliferation through phosphorylation of the retinoblastoma protein and cell cycle progression.
In concert, these studies demonstrate the presence of C-peptide in atherosclerotic lesions from diabetic patients and it is tempting to speculate that C-peptide–induced proliferation of SMC in the setting of insulin resistance and hyperinsulinemia could provide a previously unrecognized mechanism leading to accelerated atherosclerosis and its complications in patients with type 2 diabetes. The results by Walcher et al substantially improve our understanding of the role of the previously thought to be inactive C-peptide. This study together with the mentioned evidence supporting biological activity of C-peptide may require revising the recent view of C-peptide as an inert by-product of insulin synthesis. However, they also raise many new questions and leave room for further studies. For example, is there a specific cellular C-peptide receptor or a G-protein-coupled receptor activated by C-peptide? Does C-peptide induce neointimal SMC proliferation in vivo? May C-peptide promote macrovascular disease whereas having potentially beneficial effects on blood flow and microvascular disease as suggested by other evidence?20 These questions certainly deserve further molecular studies and in particular in vivo experiments using infusion or injection of C-peptide in animal models of atherosclerosis or neointimal SMC proliferation to further exploit the contribution of C-peptide to cardiovascular disease in type 2 diabetes.
Footnotes
The opinions expressed in this editorial are not necessarily those of the editors or of the American Heart Association.
Disclosures
None.
Sources of Funding
D.B. is supported by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (RO1 HL084611–01 to D. B.), the American Diabetes Association (Research Award 1–06-RA-17 to D.B.), and the American Heart Association (Scientist Development Grant 0435239N to D.B.).
References
- 1.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 2.Semenkovich CF. Insulin resistance and atherosclerosis. J Clin Invest. 2006;116:1813–1822. doi: 10.1172/JCI29024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nigro J, Osman N, Dart AM, Little PJ. Insulin resistance and atherosclerosis. Endocr Rev. 2006;27:242–259. doi: 10.1210/er.2005-0007. [DOI] [PubMed] [Google Scholar]
- 4.Ducimetiere P, Eschwege E, Papoz L, Richard JL, Claude JR, Rosselin G. Relationship of plasma insulin levels to the incidence of myocardial infarction and coronary heart disease mortality in a middle-aged population. Diabetologia. 1980;19:205–210. doi: 10.1007/BF00275270. [DOI] [PubMed] [Google Scholar]
- 5.Orchard TJ, Eichner J, Kuller LH, Becker DJ, McCallum LM, Grandits GA. Insulin as a predictor of coronary heart disease: interaction with apolipoprotein E phenotype. A report from the Multiple Risk Factor Intervention Trial. Ann Epidemiol. 1994;4:40–45. doi: 10.1016/1047-2797(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 6.Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, Faas FH, Linares E, Schaefer EJ, Schectman G, Wilt TJ, Wittes J. The Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study G. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. N Engl J Med. 1999;341:410–418. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 7.Howard G, O’Leary DH, Zaccaro D, Haffner S, Rewers M, Hamman R, Selby JV, Saad MF, Savage P, Bergman R. Insulin sensitivity and atherosclerosis. Circulation. 1996;93:1809–1817. doi: 10.1161/01.cir.93.10.1809. [DOI] [PubMed] [Google Scholar]
- 8.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis : a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, Am Heart Association. Circulation. 1995;92:1355–1374. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki LA, Poot M, Gerrity RG, Bornfeldt KE. Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: lack of direct growth-promoting effects of high glucose levels. Diabetes. 2001;50:851–860. doi: 10.2337/diabetes.50.4.851. [DOI] [PubMed] [Google Scholar]
- 10.Absher PM, Schneider DJ, Baldor LC, Russell JC, Sobel BE. The retardation of vasculopathy induced by attenuation of insulin resistance in the corpulent JCR:LA-cp rat is reflected by decreased vascular smooth muscle cell proliferation in vivo. Atherosclerosis. 1999;143:245–251. doi: 10.1016/s0021-9150(98)00295-0. [DOI] [PubMed] [Google Scholar]
- 11.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 12.Kornowski R, Mintz GS, Kent KM, Pichard AD, Satler LF, Bucher TA, Hong MK, Popma JJ, Leon MB. Increased restenosis in diabetes mellitus after coronary interventions is due to exaggerated intimal hyperplasia: a serial intravascular ultrasound study. Circulation. 1997;95:1366–1369. doi: 10.1161/01.cir.95.6.1366. [DOI] [PubMed] [Google Scholar]
- 13.Takagi T, Akasaka T, Yamamuro A, Honda Y, Hozumi T, Morioka S, Yoshida K. Impact of insulin resistance on neointimal tissue proliferation after coronary stent implantation: Intravascular ultrasound studies. J Diabetes Complications. 2002;16:50–55. doi: 10.1016/s1056-8727(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 14.Indolfi C, Torella D, Cavuto L, Davalli AM, Coppola C, Esposito G, Carriero MV, Rapacciuolo A, Di Lorenzo E, Stabile E, Perrino C, Chieffo A, Pardo F, Chiariello M. Effects of balloon injury on neointimal hyperplasia in streptozotocin-induced diabetes and in hyperinsulinemic nondiabetic pancreatic islet-transplanted rats. Circulation. 2001;103:2980–2986. doi: 10.1161/01.cir.103.24.2980. [DOI] [PubMed] [Google Scholar]
- 15.Park S-H, Marso SP, Zhou Z, Foroudi F, Topol EJ, Lincoff AM. Neointimal hyperplasia after arterial injury is increased in a rat model of non-insulin-dependent diabetes mellitus. Circulation. 2001;104:815–819. doi: 10.1161/hc3301.092789. [DOI] [PubMed] [Google Scholar]
- 16.Wang CCL, Goalstone ML, Draznin B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes. 2004;53:2735–2740. doi: 10.2337/diabetes.53.11.2735. [DOI] [PubMed] [Google Scholar]
- 17.Jonas M, Edelman ER, Groothuis A, Baker AB, Seifert P, Rogers C. Vascular neointimal formation and signaling pathway activation in response to stent injury in insulin-resistant and diabetic animals. Circ Res. 2005;97:725–733. doi: 10.1161/01.RES.0000183730.52908.C6. [DOI] [PubMed] [Google Scholar]
- 18.Walcher D, Babiak C, Poletek P, Rosenkranz S, Bach H, Betz S, Durst R, Grüb M, Hombach V, Strong J, Marx N. C-peptide induces smooth muscle cell proliferation - involvement of Src-kinase, phosphatidylinositol 3-kinase and extra-cellular signal-regulated kinase ½. Circ Res. 2006;99:1181–1187. doi: 10.1161/01.RES.0000251231.16993.88. [DOI] [PubMed] [Google Scholar]
- 19.Johansson J, Ekberg K, Shafqat J, Henriksson M, Chibalin A, Wahren J, Jornvall H. Molecular effects of proinsulin C-peptide. Biochem and Biophys Res Commun. 2002;295:1035–1040. doi: 10.1016/s0006-291x(02)00721-0. [DOI] [PubMed] [Google Scholar]
- 20.Ido Y, Vindigni A, Chang K, Stramm L, Chance R, Heath WF, DiMarchi RD, Di Cera E, Williamson JR. Prevention of vascular and neural dysfunction in diabetic rats by C-peptide. Science. 1997;277:563–566. doi: 10.1126/science.277.5325.563. [DOI] [PubMed] [Google Scholar]
- 21.Marx N, Walcher D, Raichle C, Aleksic M, Bach H, Grub M, Hombach V, Libby P, Zieske A, Homma S, Strong J. C-peptide colocalizes with macrophages in early arteriosclerotic lesions of diabetic subjects and induces monocyte chemotaxis in vitro. Arterioscler Thromb Vasc Biol. 2004;24:540–545. doi: 10.1161/01.ATV.0000116027.81513.68. [DOI] [PubMed] [Google Scholar]
- 22.Walcher D, Aleksic M, Jerg V, Hombach V, Zieske A, Homma S, Strong J, Marx N. C-peptide induces chemotaxis of human CD4-positive cells: involvement of pertussis toxin-sensitive G-proteins and phosphoinositide 3-kinase. Diabetes. 2004;53:1664–1670. doi: 10.2337/diabetes.53.7.1664. [DOI] [PubMed] [Google Scholar]
- 23.Kitamura T, Kimura K, Jung BD, Makondo K, Okamoto S, Canas X, Sakane N, Yoshida T, Saito M. Proinsulin C-peptide rapidly stimulates mitogen-activated protein kinases in Swiss 3T3 fibroblasts: requirement of protein kinase C, phosphoinositide 3-kinase and pertussis toxin-sensitive G-protein. Biochem J. 2001;355:123–129. doi: 10.1042/0264-6021:3550123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rigler R, Pramanik A, Jonasson P, Kratz G, Jansson OT, Nygren PA, Stahl S, Ekberg K, Johansson BL, Uhlen S, Uhlen M, Jornvall H, Wahren J. Specific binding of proinsulin C-peptide to human cell membranes. Proc Natl Acad Sci U S A. 1999;96:13318–13323. doi: 10.1073/pnas.96.23.13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schlessinger J. New roles for Src kinases in control of cell survival and angiogenesis. Cell. 2000;100:293–296. doi: 10.1016/s0092-8674(00)80664-9. [DOI] [PubMed] [Google Scholar]